Abstract
Glycation of proteins or other biomolecules and their further long-term degradation result in the formation of advanced glycation end products, AGEs.
AGEs and other ligands interact with their receptors, RAGEs, localized to a variety of tissues, but mainly in endothelium and vascular wall cells. This interaction triggers diverse signaling pathways that converge on the activation of NF-κB and the initiation of a local inflammatory reaction that, when prolonged, results in dysfunctional features.
Preeclampsia is a serious vascular disorder centred at the placenta–uterine interface, the placental bed, but the condition extends to the mother´s circulation. RAGEs have notorious expression in the placental bed tissues along pregnancy but, in addition, RAGEs and their ligands are expressed in the fetal membranes and are found in the amniotic fluid and the mother´s serum. Disorders complicating pregnancies and having an important vascular involvement, as preeclampsia and diabetes mellitus, have additional enhanced AGE/RAGE expression variation. This indicates that for their assessment, the assay of RAGEs or their ligands may become useful diagnostic or prognostic procedures.
Introduction
Advanced glycation end products, AGEs, result from the reaction of carbonyls from reducing sugars, as glucose, and other glycating agents with amino groups present in proteins, or with other biomolecules as phospholipids and nucleic acids, in a long-term chemical modification that is generally referred as the Maillard reaction [Citation1,Citation2]. Because it is nonenzymatic, AGEs may be found both inside living cells, in the extracellular matrix and in stored biological products such as foods.
AGEs formation through the Maillard reaction occurs in successive steps. Usually, it starts with glucose interaction with amino acid residues, mainly arginine or lysine forming Schiff bases. The compound is unstable and so this is a reversible step in case the local glucose concentration falls. However, the continued elevated glucose level and an intramolecular rearrangement may result in the formation of fructosamine and other “Amadori products” [Citation1] as hemoglobin A1C, (glycated), a frequently used clinical indicator of exposure to glucose.
During “Amadori products” rearrangements, other carbonyl containing reactive compounds, as the endogenously formed oxoaldehydes, including glyoxal, 3-deoxyglucosone, and methylglyoxal take part in the process [Citation1]. These are very important dicarbonyl intermediates produced in monocytes and neutrophils when myeloperoxidase and NADPH oxidase (NOX)-derived reactive oxygen species (ROS) oxidize target intracellular amino acids [Citation3]. Such intermediates may also result from the continued generation of mitochondria ROS acting upon polyunsaturated fatty acids or sugars, as was verified in endothelial cells grown in glucose-enriched culture media [Citation4]. Over time, their local increase and reactivity, as well as fructosamine degradation, lead to additional interaction with amino, sulfhydryl, and guanidine functional groups of proteins; moreover, through further oxidation and dehydration, there is an accumulation of negative charges and the establishment of crosslinks between reacting molecules, ending-up in chemically stable, brownish, frequently fluorescent products, the AGEs [Citation5]. The process of AGE formation is not limited to Maillard reactions because additional pathways are involved that may result in the formation of more carbonyl-containing reactive compounds and further AGEs synthesis [Citation3,Citation6].
The variety of AGEs is not surprising in view of the diversity of available pathways leading to them. The variation of their levels in samples from different human sources may also be high; for example, the red blood cell concentration of methylglyoxal and deoxyglucosone-derived hydroimidazoles is a few nmol of glycated/nonglycated residue, whereas it may be 100-fold less for Nϵ-carboxymethyl-lysine (CML); in plasma, the difference is much less-pronounced [Citation7]. The diversity also accounts for their chemical and physical characteristics: some establish cross links, as pentosidine, others do not, as the glyoxal-lysine derivative CML; some may fluoresce, as pentosidine and argpyrimidine, whereas others do not, as CML again.
AGE–RAGE interaction
Most AGE effects on cells and tissues are a consequence of the transduction process activated following AGEs interaction with its receptor (RAGE) that belongs to the immunoglobulin superfamily of receptors [Citation8]. Its encoding gene is localized to the chromosome 6 close to the Classes II and III of major histocompatibility complex [Citation9] and, interestingly, the RAGE promoter includes a sequence with binding motifs for NF-κB, thus making of this transcription factor an important regulator of RAGE [Citation10]. Its expression was noticed at the surface of various cells as endothelial cells, pericytes, monocytes, macrophages, and smooth muscle cells [Citation11], syncytio and cytotrophoblast, amnion and choridecidua (see further ahead) and even amyloid rich tissues [Citation12].
RAGE is a membrane receptor that comprises extracellular, transmembrane and intracellular regions. The 332 amino acid residues of the extracellular N-terminal region have domains that include one V (variable) immunoglobulin like domain and two “C”, constant (C1 and C2) domains. The V and C1 domains, beyond being structurally connected, appear to function together; in fact, on account of their richness in lysine and arginine residues, they possess positive charges that allow a suitable electrostatic interaction with the usually negatively charged, cross-link rich, AGEs. In addition to the short single transmembrane domain, the RAGE has an intracellular domain of 43 residues that is crucial for the transductive process to start, much like any other major receptor family variety, as the G-protein-coupled receptors or the tyrosine kinase receptors.
There is not one single isoform of this receptor but, instead, various isoforms are generated by alternative splicing and proteolytic cleavage. Beyond the full length isoform just described (RAGE), there is a membrane-bound N-terminal-truncated isoform and a C-truncated, endogenous secreted isoform, the esRAGE [Citation13]. Moreover, there is an additional circulating peptide, the cRAGE, resulting from the proteolytic cleavage of RAGE extracellular domain [Citation14]; these soluble compounds are frequently assayed together and referred as sRAGE. It is unknown why these circulating forms of RAGE are present, but they may function as decoy receptors because they are able to interact with the ligands and reduce their concentration, but unable to trigger the transductive cascade thereafter [Citation15].
RAGE oligomerizes constitutively at the cell membrane [Citation16]. It is a multiligand receptor and therefore the interaction is not exclusive for AGEs [Citation15,Citation17], although these were their ligands to be first recognized (). Moreover, it is the variability of the V region of these extracellular domains that provides for the interaction with so different AGE compounds to take place, supporting the view that the recognition relies much more in chemical pattern recognition than in ligand-specific properties. In this setting, RAGEs also bind to, and are activated by the S100 protein family that includes more than 20 members of small acidic proteins with different functions both intracellular and extracellular, and having disturbed activity in degenerative, neoplastic, and inflammatory conditions [Citation18]. High mobility group protein box 1 (HMGB1), another ligand, is either released from necrotic cells or secreted by leukocytes; through its acidic-rich residues situated at its C-terminal region, it interacts with RAGE and leads to additional attraction of inflammatory cells [Citation19]; due to its intervention in monitoring tissue injury or the presence of inflammation mediators, it was recognized as a “danger signal” [Citation20]. Beta2 integrin Mac-1, a surface receptor on macrophage is an additional RAGE ligand that, in concert with other ligands, appears to enhance leukocyte recruitment [Citation15]. Finally, RAGE also displays high affinity with the amyloid β protein when aggregated in fibrils, whereas, interestingly, no significant interactions occur with the disaggregated peptide [Citation21].
