Abstract
Advanced lipoxidation end products (ALEs) and advanced glycation end products (AGEs) have a pathogenetic role in the development and progression of different oxidative-based diseases including diabetes, atherosclerosis, and neurological disorders. AGEs and ALEs represent a quite complex class of compounds that are formed by different mechanisms, by heterogeneous precursors and that can be formed either exogenously or endogenously. There is a wide interest in AGEs and ALEs involving different aspects of research which are essentially focused on set-up and application of analytical strategies (1) to identify, characterize, and quantify AGEs and ALEs in different pathophysiological conditions; (2) to elucidate the molecular basis of their biological effects; and (3) to discover compounds able to inhibit AGEs/ALEs damaging effects not only as biological tools aimed at validating AGEs/ALEs as drug target, but also as promising drugs. All the above-mentioned research stages require a clear picture of the chemical formation of AGEs/ALEs but this is not simple, due to the complex and heterogeneous pathways, involving different precursors and mechanisms. In view of this intricate scenario, the aim of the present review is to group the main AGEs and ALEs and to describe, for each of them, the precursors and mechanisms of formation.
Introduction
Several studies indicate that advanced glycation end products (AGEs) and advanced lipoxidation end products (ALEs) have a pathogenetic role in the development and progression of different oxidative-based diseases including diabetes [Citation1–3], chronic renal failure [Citation4,Citation5], cardiovascular diseases [Citation6,Citation7] and neurological disorders [Citation8,Citation9].
From a chemical point of view, AGEs and ALEs represent a quite complex class of compounds that are formed by different mechanisms, by heterogeneous precursors and can be formed either exogenously or endogenously. In more detail, as better explained below, AGEs represent a class of covalently modified proteins generated by oxidative and non-oxidative pathways, involving sugars or their degradation products. The term ALEs includes a variety of covalent adducts which are generated by the nonenzymatic reaction of reactive carbonyl species (RCS), produced by lipid peroxidation and lipid metabolism, with the nucleophilic residues of macromolecules. AGEs and ALEs share some common properties, for example, both consist of nonenzymatic, covalently modified proteins and oxidative stress is often (although not always) involved in the mechanism of their formation. Moreover some AGEs and ALEs have the same structure, since they arise from common precursors, as in the case of carboxymethyllysine (CML) which is generated by glyoxal, which in turn is formed by both lipid and sugar oxidative degradation pathways [Citation10].
Similarly to their chemical heterogeneity and complexity, AGEs and ALEs exert their biological (damaging) activity through different and in some cases as yet not well-defined mechanisms. Some of the biological effects are due to the loss of function of the target proteins undergoing the covalent modification, such as in the case of extracellular matrix proteins that lose their elastic and mechanical functions when modified as AGEs/ALEs and in particular, when cross-links are involved [Citation11]. Other examples of a direct damaging effect of AGEs/ALEs can be ascribed to the covalent modification of enzymes and receptors that lose their activity due to the covalent modification involving the catalytic or binding site, or following a conformational change of the protein structure. AGEs and ALEs are also immunogenic and have been shown to elicit antibodies in a variety of diseases including systemic lupus erythematosus, alcoholic liver disease, diabetes mellitus, and rheumatoid arthritis as reviewed by Kurien et al. [Citation12]
AGEs also exert their damaging and pro-inflammatory effects by activating signaling cascades via specific receptors named RAGE (receptor for AGEs) [Citation13]. There are several other receptors for AGEs including P60/OST-48 protein (AGE-Rl), 80 K-H phosphoprotein (AGE-R2), galectin (AGE-R3) [Citation14]. It is still not clear whether the damaging effect of ALEs includes a receptor-mediated mechanism and in this case whether RAGE is involved.
The AGEs and ALEs’ precursors share a common electrophilic nature, which determines their marked reactivity toward the nucleophilic centers in the biomacromolecules. Indeed and as described below, all the chemical pathways leading to ALEs and AGEs can roughly be seen as condensation reactions between nucleophilic and electrophilic reactants. As discussed by LoPachin, [Citation15] these compounds cannot condense indiscriminately, but they can react, provided that they have a similar polarizability, thus generating only well-defined sets of covalent adducts.
The precursors of ALEs are generally lipophilic so they interact with the cell membrane components leading to disturbance of membrane integrity, seen as blebs formation [Citation16]. Blebs are plasma membrane vesicles, often observed in apoptosis, formed when membrane domains (lipid rafts) are detached from cytoskeleton [Citation17]. Yet blebs are active in systematic signaling during inflammation, contributing to the initiation and progression of chronic inflammatory processes in organisms, as they are released from the cell in the form of membrane vesicles [Citation18].
There is a wide interest in AGEs and ALEs involving different aspects of research, which are essentially focused on set-up and application of analytical strategies (1) to identify, characterize and quantify AGEs–ALEs in different pathophysiological conditions; (2) to elucidate the molecular basis of their biological effects; and (3) to discover compounds able to inhibit AGEs/ALEs damaging effects not only as biological tools aimed at validating AGEs/ALEs as drug target, but also as promising drugs.
All the above-mentioned research stages require a detailed knowledge of the chemical formation of AGEs/ALEs. As already mentioned, this is not simple, due to the complex and heterogeneous pathways, involving different precursors and mechanisms. In view of this intricate scenario, the aim of the present review is to group the main AGEs and ALEs and to describe, for each of them, the precursors and mechanisms of formation. While recognizing that the electrophilic precursors can also react with nucleic acids and phospholipids, generating well-established covalent adducts, this review will be almost exclusively focused on the AGEs and ALEs formation involving protein nucleophilic centers. The interested reader can refer to specialized reviews for the adducts involving other biomolecules. (e.g. [Citation19,Citation20]). Moreover, the biological effects and mechanism of AGEs and ALEs as well as the molecular strategies for their inhibition are not considered herein and for these aspects the reader can refer to the reviews published in this special issue or to some excellent reviews already published [Citation21–23].
AGEs: an introduction
The browning process, that occurs during food heating, is a well-known phenomenon which characterizes several cooked dishes and is caused by the so-called Maillard reaction, a nonenzymatic process firstly described by Maillard in 1912 (Louis Camille Maillard, 1878–1936) [Citation24]. It generates yellowish-brown colored products and involves a complex series of degradative reactions between sugars and proteins, whose chemical mechanisms were first described by Hodge 40 years later [Citation25]. For many years, studies on the Maillard reaction were focused on foods and food-like systems where it occurs during heating, processing, and storage, and plays key roles in determining color, flavor, and nutritional quality. Other studies concerned the effects of the Maillard reaction when it occurs in paper, textile, soil, and biopharmaceutical products.
Even though the first glycated protein was discovered by Kunkel and Wallenius in 1955 (a glycated HbA1C hemoglobin) [Citation26], increasing attention has been shifted to the human body only in the last three decades, namely since the pathological role of in vivo posttranslational protein glycation has become clearly evident in all its detrimental consequences. In vivo, the process can occur in tissue and body fluids, is usually associated with hyperglycemic pathophysiological conditions, and involves the generation of irreversible protein adducts and cross-links known as AGEs. After the seminal studies of Monnier and Cerami [Citation27], who postulated in the early 1980s that protein glycation could play a detrimental role in aging-related pathologies, a progressively more-detailed understanding of the individual reactions involved in AGEs generation has compellingly emphasized that their accumulation has clear pathophysiological implications [Citation28]. In vivo, several disorders appear to be exacerbated by such damaging protein adducts, especially when they involve long-lived proteins. A paradigmatic example is offered by collagen [Citation29], the glycation of which induces vascular thickening with detrimental consequences including decreased elasticity, hypertension, and endothelial dysfunctions, and induces marked accelerating effects on disorders such as atherosclerosis, nephropathy, and retinal pathologies [Citation6,Citation30,Citation31]. Moreover, glycation can also affect short-lived peptides causing detrimental changes in their biological functions as demonstrated by the accumulation of glycated insulin in the pancreatic β-cells of diabetic animal models [Citation32].
Without neglecting their biological implications, the following sections will be mainly devoted to chemical aspects of AGEs generation so as to play an introductory role for the other reviews in this special issue in which the therapeutic strategies, to reduce their formation in vivo, will be comprehensively described.