Figure 1. AGE–RAGE interaction. The transmembrane receptor RAGE oligomerizes to interact with AGEs and other ligands.
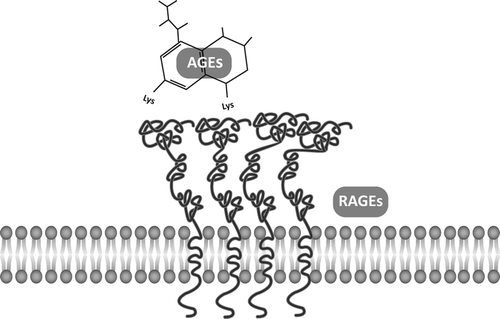
RAGEs are multiligand receptors, but AGEs do not interact exclusively with RAGEs. When bound to RAGEs, AGEs elicit a transductive response that triggers a number of different signaling pathways. In contrast, AGEs binding to other, non-RAGE proteins appear to result in AGEs clearance from the circulation, a reason why such proteins are considered as part of an AGE detoxification system [Citation22,Citation23]. Nevertheless, those proteins have been recognized as receptors. They include the AGE-R1 receptor (oligosaccharyl transferase-48), a type 1 transmembrane 48 kDa protein having an extracellular N- terminal domain, the AGE-R2, another 80–90 kDa transmembrane protein, and the AGE-R3 (galectin-3) receptor, a 32-kDa protein with affinity to AGEs through its C-terminal domain, mainly localized in the cytoplasm and nuclei, but was recognized at the surface of macrophages too [Citation24]. Additional factors with high affinity to AGEs include the macrophage scavenger receptors Class A ScR-II and the Class B CD-36 [Citation23], and the lectin-like oxidized LDL receptor 1, LOX-1, another transmembrane protein [Citation25].
AGEs in aging and diseases, in a brief overview
The diversity of RAGE ligands contrasts with the usual specificity of interaction of a receptor with its ligand. In turn, although centering RAGEs in vascular disorders, such diversity extends the role of AGEs to other conditions as inflammatory, degenerative, and neoplastic, as well as to the aging process itself.
AGEs levels increase along aging, even in healthy humans. Compared to younger, either serum CML and methylglyoxal derivatives were found elevated in older men and women [Citation26]; moreover, CML immunocytochemical detection in a variety of tissues from apparently healthy humans at different ages, revealed a clear age-related enhanced deposition; this observation included the renal tissue where, in addition to aging, diabetes promotes enhanced labeling intensity [Citation27]. Other age-related conditions as reduced muscular grip was found more severe when serum CML levels from aged individuals were higher [Citation28], suggesting a relation between specific AGEs and increased disability.
AGEs are also important contributors to disorders, frequently indistinguishable from the aging process, itself a major risk factor for disease establishment. Indeed, AGEs are not inert compounds and, apart from the receptor mediated effects, glycation is functionally disturbing for proteins [Citation29]. However, while the alterations of biological properties of glycated or AGE-modified proteins may result from changes in their secondary, tertiary, or quaternary conformation, these have not been universally established and likely depend on the protein considered [Citation29].
As the existence of a long-term, sustained, hyperglycemic environment is very important for enhanced AGEs formation, it is not surprising that the major medical condition in which they are implicated is diabetes. Yet, beyond hyperglycemia, the dyslipidemia and the increased local oxidative stress, frequently associated with diabetes, all directly promote the formation of AGEs [Citation30] and as a consequence, they are now established as outstanding factors in the pathogenesis of diabetic vascular complications.
AGEs rate of increase in serum is higher in diabetic patients compared to that of nondiabetics [Citation31] and appears to antedate their macro- and microvascular complications. AGEs contributory mechanism for atherosclerosis progression includes an augmented uptake of oxidized low-density lipoproteins (LDL) particles by vessel wall macrophages [Citation32], a condition enhanced in diabetes, as was experimentally evidenced [Citation33]. In fact, euglycemic mice treated with sRAGE, had an improvement in atherosclerotic lesions number and complexity compared to controls, but the difference was larger in streptozotocin-induced diabetic animals [Citation33]. Moreover, AGEs were noticed in atheroma lesions of coronary [Citation34] and carotid arteries [Citation35]. They were found to increase the aortic stiffness [Citation36,Citation37] and to associate with enhanced coronary disease incidence and mortality [Citation36]. In general, the increase in AGEs serum levels relates with serious cardiovascular disorders, independently of the traditional cardiovascular risk factors [Citation38]. To this are added the effects of AGEs on the development of heart failure, either indirectly, through the crosslinks created on extracellular matrix proteins, or directly, via the interaction with heart vessels or myocardial AGE receptors, thus promoting its progression and severity [Citation39].
The association of AGEs and diabetic micro-angiopathy was also recognized. One important target is the kidney, where glomerular and tubulointerstitial AGEs deposition elicits enhanced generation of ROS, more local AGEs formation and further difficulty in AGEs clearance [Citation40]. In the retina, another target, an age-related and glaucoma-related increase in AGEs was noticed as evidenced by major deposition of the compounds in and around vessel wall cells and inside Müller cells before their functional impairment [Citation41].
The relationship of AGEs formation and hypertension establishment is less clear than the AGEs effects on the microcirculation of diabetic patients; indeed, no relevant association with essential hypertension has been observed in the AGE–RAGE axis [Citation42], likely to reflect the fact that hypertension by itself is not a structural disease in contrast with the other, above mentioned conditions.
Other disorders that have been related to enhanced AGEs circulation in serum or RAGE activation include systemic lupus erythematous [Citation43], rheumatoid arthritis [Citation44], Alzheimer disease [Citation45], and even neoplasia [Citation46,Citation47]. In recent times, preeclampsia was included as another disorder for which AGEs and RAGE are intervenients, as will be addressed below.
Placentation and regulation
Preeclampsia (PE), the characteristic hypertensive disorder of pregnancy, is potentially a very serious condition. It is characterized by defined values of hypertension and proteinuria, appearing from the 20th week of pregnancy onwards and encompasses a considerable range of clinical manifestations [Citation48]. It is estimated that PE affects approximately 5% of all pregnancies and, due to the ominous consequences that arise if left untreated, it is still a common cause of maternal and fetal morbidity and mortality [Citation48]. Therefore, not only its clinical recognition is necessary for appropriate treatment, but also the knowledge of its underlying pathogenesis will help to devise strategies to prevent many of the unwanted outcomes.
Unfortunately, despite much attention and multitude of studies on the subject, no single cause for PE was recognized. Still, it is a disorder with a definite vascular involvement that appears to result from a condition referred as defective deep placentation, a process centred on the placental bed that includes the decidua, that is, the pregnancy-modified endometrium, and the adjacent myometrium. As placental bed is the interface between mother and fetal circulation, its proper establishment is critical for a successful pregnancy outcome.
Although still lacking in many details, the placental bed changes have been the subject of important research along decades. In normal conditions, for an adequate supply of nutrients and oxygen to the fetus, the terminal, narrow bore, uterine spiral arteries undergo remodeling into larger capacity vessels, much as a result of the intervention of the placenta-derived trophoblast cells. Their decisive role has been described as starting with a discrete presence of interstitial cytotrophoblasts in early pregnancy decidua that is later followed by their invasion of the spiral artery wall, deep into the myometrium, to replace apoptotic smooth muscle cells and endothelium destruction. This, dual wave, trophoblast-centred view has been challenged because features of spiral artery invasion may be seen as early as the 12th week of pregnancy [Citation49]. Moreover, remodeling mediated by decidua cells, in the absence of trophoblasts, was reported [Citation49,Citation50].
For the human placenta, it was estimated that in normal conditions, around 100 spiral arteries undergo this transformation process [Citation51]. However, when the spiral artery invasion is deficient for reasons related to the known risk factors for PE [Citation52], it is said to exist a defective placentation and the establishment of large capacity vessels is hindered for most of them. The local blood flow becomes limited, the delicate interaction at the interface ruptures, and ischemic placental phenomena may ensue, resulting in placental thrombosis and infarction [Citation53]. Apart from PE, in other major obstetrical disorders such as intrauterine growth restriction (IUGR), preterm labor with or without intact membranes, abruptio placenta, and mid-trimester abortion defective placentation was evidenced [Citation51].
The structural and functional integrity of the vascular system, including the placental bed´s, is mastered by the endothelium, the single-layered endothelial cells that line the whole system. Beyond the total amount of cells, the sort of compounds that they produce is impressive [Citation54], particularly because they include a number of vasodilators and vasoconstrictors that need a fine-tuned intracellular balance.
To such balance, an important regulatory role is exerted by ROS. In fact, when they are produced in small amounts, ROS are able to elicit a wide spectrum of responses/ signaling-pathways to include the JAK/STAT, PI3K/AKT, p38, JNK/SAPK, and Ras/MEK/ERK1/2 pathways [Citation55]. This effect is also noticed upon RAGE interaction with AGEs or other ligands [Citation56].
A considerable production of ROS with physiological importance follows NADPH oxidase activation. The distribution of NOX isoforms in tissues and a major localization in vessel wall cells and circulating phagocytes [Citation57] emphasize their value for local signaling and for the body wide consequences of their excessive activation. In fact, the endothelium expresses a variety of NOX isoforms [Citation57], considered the main source of regulatory ROS in the vasculature [Citation58]. In the placental bed, however, other cells were recognized as employing ROS as signaling molecules. These include the smooth muscle cells of myometrium, the endometrial decidualized stromal cells, and the syncytiotrophoblasts where NOX1, NOX4, and NOX5 isoforms are expressed [Citation57].
The main ROS derived from NOX activity is the superoxide anion. However, it has limited ability to diffuse across membranes, is rapidly dismutated by superoxide dismutases, and converted into the more diffusible hydrogen peroxide (H2O2), which appears to be an even more important signaling molecule [Citation59]. In the spiral artery endothelium and syncytiotrophoblast, H2O2 promotes nitric oxide (NO) synthesis by NO synthase whose consequence is local vasodilation and endothelium protection [Citation60,Citation61].
The signaling pathways activated upon ROS production depends on the cell type and the ligand employed. For example, interaction of glycated albumin and RAGEs in pulmonary artery smooth muscle cells results in ROS production, oxidation of Cys118 residue of Ras protein (formerly p21ras) and the Ras/MEK/ERK1/2 pathway activation [Citation62], whereas by a similar challenge, mesangial cells activate the p38 pathway [Citation63]. In addition, when HMGB1 interacts with RAGE in glioma cells, there is an activation of the JNK/SAPK and p38 pathways [Citation46], whereas the interaction in endothelial cells with S100/calgranulins resulted in JNK activation [Citation64]. Moreover, neutrophils from diabetic patients exhibited enhanced PKC activity and digliceride and p47phox (a NOX regulator) expression [Citation65], while the same cells, when challenged by the RAGE ligand S100B, evidenced enhanced activity of the ERK1/2 pathway [Citation66] in a mechanism recruiting phospholipase A2 (PLA2)-derived arachidonic acid [Citation67]. Mast cell, which similarly to the neutrophil has a profound involvement in the inflammatory reaction, showed enhanced ROS synthesis following activated G protein-related pathway and enhanced intracellular calcium availability, upon AGEs activated RAGEs [Citation68]. To conclude, JAK/STAT was found recruited upon RAGE activation in fibroblasts that resulted in extracellular matrix components synthesis and secretion [Citation69].
Therefore, the variety of pathways triggered upon RAGE activation is large, even within cell types having important roles in inflammation. The regulation at the endothelial cell level is thus very sensitive. As central players in vascular function, endothelial cells are also a key target in derangements when ROS production becomes excessive.
Deregulation in preeclampsia
Similarly to other disorders with important vascular involvement, there seems to be little doubt that PE is an endothelial disease [Citation70] that is centered in placental bed and extends to maternal circulation. The details of the underlying mechanism or the condition that triggers PE are unknown but it likely starts with an imbalance in the local regulation of blood perfusion that affects the endothelium.
In PE, episodes of ischemia and reperfusion occurring in the placenta [Citation71] and ill response to vasodilation [Citation72] lead to a tendency for local hypoxia. Moreover, it was proposed that late in pregnancy, enhanced oxidized LDL particles uptake by endothelial cells or increased sensitivity to angiotensin II pressor effects or vasoconstriction and vasodilation unbalance due to tumor necrosis factor alpha excess, impinges on endothelial cells to cause excessive production and release of cytokines, adhesion molecules and chemo-attractants for leukocytes, and macrophages that boost the inflammatory process [Citation73].