Maillard reaction
As shown in and following the well-known Hodge's scheme [Citation25], the first step in the Maillard reaction involves condensation between a carbonyl group from a reducing sugar (e.g. D-glucose (1.1) on which is indeed focused) and a primary amino group belonging, for example, to the lysine side chain or to the protein N-terminus. The condensation yields a carbinolamine intermediate, which dehydrates to give an unstable imine adduct which can, in turn, cyclize to generate N-substituted D-glucosylamine (1.2) [Citation33].
Figure 1. Major steps in the Maillard reaction of reducing sugars following the well-known Hodge scheme.
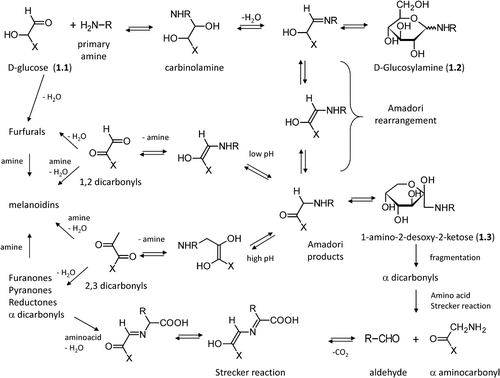
At physiological pH and room temperature, the imine derivative spontaneously undergoes the acid-catalyzed Amadori rearrangement (Mario Amadori, 1886–1941) [Citation33] and gives the corresponding N-substituted 1-amino-2-desoxy-2-ketose (1.3), which can cyclize yielding derivatives called Amadori products which are more stable compared to that of the N-substituted D-glucosylamine [Citation34] even though all these products can react with amino acids to produce brown melanoidins [Citation35]. It should be remembered that also nonreducing ketoses can undergo a similar set of reactions, which produce the corresponding N-substituted 1-amino-2-desoxy-aldoses following the so-called Heyns rearrangement [Citation36]. Formation of the Amadori products is favored at alkaline pH values and in the presence of phosphate ions.
Despite being relatively stable, the Amadori products can react following two major routes. At low pH values, they undergo enolization yielding 1,2-dicarbonyls, which then dehydrate to give furfural derivatives [Citation37]. Furfural derivatives, among which 5-hydroxymethyl furfural (2.6) is the most common, are formed quite rapidly, and indeed are usually utilized as easily detectable markers to follow food deterioration during prolonged storage [Citation38]. They quickly react with amino acids to yield brown melanoidins even though furfurals are not the main precursors leading to them [Citation39].
At higher pH values, enolization produces 2,3- dicarbonyls which then dehydrate to give reductones [Citation40]. These conjugated enediols have a moderate reducing power and can contribute to antioxidant activity. In the presence of amino acids they can generate brown melanoidins. The cyclization of 2,3 dicarbonyl intermediates can also give furanones and pyranones which include important flavor compounds with very low aroma threshold values [Citation41].
By following different pathways, Amadori products can generate α-dicarbonyls mainly through oxidative fission [Citation42] or retro-aldol fragmentation [Citation43], the former being markedly catalyzed by the presence of transition metal ions. Sugar fragmentation can also occur via a free radical-based mechanism according to the so-called Namiki pathway [Citation44]. Dicarbonyl compounds are very reactive products and their critical relevance in AGEs generation will be discussed in-depth in this review. Besides dicarbonyls, sugar fragmentation can also generate hydroxyl aldehydes and the corresponding oxidized acid analogues as depicted in . Dicarbonyl compounds can react with amino acids through the so-called Strecker degradation, in which the amino acid is transformed into the corresponding aldehyde liberating CO2 and the remaining α-aminocarbonyl compound, which can then condense to yield pyrazine derivatives, typical aroma components in heated foods [Citation39].
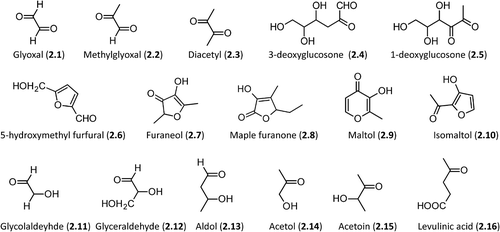
Figure 2. Relevant molecules involved in Maillard reaction. The first row includes α-dicarbolyls, the second row collects cyclization products, and the third row shows carbonyl derivatives generated by sugar fragmentation.
As shown in , all generated carbonyl products can condense with primary amines (mainly coming from amino acids) to produce polymeric melanoidins. They are high molecular mass, brown-colored adducts whose precise structure is still to be fully elucidated even though it includes nitrogen-containing heterocycles [Citation45]. Melanoidin formation increases with time and more markedly with temperature [Citation46].
The relationship between sugar structure and susceptibility to Maillard reaction has been extensively studied both in vivo and in foodstuffs [Citation47]. Although in all sugars the cyclic form is thermodynamically favored, the reactivity of monosaccharides is directly related to the abundance of their open chain form, which ranges from 0.0002% for glucose to 0.7% for fructose (6.2) [Citation48]. This easily explains why glucose, due to the mentioned stability of its pyranose form, is the least reactive sugar, while fructose whose furanosic cyclic form is somewhat less stable is about 7.5-fold more reactive than glucose [Citation49]. Notably, the Maillard reaction can also involve fructose metabolites, such as fructose-6-phosphate (4.2) or glyceraldehyde-3-phosphate (4.4), which show an extraordinary reactivity up to 200-fold compared to that of glucose. The resulting critical role of fructose in protein glycation leads to the term “fructosylation”, and the fructose effects are further exacerbated by its widespread diffusion [Citation50,Citation51]. Indeed, fructose is the most common naturally occurring monosaccharide in human diet mostly because of the high-fructose syrup produced by starch, which is usually added to beverages and baked foods [Citation52]. Despite its low reactivity, even glucose, which is present in millimolar concentration in several tissues, markedly contributes to protein glycation also because it can be converted into fructose via the polyol pathway [Citation52]. Besides the abundance of open chain forms, other factors governing susceptibility to the Maillard reaction include the sugar length and the intrinsic reactivity of the functional groups. Thus, aldoses are more reactive than ketoses, because aldehydes are more accessible and more electrophilic than ketones [Citation48].
illustrates some relevant players involved in the above-described Maillard reaction. The first row collects the major α-dicarbonyls including both short-chain derivatives (e.g. glyoxal, 2.1, and methylglyoxal, 2.2) and longer polyol dicarbonyls (e.g. glucosone analogues, 2.4 and 2.5). As mentioned before, the key role of dicarbonyls in AGEs formation will be discussed in-depth throughout the review. The second row shows the main cyclization products. Beside the already-mentioned 5-hydroxymethyl furfural (2.6), it is worth mentioning furaneol (2.7, 2,5-dimethyl-4-hydroxy-3(2H)-furanone) [Citation53], which represents an illustrative example of reductones and is one of the most flavor active volatile compounds (caramel- like odor) always present in the aroma extract from the Maillard reaction. Cyclization of 2,3-dicarbonyls can also afford flavored and colored furanones (e.g. Maple furanone, 2.8, 5-Ethyl-3-hydroxy-4-methyl-2(5H)-furanone) [Citation54] and pyranones (maltol, 2.9, 3-Hydroxy-2-methyl-4-pyrone), this latter essentially generated by the decomposition of disaccharides during caramelization [Citation55]. Lastly, isomaltol (2.10, 1-(3-hydroxy-2-furanyl)-ethanone) is another common example of furan derivatives resulting from the dehydration of 2,3-dicarbonyls which can be easily converted into the maltol analogue. It typically arises from Maillard degradation of lactose, even though it can be formed by the enzymatic degradation of starch, as well [Citation56].
The last row includes examples of hydroxyl carbonyl compounds arising from sugar fragmentation. They include aldehydes (2.11–2.13), ketones (2.14 and 2.15), and oxidized derivatives as in the case of levulinic acid (4-oxopentanoic acid, 2.16) which arises from the acid-catalyzed ring opening of the 5-hydroxymethyl furfural [Citation57]. Notably, the chemical reactivity of these last compounds decreases from left to right.
AGEs and their reactive precursors
AGE formation involves complex sequential and parallel reactions, whose precise mechanism is still debated even though at least two main pathways by which AGE can be generated are well established [Citation58,Citation59]. The first route involves irreversible rearrangements of the Amadori products following both oxidative and non-oxidative pathways (see AGEs deriving from rearrangement of the Amadori products). The second route involves condensation between the side-chain of lysine, cysteine and arginine residues, and dicarbonyls, which can be generated by enolization of Amadori products as well as by the direct degradation of aldoses and ketoses (as seen above).
Through multiple mechanisms, a large variety of AGE structures can be generated, some of which conserve completely the sugar carbon skeleton even though condensed to form aromatic rings, as in the case of crossline (8.15), while other AGEs involve oxidative degradation with loss of sugar carbon atoms. The analysis of the valence of carbon atoms shows that oxidative reactions are not necessarily required in AGE formation and, in some cases, non-oxidative pathways can be involved depending also on the considered precursors. For this reason, the term glycation is preferable to glycoxidation, which would not be broadly applied to all AGEs.
Although the following sections are clearly focused on endogenously formed AGEs, the dietary intake of food-derived AGEs may significantly contribute to the total body AGEs load [Citation60,Citation61]. Several AGEs have so far been identified in heated foods, and indeed preclinical and clinical studies have demonstrated that an elevated intake of thermally processed foods might induce diabetogenic and nephrotoxic effects, low-grade inflammation, enhance oxidative stress, and promote atherosclerosis [Citation62,Citation63]. Besides the AGEs which are also endogenously generated (e.g., carboxymethyllysine, pyrraline, pentosidine, see below) [Citation64,Citation65], some adducts are typically found only in heated foods since they derive by thermal degradation of AGEs. A well-known example is represented by furosine (N6-(2-(2-Furanyl-2-oxoethyl))-L-lysine), which arises from the heating of fuctosyl lysine in weakly acid conditions. Hence, furosine is a widely utilized marker to evaluate the nutritional quality of foods (especially for milk-based foods) by measuring the concentration of nutritionally available lysine residues [Citation66].
Finally, it is worth remembering that dicarbonyls and AGEs can react with specific residues producing several nitrogen-, sulphur-, and oxygen-containing heterocyclic derivatives, which are potent aroma compounds. They include alkylpyrazines, oxazoles, oxazolines, tetrahydropyridine, pyrroline, thiazoles, thiazolines, and pyrazinones, which are generated at high temperature via Strecker degradation followed by decarboxylation and rearrangement reactions [Citation67].
Glyoxal
Exogenous and endogenous sources. Glyoxal (2.1) is the smallest dialdehyde and appears as a yellow liquid, which forms hydrates in water solution. The hydrates tend spontaneously to condense to yield oligomeric species, the exact structures of which remain unclear [Citation68]. Sources of glyoxal can involve both exogenous and endogenous pathways. The former include dietary and environmental sources, while the latter include various metabolic and oxidative reactions all of which are able to induce cellular damage [Citation69].
Specifically, all foods, which are prepared by fermenting, roasting, baking, or frying processes, are rich dietary sources of glyoxal [Citation70]. Environmental sources include all products arising from combustion processes such as cigarette smoke, smoke from residential log fires, and vehicle exhaust, while detectable amounts of glyoxal are also found in soil, groundwater, seawater, and sediment [Citation71]. Glyoxal coming from environmental sources can be absorbed through the gut, lungs, or skin even though the specific role of each absorption route in affecting glyoxal tissue levels is still debated.
With regard to endogenous production mechanisms, glyoxal can be essentially generated starting from sugars and lipids (plus their metabolites) [Citation72]. Sugars can produce glyoxal through two major pathways. The first includes direct autoxidation reactions, which appear to be promoted by the presence of phosphates and transition metal ions, although the direct conversion of sugars in glyoxal can also involve retroaldol fragmentation [Citation73,Citation74]. The second pathway involves the Maillard reaction and occurs via Amadori products, which can be transformed into glyoxal by enolization as described above [Citation75].
Although sugars are often seen as the main source, glyoxal can also arise from lipid peroxidation. Indeed, the nonenzymatic peroxidation of polyunsaturated fatty acids, such as linoleic and linolenic acids, generates peroxide intermediates, which then degrade to yield a variety of oxidative products including glyoxal [Citation76]. Lastly, ascorbate can also spontaneously hydrolyze into glyoxal through an unknown mechanism [Citation77].
Concerning the metabolic fate of glyoxal, the large majority is enzymatically converted in glycolate by GSH-dependent glyoxalase system, which comprises two isozymes differing for tissue and subcellular localization [Citation78]. When GSH is depleted, as happens in many oxidative-based disorders, other enzymes including aldehyde reductase, aldose reductase, carbonyl reductase, aldehyde dehydrogenase, and 2-oxoaldehyde dehydrogenase can contribute to the glyoxal metabolism [Citation79]. Finally, several lines of evidence have revealed that glyoxal catabolism could be involved in oxalate formation as suggested, for example, by studies on diabetics showing that they have increased levels of both plasma glyoxal and urinary oxalate [Citation80]. Such a pathway is catalyzed by glycolate oxidase and lactate dehydrogenase, involves glycolate as a key oxidized intermediate, and may influence calcium oxalate stone formation [Citation81].
Glyoxal-derived AGEs. The remarkable activity of glyoxal is clearly evidenced by the observation that more than 90% of glyoxal present in biological matrices is covalently bound to Cys, Lys, and Arg residues thus having a key role in protein glycation [Citation82,Citation83].
As depicted by , glyoxal can react with the lysine side chain to yield Nϵ-(carboxymethyl)lysine (CML, 3.1) through an aldimine intermediate, and with the cysteine thiol group to give an irreversible end product, S-(carboxymethyl)cysteine (CMC, 3.2), the formation of which involves a Cannizzaro-like rearrangement of the corresponding thioacetal intermediate [Citation84]. Notably, the single modified CMC amino acid has long been known as a mucolytic agent, and has attracted recent interest in the long-term treatment of chronic obstructive pulmonary disease (COPD) for its capacity to decrease the adhesion of bacteria to the upper respiratory tract [Citation85].
Figure 3. Major pathways for the formation of the glyoxal-derived AGEs in vivo.
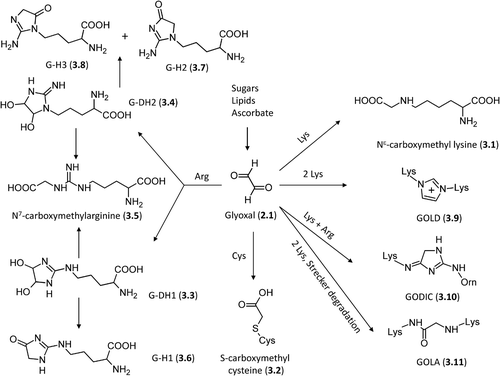
shows that the in vivo reactions between the arginine side chain and glyoxal are clearly more complex [Citation86], since they involve the formation of two possible dihydroxyimidazolidine intermediates: G-DH1 (3.3), Nδ- (3,4-dihydroxy-1-imidazolidin-2-yl) ornithine and its isomer G-DH2 (3.4), (5-(4,5-dihydroxy-2-imino-1-imidazolidinyl) norvaline. Both intermediates can (1) open to give the N7-(carboxymethyl) arginine (CMA, 3.5) adduct [Citation87] or (2) dehydrate to yield three different imidazolone derivatives, that is, Nδ- (5-hydro-4-imidazolon- 2-yl) ornithine, G-H1 (3.6), and its isomers 5-(2-amino- 5-hydro-4-imidazolon-1-yl) norvaline G-H2 (3.7) and 5-(2-amino-4-hydro-5-imidazolon-1-yl)norvaline, G-H3 (3.8). Among the possible arginine adducts, the dihydroxyimidazolidines, despite acting as intermediates, are the most abundant followed by the imidazolones and CMA [Citation88]. While considering that each protein residue possesses a specific reactivity depending on its microenvironment, comparison of the possible adducts of lysine and arginine residues shows that the latter are usually more abundant thus emphasizing the greater susceptibility of arginine residues in producing AGEs [Citation89]. Notably, all arginine-derived AGEs can degrade to give an ornithine residue, which can in turn react with glyoxal generating a set of ornithine-based AGEs, similar to those observed for lysine as exemplified by the formation of carboxymethylornithine (CMO) [Citation90].