These conditions favor additional local production of ROS through enhanced NOX activity, increased NF-κB expression, release of superoxide anion and its effect on NO, generating peroxynitrite; the rise in the local pro-oxidant status and the continued oxidative stress insult favor placental bed cell apoptosis or necrosis, and a deficient local remodeling. In addition, it is thought that locally produced cell debris enter the maternal circulation and extend the process to cause generalized endothelial disturbance, more vasoactive mediators release, more inflammation, and additional maternal endothelial dysfunction [Citation74]. The variety of organs that may be seriously affected by PE (brain, eye, liver, heart, kidney, bone marrow, and lung) results in a systemic condition with a diversity of clinical manifestations but having a common vascular involvement that is centered on endothelial functional impairment.
It is interesting that the pathophysiology of other vascular disorders has important similarities to PE. The pathological basis of most cardiovascular disorders is atherosclerosis for which aging, diabetes, hypertension, and obesity are well-known risk factors. A fundamental vascular change that antedates atherogenesis is endothelial dysfunction too, itself consequent to reduction of local availability of NO and diminished vascular relaxation [Citation75]. This is likely consequent to the conversion of hydrogen peroxide into the hydroxyl anion, another ROS that reacts strongly with most biomolecules. Therefore, while a small production of ROS is useful, a sustainable or enhanced ROS production is able to shift their signaling properties into molecular and cellular damaging effects [Citation55].
The interaction of AGEs with their receptors appears to be a contributing condition for PE triggering, as their continuous activation favors the establishment of various disorders [Citation76]. In endothelial cells [Citation77] or vascular smooth muscle cells [Citation62] the interaction of RAGE with AGEs or other RAGE ligands [Citation78] promotes oxidative stressful conditions, eventually through NOX activation [Citation79], that together with the triggering of ERK1/2-MAPK pathway, the PI3K-AKT or the p38-SAPK/JNK pathways converge on NF-κB transcription factor activation () and nuclear translocation, where it switches on a number of genes with a variety of effects on tissues [Citation76,Citation80,Citation81]. These include local inflammation, vascular cell proliferation or apoptosis, extracellular matrix turnover, and accumulation of AGE adducts [Citation82].
Figure 2. AGE–RAGE interaction recruits NADPH oxidase, enhances reactive oxygen species production, and initiates a transductive cascade that may follow different pathways. They converge on the activation of the transcription factor NF-κB that promotes the expression of a variety of genes involved in inflammation, vascular cell proliferation or apoptosis and extracellular matrix turnover.
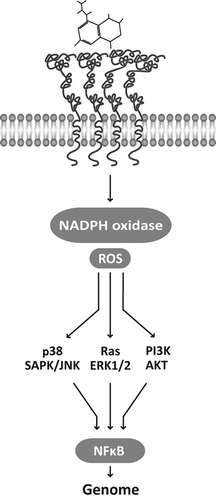
In the prolongation of the process, NF-κB activation appears to feed back further RAGE expression through the NF-κB binding motifs of RAGE promoter, and convert the initial inflammatory reaction into a sustained chronic state [Citation15] with tissue derangement and a clinically expressive vascular disorder.
AGEs and RAGE in pregnancy
The study of the involvement of AGEs and other RAGE ligands and their receptors in normal pregnancy and pregnancy complicated by PE is a fairly recent issue. However, it already attracted considerable attention because it appears that some of the factors involved exhibit a biomarker behaviour.
RAGE in placental bed and placenta
RAGE receptor expression was recognized in most tissues related to the placental bed. Important labelling intensity was detected in decidua [Citation83–85] and in myometrium smooth muscle and endothelium cells, which further showed enhanced expression in PE [Citation86].
In placenta, RAGEs were evidenced in syncytiotrophoblat and cytotrophoblast of normal pregnancy at term [Citation84,Citation85,Citation87], but RAGE expression appears to start earlier and becomes more intense in the succeeding trimesters [Citation88]. In PE, however, the reports are conflicting. In fact, Holmlund et al. did not report significant differences when samples from normal and PE pregnancies at term or near term were compared [Citation83], but in other studies enhanced RAGE levels in placental extracts from PE patients were reported [Citation85,Citation87,Citation89].
Available data thus indicate that in normal pregnancies, RAGE is found at a considerable amount in tissues related to the placental bed. Moreover, its expression may increase in PE, supporting their role in placental disorders establishment.
Soluble RAGE
In pregnancy, the soluble components of RAGE, including the esRAGE and cRAGE, can be assessed as sRAGE. Compared to nonpregnant women, sRAGE serum level was found reduced in pregnancy [Citation90,Citation91], but its course along it produced conflicting results. Either an increase toward the mid-pregnancy and a decrease thereafter [Citation30,Citation90,Citation92] as well as, inversely, a mid-gestation through [Citation85], were described. In PE, the scarcity of data does not allow a sound conclusion regarding its variation, but sRAGE appears to be increased [Citation85,Citation90,Citation92,Citation93].
In contrast, sRAGE was found reduced in preterm delivery [Citation92] and the level may have prognostic value. In fact, among the preterm deliveries, it was recognized that in the event it resulted in neonatal sepsis, maternal sRAGE serum levels at the admission to the hospital were lower than in those cases where preterm did not result in sepsis [Citation94].
Therefore, both in situ and in blood, RAGEs are sensitive to PE and respond by enhancing its expression.
AGEs and other RAGE ligands
These compounds follow closely the expression of the receptor as they may be detected in placental bed tissues or circulating in the blood as well. AGEs were noticed in the syncytiotrophoblast, endothelial cells, and macrophages of placenta in normal pregnancies; they exhibited an enhanced intensity of labelling in those complicated by PE [Citation87], but not all data agree [Citation95].
PE also appears to promote HMGB1 expression. In fact, by immunocytochemistry and in situ hybridization, HMGB1 expression was localized to the syncytiotrophoblast as well as the decidua and tended to be higher in PE patients compared to normal pregnancies [Citation83]. Serum HMGB1, considered a “danger signal” molecule, had similar levels along the pregnancy [Citation96,Citation97] but, in severe PE it was found significantly increased [Citation96].
Serum CML, a major monolysyl adduct RAGE ligand, did not evidence changes along normal pregnancy or when it complicated with severe PE [Citation96], but the finding is not consensual as enhanced levels were found in PE patients [Citation87]. S100A12, a member of the S100 protein family, evidenced a significant serum decrement along the normal pregnancy, but in PE it raised to high and significant levels [Citation96]. Its expression in the placental bed is rather small in normal conditions but became intense in inflammatory cells in chorioamnionitis [Citation84].
Therefore, these data suggest that while uneventful pregnancies may course with smaller variations in these RAGE ligands, a systemic condition as PE associates with their upward shift, similarly with RAGEs.
Diabetes in pregnancy
Diabetes, a condition implicated in enhanced AGE–RAGE interaction consequences, also affects adversely the pregnancy outcome. AGEs, as measured by skin auto-fluorescence, increase along pregnancies of diabetic women, in contrast with normal pregnancies [Citation98] and may remain high despite glucose control [Citation99]. AGEs were found elevated in plasma of diabetic women [Citation95,Citation100], together with sRAGE levels reduction [Citation100].
Diabetic pregnant women also exhibited higher serum levels of other AGEs, as CML and methylglyoxal-derived hydroimidazolone, when compared to that of non-diabetic patients, a difference that was more evident in those women who later developed PE [Citation30]. This may reflect their higher sensitivity to serum glucose levels as it was noticed that an acute loading of 50 g glucose, as frequently used in the screening of gestational diabetes, results in enhanced levels of methylglyoxal and CML [Citation101]. Interestingly, most of the time, sRAGE coursed with lower levels in diabetic women who became pregnant, especially in those who developed PE, indicating an association between low sRAGE and PE development [Citation30].
Amniotic fluid and membranes
The main structural changes accompanying PE are centred at the placenta and decidua vascular interface. However, PE may have other targets, including the fetus as already indicated, for which RAGE and RAGE ligand interaction may be a contributor factor.
RAGE is expressed in amniotic membranes [Citation83–85], particularly under PE [Citation85]. By immunocytochemistry and in situ hybridization, HMGB1 gene expression was also localized there and the labelling was more intense in PE patients [Citation83].
The enhanced expression of RAGE in PE may be related to the high levels of soluble sRAGE found in amniotic fluid, higher than mother or infant's serum [Citation85]. The scarcity of data does not allow a sound conclusion on the biological meaning of this finding but they indicate that high sRAGE levels are important to afford some kind of protection. In chorioamnionitis at term, an intense inflammatory and infectious condition, amniotic fluid sRAGE is decreased, in contrast to the enhanced levels of the alarmin HMGB1 [Citation102]. In addition, in positive amniotic fluid cultures, sRAGE amniotic fluid was found reduced whereas RAGE ligand S100A12 level was elevated [Citation84]. Moreover, it was recognized that low cord blood sRAGE level and high S100B ligand concentration in preterm infants associated with enhanced risk to develop cerebral palsy or die within a year [Citation103]. That is, sRAGE, the decoy receptor of RAGE may work as a neutralizing molecule of RAGE ligands and prevent their interaction with the active, canonical RAGE, thus precluding the start of an inflammatory reaction with serious implications.
Future directions in preeclampsia research
In modern societies, the fetal and maternal mortality and morbidity reduced substantially due to improved obstetrical care. However, preeclampsia, a vascular disorder centered at the placental bed, is still a condition of much concern. Its pathophysiology has similarities to other frequent vascular entities that course with local inflammation. Their meeting point appears to be the endothelial cell and its dysfunction long before the disease is clinically diagnosed ().
Figure 3. In the course of normal pregnancy, the trophoblast cell invasion of the uterine spiral arteries results in the replacement of their endothelium and wall cells, and consequent change of narrow arteries into large bore capacitance vessels. Adequate functioning of the new endothelium by appropriate signaling and the local microcirculation assures normal blood supply to the placental bed throughout the pregnancy.
In preeclampsia (PE), there is a deficient trophoblast invasion of the spiral arteries and reduced blood perfusion. Hypoxic conditions lead to endothelial cell dysfunction and frequently are a cause of small vessel occlusion that leads to further local hypoxia, inflammatory reaction, and arrival of monocytes and neutrophils that together promote oxidative stress. Its persistence amplifies the local condition into a systemic endothelial dysfunction and to enhanced AGEs production, which feeds additional endothelium dysfunction and becomes clinically evident PE.
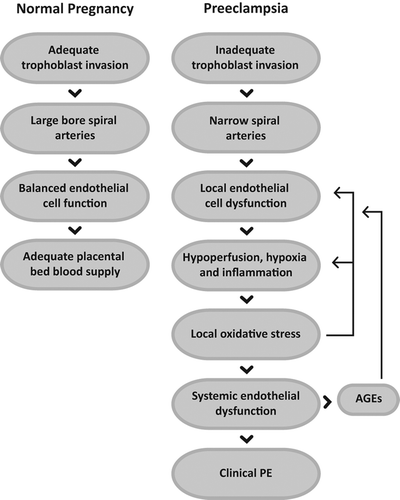
AGEs are ligands that upon interacting with cognate receptors elicit a local inflammatory reaction that enhances ROS production, attracts cytokine secretory cells, and prolong the inflammation. AGE receptors are well expressed at the placental bed where they initiate a similar reaction when activated in PE although exhibiting a different intensity scale. In fact, while their role in diabetic complications accompanies a slow process of vascular derangement, in PE it associates to a fast-moving disorder that extends to the mother´s circulation and has a grim outcome if untreated. It is most interesting that the treatment of PE may be as simple as to bring pregnancy to the end, in other words, to interrupt the placenta–uterine interaction.
RAGE ligands enhanced expression in PE may alert for impending vascular inflammatory disorder. Moreover, the possibility that AGEs or other RAGE ligands have diagnostic or predictive value for pregnancy outcome, cannot be neglected. Recent data suggest so, foreseeing more evidence in the near future. Acting at the very center of the disease, the endothelium of the placental bed, they are even able to provide an answer to the so long quest for the cause of PE.
Acknowledgements
We thank Sérgio Evangelista, Laboratório de Iconografia, Faculdade de Medicina da Universidade do Porto, for illustrations.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
This work was supported in part by “Prémio Crioestaminal” (Federação das Sociedades Portuguesas de Ginecologia e Obstetrícia, Portugal) and by COST Action CM1001: Chemistry of non-enzymatic protein modification—modulation of protein structure and function (European Union).
L. Matos and E. Silva were supported by Fundação para a Ciência e Tecnologia (FCT), Portugal, grants SFRH/BD/61820/2009 and SFRH/BPD/72536/2010 respectively.
References
- Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 1999;344: 109–116.
- Thornalley PJ. The enzymatic defence against glycation in health, disease and therapeutics: a symposium to examine the concept. Biochem Soc Trans 2003;31:1341–1342.
- Uribarri J, Tuttle KR. Advanced glycation end products and nephrotoxicity of high-protein diets. Clin J Am Soc Nephrol 2006;1:1293–1299.
- Nishikawa T, Edelstein D, Du XL, Yamagishi S, Matsumura T, Kaneda Y, et al. Normalizing mitochondrial superoxide production blocks three pathways of hyperglycaemic damage. Nature 2000;404:787–790.