Due to its dicarbonyl structure, glyoxal can also react with two lysine or arginine residues yielding imidazol cross-linked adducts. As represented in , glyoxal can react with two lysine residues giving the corresponding glyoxal–lysine dimer (GOLD, 3.9, 6- {1-[(5S)-5-ammonio-6-oxido-6-oxohexyl]imidazolium-3-yl}-L-norleucine) whose mechanism of formation is common to the corresponding methylglyoxal containing dimer (see below) and involves an initial bis lysyl diimine intermediate, which reacts with a second glyoxal molecule through a Cannizzaro-type reaction, and, after dehydration, condenses to give the final imidazolium dimer [Citation91].
The cross-linking reaction between arginine, lysine, and glyoxal gives the GODIC product (3.10, N6-(2-{[(4S)-4-ammonio-5-oxido-5-oxopentyl]amino}-3,5- dihydro-4H-imidazol-4-ylidene)-L-lysine) which involves the initial formation of an imine intermediate coming from lysine plus glyoxal and then reacts with the arginine's guanidine group to yield, after dehydration, the final 4,5-dihydro-1H-imidazol derivative [Citation92]. Lastly, two lysine residues can react with glyoxal via Strecker degradation to yield the amide adduct, GOLA (3.11, N6-(2-{[(5S)-5-ammonio-6-oxido-6-oxohexyl] amino}-2-oxoethyl)-L-lysine) which can be derived also through oxidative fragmentation of the Amadori products and which appears to be favored with increased glyoxal concentrations [Citation93,Citation94].
Methylglyoxal
Exogenous and endogenous sources. Methylglyoxal (2.2), also called pyruvaldehyde or 2-oxopropanal (CH3–CO–CHO or C3H4O2), is a yellow liquid with a characteristic pungent odor. It is ubiquitous in living cells and therefore almost all foods contain methylglyoxal, the concentration of which is increased by processes such as cooking [Citation95], fermentation, and prolonged storage [Citation96]. In foods and beverages, the main sources of methylglyoxal are represented by sugars and lipids [Citation70]. Sugars can generate methylglyoxal through retro-aldol reaction or auto-oxidative fragmentation and, although these processes are greatly favored by alkaline conditions, they can also occur during caramelization by the heating of mono-, oligo-, and polysaccharides [Citation97,Citation98]. Besides heating processes, also prolonged storage can degrade sugars to generate methylglyoxal through a process which is especially favored in foods with a high content of simple carbohydrates [Citation99]. Due to industrial manufacturing and storage, also lipids can fragment inducing accumulation of methylglyoxal [Citation100]. Thus, heating and photodegradation are two well-known processes with which lipids can yield methylglyoxal as a degradation product. Lastly, many microorganisms produce and release methylglyoxal and thus fermentation can cause an increase of this dicarbonyl product in alcoholic drinks and fermented foods [Citation101]. Regarding environmental sources and similarly to what has already been seen with glyoxal, combustion processes can generate methylglyoxal, as in the case of cigarette smoke, which indeed is one of the major sources of air contamination by this toxic dicarbonyl compound [Citation102]). Drinking water can contain methylglyoxal commonly because of the applied purification processes [Citation103], while rainwater can absorb methylglyoxal from polluted air and conveys it to the soil [Citation71].
Endogenous sources of methylglyoxal involve many pathways that can be subdivided into enzymatic or nonenzymatic reactions [Citation104–107]. As represented by , the former includes reactions catalyzed by methylglyoxal synthase, cytochrome P450 2E1, myeloperoxidase, and amino oxidase, whereas the latter include the spontaneous decomposition of dihydroxyacetone-phosphate, the Maillard reaction, the oxidation of acetol, and lipid peroxidation [Citation106]. In more detail, the main route leading to methylglyoxal involves the enzymatic or nonenzymatic degradation of the triose phosphate intermediates originating from the glycolytic processes [Citation105]. The intermediates include glyceraldehyde 3-phosphate (4.4) and dihydroxyacetone-phosphate (4.5), which are in equilibrium due to the activity of triosephosphate isomerase [Citation108,Citation109]. Thus, dihydroxyacetone-phosphate can yield methylglyoxal by both spontaneous nonenzymatic elimination of the phosphate group and by the effect of methylglyoxal synthase, an enzyme found in procaryotic and mammalian systems and inhibited by inorganic phosphate ions [Citation101,Citation110]. Similarly to what was observed during food cooking, methylglyoxal can also derive via the Maillard reaction in vivo under physiological conditions.
Figure 4. Schematized pathways for MGO formation and catabolism (dotted lines indicate nonenzymatic MGO formation). Adapted from [Citation105].
![Figure 4. Schematized pathways for MGO formation and catabolism (dotted lines indicate nonenzymatic MGO formation). Adapted from [Citation105].](/cms/asset/b0e83dea-b99e-417f-892a-0d9d6206504c/ifra_a_815348_f0004_b.gif)
Lipids can take part in the methylglyoxal generation through the acetone metabolism [Citation111]. In detail, acetone (4.7) derives from acetoacetate (4.6) by myeloperoxidase activity and is converted to methylglyoxal by the cytochrome P450 2E1 via acetol (2.14) as intermediate [Citation112]. Particular pathophysiological conditions, such as ketosis and diabetic ketoacidosis, significantly increase methylglyoxal generation from acetone [Citation113]. Yet again, glycerol (4.8) resulting from triacylglycerol hydrolysis can be transformed into methylglyoxal through the glycerolphosphate (4.9) intermediate which is produced by a specific glycerol kinase [Citation114]. Threonine and glycine can also generate methylglyoxal through the aminoacetone intermediate (4.10) [Citation115]. This metabolic pathway is mediated by semicarbazide sensitive amine oxidase (SSAO) and appears to be exacerbated in low coenzyme A states [Citation116,Citation117].
Different enzymes contribute to methylglyoxal detoxification among which the glyoxalase system, aldose reductase (ALR), betaine aldehyde dehydrogenase, and 2-oxoaldehyde dehydrogenase play a key role [118– 120]. The above-described glyoxalase system catalyzes the biotransformation of methylglyoxal to D-lactate (4.11), which in turn is converted into pyruvate (4.12) by D-lactate dehydrogenase. Aldose reductase (ALR2, AKR1B1) catalyzes the NADPH-dependent reduction of methylglyoxal into lactaldehyde (4.13) or into acetol when GSH is depleted [Citation121]. ALR then transforms both products into 1,2-propandiol (4.14) even though this final reaction is markedly more favored for lactaldehyde compared to acetol which consequently tends to accumulate in the absence of GSH. Since acetol spontaneously converts into methylglyoxal, the reduction of methylglyoxal in the absence of GSH could result in a vicious circle with nil effect [Citation122]. Nonspecific aldehyde dehydrogenases can recognize substrates only in their anhydrate forms and thus they cannot detoxify methylglyoxal and other α-oxoaldehydes, which are completely hydrated under physiological conditions [Citation123]. However, methylglyoxal is substrate for betaine aldehyde dehydrogenase (ALDH9 or E3) which indeed contributes to its detoxification [Citation124]. Moreover, the missing activity of nonspecific aldehyde dehydrogenases is counterbalanced by the specific 2-oxoaldehyde dehydrogenase (2-ODH) which catalyzes the NAD/NADPH-dependent oxidation of methylglyoxal into pyruvate (4.12). The enzyme also requires an activator: vicinal aminoalcohols, aminothioles, and glycine were found to act as activators even though the physiological activator is still unknown [Citation125].