- Goldin A, Beckman JA, Schmidt AM, Creager MA. Advanced glycation end products: sparking the development of diabetic vascular injury. Circulation 2006;114:597–605.
- Del Turco S, Basta G. An update on advanced glycation endproducts and atherosclerosis. Biofactors 2012;38: 266–274.
- Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem J 2003;375:581–592.
- Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 1992;267:14998–5004.
- Sugaya K, Fukagawa T, Matsumoto K, Mita K, Takahashi E, Ando A, et al. Three genes in the human MHC class III region near the junction with the class II: gene for receptor of advanced glycosylation end products, PBX2 homeobox gene and a notch homolog, human counterpart of mouse mammary tumor gene int-3. Genomics 1994;23:408–419.
- Li J, Schmidt AM. Characterization and functional analysis of the promoter of RAGE, the receptor for advanced glycation end products. J Biol Chem 1997;272:16498–16506.
- Brett J, Schmidt AM, Yan SD, Zou YS, Weidman E, Pinsky D, et al. Survey of the distribution of a newly characterized receptor for advanced glycation end products in tissues. Am J Pathol 1993;143:1699–1712.
- Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer's disease. Nature 1996;382:685–691.
- Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J 2003; 370:1097–1109.
- Raucci A, Cugusi S, Antonelli A, Barabino SM, Monti L, Bierhaus A, et al. A soluble form of the receptor for advanced glycation endproducts (RAGE) is produced by proteolytic cleavage of the membrane-bound form by the sheddase a disintegrin and metalloprotease 10 (ADAM10). FASEB J 2008;22:3716–3727.
- Fritz G. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci 2011;36:625–632.
- Xie J, Reverdatto S, Frolov A, Hoffmann R, Burz DS, Shekhtman A. Structural basis for pattern recognition by the receptor for advanced glycation end products (RAGE). J Biol Chem 2008;283:27255–27269.
- Hegab Z, Gibbons S, Neyses L, Mamas MA. Role of advanced glycation end products in cardiovascular disease. World J Cardiol 2012;4:90–102.
- Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature). Biochem Biophys Res Commun 2004;322:1111–1122.
- Bianchi ME, Manfredi AA. High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev 2007;220:35–46.
- Zhang S, Zhong J, Yang P, Gong F, Wang CY. HMGB1, an innate alarmin, in the pathogenesis of type 1 diabetes. Int J Clin Exp Pathol 2009;3:24–38.
- Haupt C, Bereza M, Kumar ST, Kieninger B, Morgado I, Hortschansky P, et al. Pattern recognition with a fibril- specific antibody fragment reveals the surface variability of natural amyloid fibrils. J Mol Biol 2011;408:529–540.
- Vlassara H. The AGE-receptor in the pathogenesis of diabetic complications. Diabetes Metab Res Rev 2001;17: 436–443.
- Busch M, Franke S, Rüster C, Wolf G. Advanced glycation end-products and the kidney. Eur J Clin Invest 2010;40: 742–755.
- Vlassara H, Li YM, Imani F, Wojciechowicz D, Yang Z, Liu FT, Cerami A. Identification of galectin-3 as a high- affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med 1995;1:634–646.
- Jono T, Miyazaki A, Nagai R, Sawamura T, Kitamura T, Horiuchi S. Lectin-like oxidized low density lipoprotein receptor-1 (LOX-1) serves as an endothelial receptor for advanced glycation end products (AGE). FEBS Lett 2002;511:170–174.
- Uribarri J, Cai W, Peppa M, Goodman S, Ferrucci L, Striker G, Vlassara H. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci 2007;62:427–433.
- Schleicher ED, Wagner E, Nerlich AG. Increased accumulation of the glycoxidation product N(epsilon)-(carboxymethyl)lysine in human tissues in diabetes and aging. J Clin Invest 1997;99:457–468.
- Dalal M, Ferrucci L, Sun K, Beck J, Fried LP, Semba RD. Elevated serum advanced glycation end products and poor grip strength in older community-dwelling women. J Gerontol A Biol Sci Med Sci 2009;64:132–137.
- Yeboah FK, Alli I, Yaylayan VA, Yasuo K, Chowdhury SF, Purisima EO. Effect of limited solid-state glycation on the conformation of lysozyme by ESI-MSMS peptide mapping and molecular modeling. Bioconjug Chem 2004; 15:27–34.
- Yu Y, Hanssen KF, Kalyanaraman V, Chirindel A, Jenkins AJ, Nankervis AJ, et al. Reduced soluble receptor for advanced glycation end-products (sRAGE) scavenger capacity precedes pre-eclampsia in Type 1 diabetes. BJOG 2012; 119:1512–1520.
- Kilhovd BK, Berg TJ, Birkeland KI, Thorsby P, Hanssen KF. Serum levels of advanced glycation end products are increased in patients with type 2 diabetes and coronary heart disease. Diabetes Care 1999;22:1543–1548.
- Zieman S, Kass D. Advanced glycation end product cross-linking: pathophysiologic role and therapeutic target in cardiovascular disease. Congest Heart Fail 2004;10:144–149; quiz 150–151.
- Park L, Raman KG, Lee KJ, Lu Y, Ferran LJ, Chow WS, et al. Suppression of accelerated diabetic atherosclerosis by the soluble receptor for advanced glycation endproducts. Nat Med 1998;4:1025–1031.
- Obayashi H, Nakano K, Shigeta H, Yamaguchi M, Yoshimori K, Fukui M, et al. Formation of crossline as a fluorescent advanced glycation end product in vitro and in vivo. Biochem Biophys Res Commun 1996;226:37–41.
- Cipollone F, Iezzi A, Fazia M, Zucchelli M, Pini B, Cuccurullo C, et al. The receptor RAGE as a progression factor amplifying arachidonate-dependent inflammatory and proteolytic response in human atherosclerotic plaques: role of glycemic control. Circulation 2003;108:1070–1077.
- Yoshida N, Okumura K, Aso Y. High serum pentosidine concentrations are associated with increased arterial stiffness and thickness in patients with type 2 diabetes. Metabolism 2005;54:345–350.
- Sims TJ, Rasmussen LM, Oxlund H, Bailey AJ. The role of glycation cross-links in diabetic vascular stiffening. Diabetologia 1996;39:946–951.
- Nin JW, Jorsal A, Ferreira I, Schalkwijk CG, Prins MH, Parving HH, et al. Higher plasma levels of advanced glycation end products are associated with incident cardiovascular disease and all-cause mortality in type 1 diabetes: a 12-year follow-up study. Diabetes Care 2011;34:442–447.