Methylglyoxal-derived AGEs. It has long been known that methylglyoxal is able to induce irreversible modifications in proteins under physiological conditions [Citation126–128]. The formed adducts can be grouped into fluorescent and nonfluorescent products. Specifically and as schematized by , methylglyoxal reacts primarily with arginine residues yielding cyclic imidazolone adducts [Citation129], whose formation mechanism involves firstly the condensation between a carbonyl group and the N7 guanidino atom to give the corresponding carbinolamine which then cyclizes to form the diol intermediates and to yield the final adducts through two successive dehydrations [Citation130]. From a kinetic point of view, cyclization is a fast reaction while the two eliminations represent the slow and rate-determining steps [Citation131]. Depending on the nitrogen atoms involved in the cyclization and on environmental pH value, three different adducts are possible, as represented in , namely Nδ-(5-methyl-4-imidazolon-2-yl)-L-ornithine (MG-H1, 5.1), 2-amino-5-(2-amino-5-hydro-5-methyl-4-imidazolon-1-yl)pentanoic acid (MG-H2, 5.2), 2-amino-5-(2-amino-4-hydro-4-methyl-5-imidazolon-1-yl)pentanoic acid (MG-H3, 5.3). These three imidazolone adducts are in equilibrium since they can open giving the carboxyethylarginine (CEA, 5.4) adduct which in turn re-cyclizes allowing mutual interconversion between the imidazolone adducts [Citation132].
Figure 5. Major pathways for the formation of the methylglyoxal-derived AGEs in vivo.
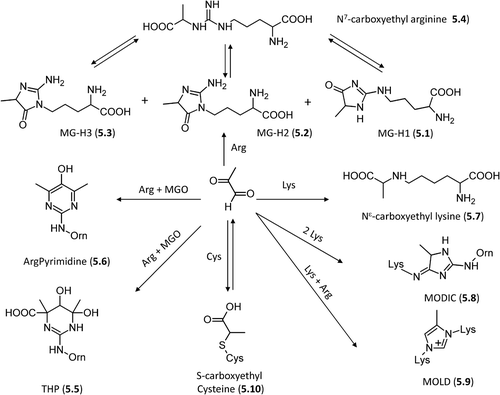
Besides the mentioned ring opening, the imidazolone adducts can undergo other reactions as depicted by . In more detail, they can add a second methylglyoxal molecule yielding either THP (5.5, Nδ-(4-carboxy-4,6-dimethyl-5,6-dihydroxy-1,4,5,6-tetrahydropyrimidine-2-yl)-L-ornithine) through an open intermediate [Citation133] and a final re-cyclization reaction or argpyrimidine, (5.6, Nδ-(5-hydroxy-4,6-dimethylpyrimidine-2-yl)-l-ornithine), a fluorescent derivative via decarboxylation and dehydration [Citation134]. Although THP would theoretically give argpyrimidine through a reaction triggered by the gained aromaticity, no experimental evidence has hitherto been found to support such a pathway.
Methylglyoxal can react with lysine residues generating the carboxylethyllysine (CEL, 5.7) adduct through a mechanism, which involves an aldimine intermediate and brings to mind the already mentioned pathway for the formation of the nor-analogue carboxymethyl lysine (3.1) [Citation135]. Notably, the aldimine intermediates can oxidize through a metal-catalyzed process giving deaminated allysine (adipic semialdehyde). This latter further oxidizes to yield 2-amino adipic acid, which indeed accumulates in oxidative-based disorders [Citation136].
As already seen for glyoxal, the aldimmine intermediate can react with the arginine guanidine group yielding a cyclic intermediate that, after dehydration and rearrangement, produces the final cross-link imidazol MODIC adduct (5.8, 2-ammonio-6-({2–[4-ammonio-5-oxido-5-oxopently)amino]-4-methyl-4,5-dihydro-1H-imidazol-5-ylidene}amino)hexanoate) with an overall mechanism almost identical to that described for GODIC (3.10) formation, even though this latter appears to be favored as suggested by experimental and computational results [Citation91,Citation92].
Yet again, similarly to what was described for glyoxal, methylglyoxal can react with two lysine residues forming an initial diimine intermediate, which then adds a second methylglyoxal molecule and, via an intermolecular Cannizzaro-type reaction, yields the final imidazolium MOLD adduct (5.9, 6-{1-[(5S)-5-ammonio-6-oxido-6-oxohexyl]-4-methyl- imidazolium-3-yl}-L-norleucine) [Citation137]. This cross-link reaction occurs more efficiently with methylglyoxal compared to glyoxal and this difference is in agreement with the proposed mechanism since the required fragmentation reaction is more favorable for methylglyoxal, due to increased relative stability of the leaving group [Citation138]. Lastly, methylglyoxal can react with cysteine residues giving reversible hemithioacetal adducts (5.10) [Citation130] and with tryptophan residues yielding β carboline derivatives, a particularly common reaction during prolonged food storage [Citation96,Citation139].
3-Deoxyglucosone
Exogenous and endogenous sources
3-Deoxyglucosone [3-DG, (4S,5R)-4,5,6-trihydroxy-2-oxohexanal] is a highly reactive dicarbonyl compound that, as schematized in , mostly arises from the rearrangement of the Amadori products [Citation140]. These latter are formed via Maillard reaction and then undergo enolyasation and deydratation reactions to generate dicarbonyl species, among which 3-Deoxyglucosone (3-DG) is one of the most reactive products in condensing with free amino groups in proteins to form AGEs during the late stage of the Maillard reaction [Citation141,Citation142]. The formation of 3-DG via Amadori products was found to be increased in nonaerobic conditions and in the presence of phosphate ions.
Figure 6. Schematized pathways for 3-DG formation and catabolism.
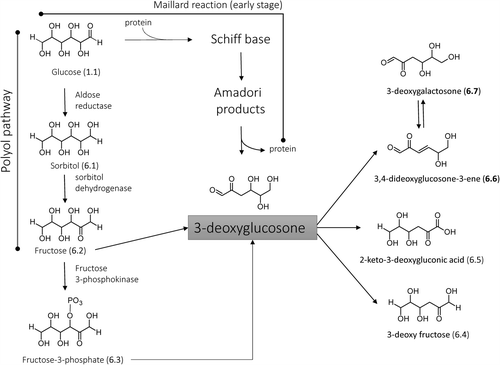
With regard to 3-DG dietary sources, all foods, which are particularly rich in sugar content and the preparation of which involves cooking, ripening, and fermentation, are significant dietary sources of 3-DG (e.g. [Citation143]). In more detail, 3-DG is found at significant concentrations in all alcoholic and nonalcoholic drinks whose sugar content is due to the presence of monosaccharides, like glucose and fructose, artificially added as syrup or deriving from the raw material [Citation144]. Conversely, sucrose-containing beverages show markedly lower 3-DG concentrations because of the greater stability of the disaccharide's acetal structure. Vinegars, especially balsamic vinegars, and soy sauces show very high 3-DG levels, a result which is easily explainable considering their manufacturing processes, which involve, as in the case of balsamic vinegars, prolonged heating and fermentation of must. Since dicarbonyls are well-known precursors of aroma ingredients, the stated 3-DG content should play a crucial role in the aromatizing capabilities of these condiments [Citation145]. The reported data should be seen as examples showing the vast presence of 3-DG in foodstuffs and the interested reader can refer to specialized reviews to analyze the 3-DG content in common foods [Citation70].
Beside the already discussed Maillard reaction, the endogenous pathways leading to the 3-DG formation include the polyol pathway [Citation146]. In detail, 3-DG is nonenzymatically generated in the absence of amino groups by the oxidation of fructose (6.2) [Citation147]. This latter, as described in , can also arise from glucose (1.1) via sorbitol (6.1) as intermediate due to the enzymatic activity of ALR and sorbitol dehydrogenase, a metabolic route particularly active in diabetes [Citation148]. Again, 3-DG can derive from the hydrolysis of fructose-3-phosphate (6.3) which is enzymatically generated starting from fructose by fructosamine 3-kinase [Citation149], and was found to be accumulated in diabetic animal models [Citation150].
The formed 3-DG can permeate the cellular membrane and thus is taken up by cells, where it is reduced to 3-deoxy fructose (6.4) by ALR as confirmed by the observation that aldose reductase inhibitors (ARI) exacerbate the 3-DG induced protein glycation [Citation151]. Moreover, 3-DG can be enzymatically oxidized into a nonreactive 2-keto-3-deoxygluconic acid (6.5) by the effect of aldehyde dehydrogenase 1A1 (ALDH1A1), which is a NAD-dependent dehydrogenase that acts at neutral pH and is known to catalyze the oxidation of retinaldehyde to retinoic acid [Citation152]. Unfortunately, a fraction of 3-DG can nonenzymatically oxidize into very reactive and toxic 3,4-dideoxyglucosone-3-ene (3,4-DGE, 6.6) which combines the dicarbonyl reactivity with that of α,β unsaturated carbonyls and which is generated under very oxidative pathophysiological conditions such as those observed during peritoneal dialysis [Citation153,Citation154]. This very reactive compound can also hydrate giving 3-deoxygalactosone (3-DGal, 6.7) which is thus in equilibrium with 3DG through the 3,4-DGE derivative as common intermediate [Citation155].
Deoxygluocosone-derived AGEs
Several studies evidenced that 3-DG quickly reacts with residues in proteins forming a variety of AGE structures among which imidazolones and carboxymethyl lysine appear to be the most abundant ones [Citation156]. As schematized in , -DG can easily attack arginine residues forming dihydroimidazole derivatives, which dehydrate to yield the final imidazolone adducts [Citation157,Citation158]. As seen for glyoxal and methylglyoxal, also 3-DG can generate three distinct imidazolone adducts, namely, 3DG-H1, (7.2, Nδ-[5-hydro-5-(2,3,4-trihydroxybutyl)-4-imidazolon-2-yl] ornithine, 3DG-H2 (5-[2-amino-5-hydro-5-(2,3,4-trihydroxybutyl)-4-imidazolon-1-yl] norvaline), and 3DG-H3, (5-[2-amino-4-hydro-4-(2,3,4-trihydroxy butyl)-5-imidazolon-1-yl] norvaline), although in only 3DG-H1 is displayed for the sake of simplicity. In fact and under physiological conditions, the reaction between 3-DG and arginine leads to the accumulation of the initial dihydroimidazole adducts, while the formation of the final imidazolones is slow and appears to be favored only at high temperatures. Nevertheless, several lines of evidence reveal that imidazolone adducts are a common marker of 3-DG-modified proteins and are present in vivo at high concentrations in diabetic animal models [Citation159].
Figure 7. Major pathways for the formation of the 3-DG-derived AGEs in vivo.
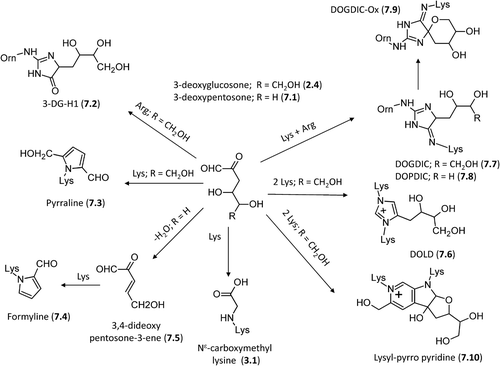
3-Deoxyglucosone, and more generally all 3-deoxyosones [Citation160], can react with a lysine residue forming, through a Paal–Knorr reaction, a pyrrole containing adduct called pyrraline (7.3) or pyrrole lysine, 6-(2-formyl-5-hydroxymethyl-1-pyrrolyl)-L-norleucine [Citation161,Citation162]. A similar cyclization product, formyline (7.4), has recently been identified from the reaction between lysine and 3-deoxypentosone (7.1). Formyline, or (6-(2-formyl-1-pyrrolyl)-L-norleucine, is generated by the dehydratation of 3-deoxypentosone to yield 3,4-dideoxypentosone-3-ene (7.5) which then reacts with lysine to generate this cyclic adduct [Citation163]. Pyrrole containing adducts can also arise from the lipid peroxidation reactions of 4,5-epoxy-2- alkenals which are degradation products of the ω-3 and ω-6 fatty acids [Citation164]. In vivo and under oxidative conditions, pyrraline can induce protein cross-linking by generating ether bonds with cysteinyl thiol function or other pyrraline adducts [Citation165]. Interestingly, pyrraline containing dipeptides are recognized and transported at intestinal level by hPepT1, a di/tripeptide transporter, which therefore would be responsible for the intestinal absorption of pyrraline dipeptides after food intake [Citation166]. However and as mentioned above, the major adduct between 3-DG and lysine residues is represented by carboxymethyl lysine (CML, 3.1) which derives from degradation of the initial 3DG-Lys adduct thus indicating that the 3-DG can significantly contribute along with glyoxal to the CML adducted proteins as observed in in vivo studies [Citation146,Citation167].
Regarding the cross-linking adducts, 3-Deoxyglucosone can react with two lysine residues yielding a 3-deoxy glucosone lysine dimer, DOLD (7.6, 1,3-di(Nϵ-lysino) -4-(2,3,4-trihydroxybutyl)-imidazolium), a cross-link resulting from the reaction between hippuryl-lysine and 3-deoxyglucosone as characterized by Skovsted et al. [Citation135,Citation168]. As depicted in , -DG can cross-link arginine and lysine residues forming an imidazol adduct N6-{2-{[(4S)-4-ammonio-5-oxido-5-oxopentyl]amino}-5-[(2S,3R)-2,3,4-trihydroxybutyl]-3,5-dihydro-4H-imidazol-4-ylidene}-L-lysinate, which exists as a mixture of two diastereoisomers and has been named DOGDIC (7.7, aka DODIC) in accordance with the acronyms GODIC and MODIC already seen for the corresponding glyoxal and methylglyoxal adducts [Citation169]. A similar adduct is also formed by 3-deoxypentosone which produces the corresponding DOPDIC product (7.8, N6-{2-{[(4S)-4- ammonio-5-oxido-5-oxopentyl] amino}- 5-[(2S)-2,3- dihydroxypropyl]-3,5-dihydro-4H-imidazol-4-ylidene}-L-lysinate [Citation170]. Reihl et al. evidenced that DOGDIC, DOPDIC, and more generally all imidazolone adducts can further oxidize under physiological conditions to give the corresponding spiro [Citation4,Citation4] amino imidazol analogues (e.g. DOGDIC-Ox, 7.9) [Citation171]. Comparative analyses showed that imidazolones and CML are produced in similar high concentrations in the presence of 3-DG, while pyrraline and DOGDIC are generated in a markedly lower amount and accumulate only at high (nonphysiological) temperatures [Citation156]. Lastly, 3-DG can react with two lysine residues forming the cross-linking lysyl-pyrropyridine adduct (7.10), the level of which is increased in diabetic rats thus suggesting that its formation might be related to the progression of diabetic complications [Citation45].
AGEs deriving from rearrangement of the Amadori products
As described before and due to the instability of its acetal form, fructose (6.2) is particularly reactive in condensing with protein amino groups to give the corresponding Amadori products. In detail, shows that fructose can condense with the free N-terminus or more frequently with the lysine side chain to yield the N-ϵ-fructosyl lysine adduct (8.1) [Citation172]. Despite being less reactive, glucose can also condense with lysine residues giving the corresponding N-ϵ-glucosyl lysine adduct (8.2) [Citation173]. Under physiological conditions, these Amadori products can oxidize and rearrange to give the corresponding reactive dideoxyosones (namely, 8.3, N6-(2-hydroxy-4,5-dioxopentyl)-L-lysine for N-ϵ-fructosyl lysine and N6-(2,3-dihydroxy-5,6-dioxohexyl)-L-lysine for N-ϵ-glucosyl lysine, 8.4) [Citation174].
Figure 8. Major pathways for the formation in vivo of the AGEs deriving from direct rearrangement of the Amadori products.
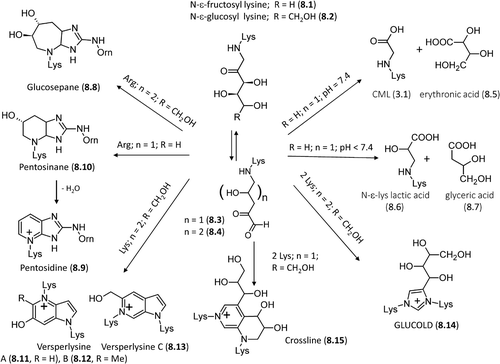
At physiological pH, N-ϵ-fructosyl lysine can further oxidize to form carboxymethyl lysine (CML, 3.1) and erythronic acid (8.5), while in slightly more acid conditions it yields N-ϵ-lysine lactic acid (8.6) plus glyceric acid (8.7), thus suggesting that the chemical fate of N-ϵ-fructosyl lysine strongly depends on the environmental pH [Citation175].