- Hartog JW, Voors AA, Bakker SJ, Smit AJ, van Veldhuisen DJ. Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail 2007;9:1146–1155.
- Fukami K, Yamagishi S, Ueda S, Okuda S. Role of AGEs in diabetic nephropathy. Curr Pharm Des 2008;14: 946–952.
- Milne R, Brownstein S. Advanced glycation end products and diabetic retinopathy. Amino Acids 2011;44:1397–1407.
- Baumann M. Role of advanced glycation end products in hypertension and cardiovascular risk: human studies. J Am Soc Hypertens 2012;6:427–435.
- de Leeuw K, Graaff R, de Vries R, Dullaart RP, Smit AJ, Kallenberg CG, Bijl M. Accumulation of advanced glycation endproducts in patients with systemic lupus erythematosus. Rheumatology (Oxford) 2007;46:1551–1556.
- de Groot L, Posthumus MD, Kallenberg CG, Bijl M. Risk factors and early detection of atherosclerosis in rheumatoid arthritis. Eur J Clin Invest 2010;40:835–842.
- Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, Münch G. Advanced glycation endproducts and their receptor RAGE in Alzheimer's disease. Neurobiol Aging 2011;32:763–777.
- Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, et al. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature 2000;405:354–360.
- Rojas A, Figueroa H, Morales E. Fueling inflammation at tumor microenvironment: the role of multiligand/RAGE axis. Carcinogenesis 2010;31:334–341.
- Sibai BM, Stella CL. Diagnosis and management of atypical preeclampsia-eclampsia. Am J Obstet Gynecol 2009;200: 481.e1–7.
- Smith SD, Dunk CE, Aplin JD, Harris LK, Jones RL. Evidence for immune cell involvement in decidual spiral arteriole remodeling in early human pregnancy. Am J Pathol 2009;174:1959–1971.
- Hazan AD, Smith SD, Jones RL, Whittle W, Lye SJ, Dunk CE. Vascular-leukocyte interactions: mechanisms of human decidual spiral artery remodeling in vitro. Am J Pathol 2010;177:1017–1030.
- Brosens I, Pijnenborg R, Vercruysse L, Romero R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am J Obstet Gynecol 2011; 204:193–201.
- Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet 2005;365:785–799.
- Roberts DJ, Post MD. The placenta in pre-eclampsia and intrauterine growth restriction. J Clin Pathol 2008;61: 1254–1260.
- Barton M. Obesity and aging: determinants of endothelial cell dysfunction and atherosclerosis. Pflugers Arch 2010; 460:825–837.
- Martindale JL, Holbrook NJ. Cellular response to oxidative stress: signaling for suicide and survival. J Cell Physiol 2002;192:1–15.
- Sorci G, Riuzzi F, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta 2013;1833:101–109.
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev 2007;87:245–313.
- Lambeth JD. Nox enzymes, ROS, and chronic disease: an example of antagonistic pleiotropy. Free Radic Biol Med 2007;43:332–347.
- Takac I, Schröder K, Brandes RP. The Nox family of NADPH oxidases: friend or foe of the vascular system?Curr Hypertens Rep 2012;14:70–78.
- Thomas SR, Chen K, Keaney JF. Hydrogen peroxide activates endothelial nitric-oxide synthase through coordinated phosphorylation and dephosphorylation via a phosphoinositide 3-kinase-dependent signaling pathway. J Biol Chem 2002;277:6017–6024.
- Lyall F. Mechanisms regulating cytotrophoblast invasion in normal pregnancy and pre-eclampsia. Aust N Z J Obstet Gynaecol 2006;46:266–273.
- Lander HM, Tauras JM, Ogiste JS, Hori O, Moss RA, Schmidt AM. Activation of the receptor for advanced glycation end products triggers a p21(ras)-dependent mitogen-activated protein kinase pathway regulated by oxidant stress. J Biol Chem 1997;272:17810–17814.
- Chang PC, Chen TH, Chang CJ, Hou CC, Chan P, Lee HM. Advanced glycosylation end products induce inducible nitric oxide synthase (iNOS) expression via a p38 MAPK-dependent pathway. Kidney Int 2004;65:1664–1675.
- Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest 2008;118:183–194.
- Karima M, Kantarci A, Ohira T, Hasturk H, Jones VL, Nam BH, et al. Enhanced superoxide release and elevated protein kinase C activity in neutrophils from diabetic patients: association with periodontitis. J Leukoc Biol 2005;78:862–870.
- Omori K, Ohira T, Uchida Y, Ayilavarapu S, Batista EL, Yagi M, et al. Priming of neutrophil oxidative burst in diabetes requires preassembly of the NADPH oxidase. J Leukoc Biol 2008;84:292–301.
- Ayilavarapu S, Kantarci A, Fredman G, Turkoglu O, Omori K, Liu H, et al. Diabetes-induced oxidative stress is mediated by Ca2+-independent phospholipase A2 in neutrophils. J Immunol 2010;184:1507–1515.
- Sick E, Brehin S, André P, Coupin G, Landry Y, Takeda K, Gies JP. Advanced glycation end products (AGEs) activate mast cells. Br J Pharmacol 2010;161:442–455.
- Huang JS, Guh JY, Chen HC, Hung WC, Lai YH, Chuang LY. Role of receptor for advanced glycation end-product (RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen production in NRK-49F cells. J Cell Biochem 2001;81:102–113.
- Poston L. Endothelial dysfunction in pre-eclampsia. Pharmacol Rep 2006;58:69–74.
- Hung TH, Skepper JN, Burton GJ. In vitro ischemia- reperfusion injury in term human placenta as a model for oxidative stress in pathological pregnancies. Am J Pathol 2001;159:1031–1043.
- Knock GA, Poston L. Bradykinin-mediated relaxation of isolated maternal resistance arteries in normal pregnancy and preeclampsia. Am J Obstet Gynecol 1996;175:1668–1674.
- Sankaralingam S, Arenas IA, Lalu MM, Davidge ST. Preeclampsia: current understanding of the molecular basis of vascular dysfunction. Expert Rev Mol Med 2006;8:1–20.
- Sargent IL, Germain SJ, Sacks GP, Kumar S, Redman CW. Trophoblast deportation and the maternal inflammatory response in pre-eclampsia. J Reprod Immunol 2003;59: 153–160.
- Mateos-Cáceres PJ, Zamorano-León JJ, Rodríguez-Sierra P, Macaya C, López-Farré AJ. New and old mechanisms associated with hypertension in the elderly. Int J Hypertens 2012;2012:150107.
- Han SH, Kim YH, Mook-Jung I. RAGE: the beneficial and deleterious effects by diverse mechanisms of actions. Mol Cells 2011;31:91–97.
- Yan SD, Schmidt AM, Anderson GM, Zhang J, Brett J, Zou YS, et al. Enhanced cellular oxidant stress by the interaction of advanced glycation end products with their receptors/binding proteins. J Biol Chem 1994;269: 9889–9897.
- Arumugam T, Simeone DM, Schmidt AM, Logsdon CD. S100P stimulates cell proliferation and survival via receptor for activated glycation end products (RAGE). J Biol Chem 2004;279:5059–5065.
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001;280:E685–E694.
- Wautier JL, Schmidt AM. Protein glycation: a firm link to endothelial cell dysfunction. Circ Res 2004;95:233–238.
- Rojas A, Delgado-López F, González I, Pérez-Castro R, Romero J, Rojas I. The receptor for advanced glycation end-products: a complex signaling scenario for a promiscuous receptor. Cell Signal 2013;25:609–614.
- Stitt AW, Curtis TM. Diabetes-related adduct formation and retinopathy. J Ocul Biol Dis Infor 2011;4:10–18.
- Holmlund U, Wähämaa H, Bachmayer N, Bremme K, Sverremark-Ekström E, Palmblad K. The novel inflammatory cytokine high mobility group box protein 1 (HMGB1) is expressed by human term placenta. Immunology 2007; 122:430–437.
- Buhimschi IA, Zhao G, Pettker CM, Bahtiyar MO, Magloire LK, Thung S, et al. The receptor for advanced glycation end products (RAGE) system in women with intraamniotic infection and inflammation. Am J Obstet Gynecol 2007;196:181.e1–13.
- Oliver EA, Buhimschi CS, Dulay AT, Baumbusch MA, Abdel-Razeq SS, Lee SY, et al. Activation of the receptor for advanced glycation end products system in women with severe preeclampsia. J Clin Endocrinol Metab 2011; 96:689–698.
- Cooke CL, Brockelsby JC, Baker PN, Davidge ST. The receptor for advanced glycation end products (RAGE) is elevated in women with preeclampsia. Hypertens Pregnancy 2003;22:173–184.
- Chekir C, Nakatsuka M, Noguchi S, Konishi H, Kamada Y, Sasaki A, et al. Accumulation of advanced glycation end products in women with preeclampsia: possible involvement of placental oxidative and nitrative stress. Placenta 2006; 27:225–233.
- Konishi H, Nakatsuka M, Chekir C, Noguchi S, Kamada Y, Sasaki A, Hiramatsu Y. Advanced glycation end products induce secretion of chemokines and apoptosis in human first trimester trophoblasts. Hum Reprod 2004; 19:2156–2162.
- Feng C, Tao Y, Shang T, Yu M. Calprotectin, RAGE and Tᵏα in hypertensive disorders in pregnancy: expression and significance. Arch Gynecol Obstet 2011;283: 161–166.
- Kwon JH, Kim YH, Kwon JY, Park YW. Clinical significance of serum sRAGE and esRAGE in women with normal pregnancy and preeclampsia. J Perinat Med 2011;39: 507–513.
- Hájek Z, Germanová A, Koucký M, Zima T, Kopecký P, Vítkova M, et al. Detection of feto-maternal infection/ inflammation by the soluble receptor for advanced glycation end products (sRAGE): results of a pilot study. J Perinat Med 2008;36:399–404.
- Germanová A, Koucký M, Hájek Z, Parízek A, Zima T, Kalousová M. Soluble receptor for advanced glycation end products in physiological and pathological pregnancy. Clin Biochem 2010;43:442–446.
- Fasshauer M, Seeger J, Waldeyer T, Schrey S, Ebert T, Lossner U, et al. Endogenous soluble receptor for advanced glycation endproducts is increased in preeclampsia. J Hypertens 2008;26:1824–1828.
- Bastek JA, Brown AG, Foreman MN, McShea MA, Anglim LM, Adamczak JE, Elovitz MA. The soluble receptor for advanced glycation end products can prospectively identify patients at greatest risk for preterm birth. J Matern Fetal Neonatal Med 2012;25:1762–1768.
- Harsem NK, Braekke K, Torjussen T, Hanssen K, Staff AC. Advanced glycation end products in pregnancies complicated with diabetes mellitus or preeclampsia. Hypertens Pregnancy 2008;27:374–386.
- Naruse K, Sado T, Noguchi T, Tsunemi T, Yoshida S, Akasaka J, et al. Peripheral RAGE (receptor for advanced glycation endproducts)-ligands in normal pregnancy and preeclampsia: novel markers of inflammatory response. J Reprod Immunol 2012;93:69–74.
- Wang B, Koga K, Osuga Y, Hirata T, Saito A, Yoshino O, et al. High mobility group box 1 (HMGB1) levels in the placenta and in serum in preeclampsia. Am J Reprod Immunol 2011;66:143–148.
- de Ranitz-Greven WL, Kaasenbrood L, Poucki WK, Hamerling J, Bos DC, Visser GH, et al. Advanced glycation end products, measured as skin autofluorescence, during normal pregnancy and pregnancy complicated by diabetes mellitus. Diabetes Technol Ther 2012;14:1134–1139.
- Guosheng L, Hongmei S, Chuan N, Haiying L, Xiaopeng Z, Xianqiong L. The relationship of serum AGE levels in diabetic mothers with adverse fetal outcome. J Perinatol 2009;29:483–488.
- Pertyńska-Marczewska M, Głowacka E, Sobczak M, Cypryk K, Wilczyński J. Glycation endproducts, soluble receptor for advanced glycation endproducts and cytokines in diabetic and non-diabetic pregnancies. Am J Reprod Immunol 2009;61:175–182.
- Williams MA, Enquobahrie DA, Zimmer J, Qiu CF, Hevner K, Abetew D, et al. Maternal plasma advanced glycation end products concentrations in response to oral 50-gram glucose load in mid-pregnancy: a pilot study. Clin Lab 2012; 58:1045–1050.
- Romero R, Chaiworapongsa T, Savasan ZA, Hussein Y, Dong Z, Kusanovic JP, et al. Clinical chorioamnionitis is characterized by changes in the expression of the alarmin HMGB1 and one of its receptors, sRAGE. J Matern Fetal Neonatal Med 2012;25:558–567.
- Costantine MM, Weiner SJ, Rouse DJ, Hirtz DG, Varner MW, Spong CY, et al. Umbilical cord blood biomarkers of neurologic injury and the risk of cerebral palsy or infant death. Int J Dev Neurosci 2011;29:917–922.