As schematized in , N-ϵ-glucosyl lysine can react with arginine side chains forming a bicyclic non-fluorescent cross-linking adduct, glucosepane (8.8, 2-acetylamino-5-[(4-butyl-6,7-dihydroxy-4,5,6,7,8,8a-hexahydroimidazo[4,5-b]azepin-2-yl)amino]-pentanoic acid) [Citation176], whose mechanism of formation is still debated. Its first step involves the conversion of the Amadori product into dideoxyosone (8.4) via dehydration and rearrangement reactions. Afterward, dideoxyosone firstly cyclizes to form an azepinone intermediate (i.e. 6-(3,4-dihydroxy-6-oxo-3,4,5,6-tetrahydro-2H-azepinium-1-yl) nor-leucine) which then condenses with the arginine's guanidino group to generate the final glucosepane adduct. An alternative route should also be possible in which the Amadori product firstly condenses with the arginine side chain to yield an imidazol intermediate, which then cyclizes and forms the glucosepane product [Citation177]. Glucosepane can exist in four diastereoisomeric pairs, whose relative abundance strongly depends on the protein environment in which they are formed [Citation174]. Whatever the mechanism of formation is, glucosepane is the most abundant cross-linking AGE found in senescent extracellular matrix accounting for the structural and cellular dysfunctions observed in aging and diabetes [Citation170,Citation178]. As already seen for other AGEs, also glucosepane can be transformed into ornithine thus confirming that all arginine-containing AGEs can be seen as in vivo nonenzymatic sources of ornithine [Citation179].
As shown in , pentose-derived Amadori products can undergo a quite similar reaction scheme giving the fluorescent adduct pentosidine (8.9, 6-[2-[[(4S)-4-amino-5-hydroxy-5-oxopentyl] amino]-4-imidazo [4,5-b]pyridinyl]-L-norleucine) [Citation180–182] the formation of which involves a nonaromatic intermediate pentosinane, (8.10, 6-((6S)-2-{[(4S)-4- ammonio-5- oxido-5- oxopentyl] amino}-6-hydroxy-5,6,7,7 a-tetra hydro -4H imidazo [4,5-b]pyridin-4-yl)-L-norleucine), which then dehydrates and aromatizes to give the final pentosidine [Citation183]. Under oxidative conditions, pentosidine can also result from reactions of lysine and arginine residues with hexoses, ascorbate, 3-deoxyglucosone (3-DG), glyceraldehydes and glycolaldehyde [Citation184]. Recent experimental and computational studies have demonstrated that pentosidine can also be generated by the reaction of lysine and arginine residues with two molecules of short-chain α-oxoaldehydes, such as glyoxal (GO) and methyl glyoxal (MGO), as observed in uremic patients [Citation185]. The reaction between lysine-containing Amadori products and arginine can finally generate two already-described imidazole adducts, namely DOGDIC (7.7) from hexoses and DOPDIC (7.8) from pentose sugars [Citation170,Citation178].
Glucose-derived, lysine-containing Amadori products can react with a second lysine residue forming three possible fluorescent cross-linking pyrrolopyridinium adducts, named vesperlysines A (8.11), B (8.12), and C (8.13) [Citation186]. The original six carbon atoms of glucose are not completely incorporated into vesperlysines thus suggesting that they are glyco-oxidation adducts derived from two glucose molecules through dehydration, oxidative fragmentation, and condensation of the sugar moieties. Moreover, they can be formed from shorter chain sugars, for example, ribose and glyceraldehyde [Citation187].
Glucose-derived lysine-containing Amadori products can also react with each other giving dimer cross-link adducts which can form directly or via the 1,4-dideoxy-5,6-dioxoglucosone intermediate. In the first case, they form a recently identified imidazolium adduct named GLUCOLD (8.14, 1,3-bis-(5-amino-5-carboxypentyl)-4-(1,2,3,4-tetrahydroxybutyl)-3H-imidazolium) [Citation188]. In the second case, the cited intermediate cyclizes to give a tetrahydropyridinium ring which adds a second Amadori product and after dehydration and rearrangements yields the final crossline adduct (8.15) [Citation189]. Regardless of the protein content of lysine and arginine residues, dimer cross-links are found in negligible amounts compared to glucosepane (21, 1 and 611 pmol/mg, for GLUCOLD, crossline and glucosepane, respectively). Mechanistic studies have revealed that the rapid formation of the N6-1,4-dideoxy-5,6-dioxoglucosone intermediates and their facile reaction with the guanidino group can easily explain the vast majority of arginine-containing adducts [Citation188].
ALEs: an introduction
The term ALEs (advanced lipoxidation end products) includes a variety of adducts and cross-links which are generated by the nonenzymatic reaction of RCS produced by lipid peroxidation and the lipid metabolism with the nucleophilic sites of macromolecules such as proteins, DNA, and aminophospholipids, leading to their irreversible modification [Citation190]. The most-studied ALEs are those arising from the reaction of RCS break-down products of the lipid peroxidation cascade with the nucleophilic residues of proteins, mainly Cys, Lys, His, and Arg residues. As schematized in , the RCS arising from the lipid peroxidation cascade are quite heterogeneous and can be divided into three main classes, namely : (1) the α,β-unsaturated aldehydes, including hydroxylated, such as 4-hydroxy-2-nonenal (HNE, 9.1) and 4-hydroxy hexenal (HHE, 9.2), and nonhydroxylated derivatives, nonenal (9.3) and acrolein (ACR, 9.4); (2) di-aldehyde, including the well-known lipid peroxidation by product malondialdehyde (9.5) and glyoxal (2.1); 3) cheto-aldehyde such as methylglyoxal (2.2), 4-oxo-nonenal (ONE, 9.6), and the isoketals also called levuglandins (LGs, as exemplified in by LGD2, 9.7) which are gamma-ketoaldehydes formed via the isoprostane pathway of arachidonic acid peroxidation.
Figure 9. Structures of the most reactive carbonyl species arising from lipidoxidation break-down and prostaglandin metabolism.
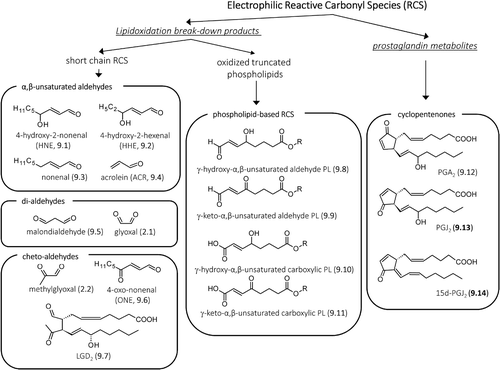
The RCS acting as precursor of ALEs, besides including the short-chain carbonyl derivatives generated by the break-down of nonenzymatic lipoxidation-derived hydroperoxides, also include the oxidized truncated phospholipids, where the electrophilic moiety remains covalently linked to the phospholipid [Citation191]. shows some exemplificative phospholipid-based RCS, including γ-hydroxy-α,β-unsaturated aldehyde phospholipid (9.8), γ-keto-α,β-unsaturated aldehyde phospholipid (9.9), and γ-keto-α,β-unsaturated carboxylic phospholipid (9.11) [Citation192]. As in the case of short-chain RCS, also the oxidized phospholipids containing electrophilic moieties are able to covalently react with the nucleophilic sites of proteins forming Michael adducts or Schiff bases. Although the ALEs formed by short-chain RCS have been much more studied in respect to those generated by oxidized phospholipid, some protein and peptide targets of oxidized phospholipid have already been identified, such as cathepsin B [Citation193] and angiotensin II [Citation194].
It should be noted that although the chemical reactions of RCS in free form or bound to phospholipid are practically the same and mainly based on the formation of Michael adduct or Schiff base as below described, the protein targets might be different since the two classes of compounds have a quite different lipophilicity and consequently a different cellular distribution.
The above-mentioned RCS are generated by a common lipid-peroxidation cascade, which can involve free polyunsatured fatty acids (PUFAs) or phospholipids. An additional class of RCS able to form protein adducts is represented by electrophilic prostaglandin metabolites, which are characterized by an α,β-unsaturated carbonyl moiety (cyclopentenone). Among these RCS, it is worth mentioning PGA2 (9.12) generated from the dehydration of PGE2 and 15-deoxy-Δ12,14-prostaglandin J2 (15d-PGJ2, 9.14) which is generated as a consequence of dehydration of PGD2, a principal COX-2 product formed in various cells and tissues during the inflammatory processes [Citation195,Citation196].
It should be stressed that some RCS such as glyoxal and methylglyoxal are generated by the oxidative pathways involving both lipids and sugars, thus the corresponding reaction products with proteins can be named both as ALEs and AGEs or as EAGLEs (either advanced glycation or lipoxidation endproducts).
α,β-unsaturated carbonyls
Exogenous and endogenous sources
Regarding the endogenous sources of α,β-unsaturated carbonyls, lipid peroxidation represents the major mechanism by which they can be generated in vivo [Citation197,Citation198]. Lipid peroxidation is initiated by the generation of free radicals close to cellular membranes, which represent rich sources of PUFAs. Hence, the levels of α,β-unsaturated carbonyls is strictly related to radical formation and consequently to oxidative stress conditions. It should be noted that moderate concentrations of free radicals are generated also under physiological conditions because of oxidase activity, imperfection in mitochondrial electron transport, and/or as a part of different metabolic processes. However, cells maintain redox homeostasis through scavenging enzyme cascades (i.e., superoxide dismutase, catalase, and glutathione peroxidase) as well as protective factors (i.e., GSH, vitamin C, and vitamin E) [Citation199]. Pro-oxidant conditions can dysregulate oxidative enzymes inducing an overproduction of free radicals which are no longer counteracted by redox homeostasis thus resulting in cellular damage caused by both free radical species and the so generated nonradical oxidants. In detail, radical species (mainly, HO• and ONOO−) attack PUFA molecules by abstracting an allylic hydrogen atom [Citation200]. This generates a less-reactive carbon- centered lipid radical (L•) which in turn reacts with molecular oxygen to yield lipid peroxyl (LOO•) and lipid alkoxyl radicals (LO•), both of which can generate reactive α,β-unsaturated aldehydes [Citation201].
With regard to ACR (9.4) endogenous generation, it also involves the myeloperoxidase-mediated degradation of threonine as well as the amine oxidase-mediated degradation of spermine and spermidine, reactions which are particularly relevant in situations of oxidative stress and inflammation [Citation202,Citation203].
The α,β-unsaturated carbonyls are readily detoxified by glutathione via a Micheal addition which can occur spontaneously but is one hundred times faster when catalyzed by glutathione-S-transferases (GSTs) [Citation204]. Other metabolic reactions can involve the carbonyl function that can be either reduced into alcohol or oxidized into acid, involving alcohol dehydrogenase or aldo/keto reductases (AKR) and aldehyde dehydrogenase, respectively [Citation205]. Notably, these redox reactions can also involve GSH conjugates and indeed the reduced mercapturic acid of HNE, namely 1,4-dihydroxy nonane mercapturic acid (DHN-MA), is the major metabolite found in urine [Citation206].
While HNE (9.1) is almost exclusively formed endogenously, ACR (9.4) is ubiquitously present in foods and in the environment, which might help understanding of the mostly cytotoxic effects of ACR on the one hand and cytotoxic and regulatory effects of HNE on the other [Citation207,Citation208].
It is generated by heat-induced dehydration and decomposition reactions of sugars, lipids, and amino acids during food cooking [Citation209]. Environmental sources include combustion of petroleum fuels and biodiesel [Citation210] and industrial manufacturing since ACR is an important intermediate for the production of acrylic acid and plastics [Citation211]. Nonetheless, smoking of tobacco products represents the major source of the total human exposure to ACR [Citation212,Citation213] as demonstrated by the level of its main metabolite, namely 3-hydroxypropyl mercapturic acid, in urine of smokers, which is about twice than that of nonsmokers [Citation214].
ALEs formed by α,β-unsaturated carbonyls
Most of the lipid-derived RCS are characterized by a carbonyl group (a keto or an aldehyde function) which is conjugated to a double bond and which regulates the electrophilic reactivity toward the nucleophilic moieties. In particular, the carbonyl group acts as an electron-withdrawing group, which decreases the electron-richness of the conjugated alkene group. The combination of polarizable mobile electrons and the electron-withdrawing capacity of the carbonyl group thus creates an area of electron deficiency at the alkene β-carbon atom of the α,β-unsaturated carbonyl derivatives [Citation215]. Hence, the β-carbon atom shows an electrophilic character, which in the case of γ-hydroxylated derivatives, such as HNE and HHE is further enhanced by the inductive effect of the vicinal hydroxyl group. It covalently reacts with the nucleophilic (electron-rich species) side chains on amino acid residues of proteins, such as the thiol group of cysteine, the imidazolic nitrogen of histidine, and the epsilon amino group of Lys, forming the corresponding Michael adduct. However, although the Michael adducts formed with Cys and His are stable in isolation, Michael adducts to Lys ϵ-amino groups are formed reversibly and can be isolated only following reductive trapping with NaBH4 [Citation216,Citation217]. If the resulting Michael adduct contains an γ-hydroxyaldehyde, it can further react intra-molecularly forming cyclic hemiacetals (). Another general reaction involving α,β-unsaturated aldehydes consists of the reaction of the aldehydic carbonyl group with the primary amino group of Lys or with the terminal amino group, forming the corresponding Schiff base. In more detail, the reaction starts with the formation of a carbinolamine intermediate that rearranges and loses water to yield a Schiff base. It should be noted that the Schiff base is a reversible adduct which undergoes a hydrolytic cleavage and hence it is more stable when the reaction involves the Lys residues embedded in lipohilic and anhydrous protein pockets. (in particular the right side) summarizes the general reaction mechanisms for hydroxylated and nonhydroxylated α,β-unsaturated aldehydes.
Figure 10. General reaction mechanisms of ALEs formation for hydroxylated and nonhydroxylated α,β-unsaturated aldehydes with specific focus to HNE-derived ALEs.
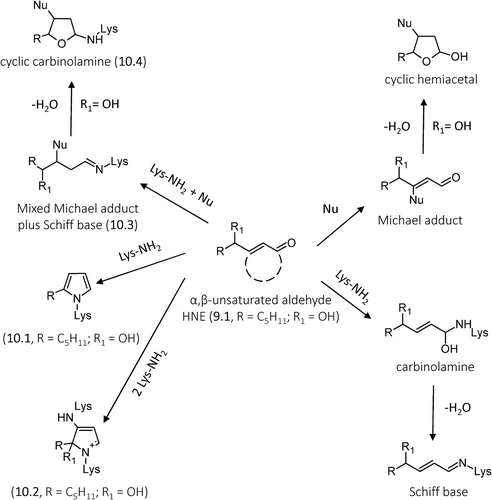
The Michael adducts and Schiff bases are the early reaction products of α,β-unsaturated carbonyl that for most of the RCS further react and rearrange forming additional reaction products such as cross-links and which are specific for each RCS.
For example, the left side of shows that HNE (one of the most abundant and reactive lipid-derived RCS, generated through the β-cleavage of hydroperoxides from PUFAs containing ω-six chains, such as linoleic acid and arachidonic acid, 13.6) [Citation218], besides forming Michael adduct reaction products with His, Cys, and Lys residues as well as Schiff bases with Lys residues, also forms other different ALEs, such as the HNE-derived 2-pentylpyrroles (10.1) and the fluorescent four-electron oxidation product, 2-hydroxy-2-pentyl-1,2-dihydropyrrol-3-one iminium (10.2) [Citation219]. HNE, since possessing both an aldehyde and an electrophilic carbon, can form cross-links, involving condensation with Lys at carbonyl carbon atom (Schiff base) and the Michael adduct at β-carbon atom with nucleophilic residues (10.3). This adduct can then dehydrate yielding the cyclic carbinolamine (10.4) [Citation220].
The 4-keto derivative of HNE, 4-oxo-nonenal (ONE, 9.6), has been demonstrated to be a direct product of lipid oxidation [Citation221,Citation222], arising independently and not from oxidation of HNE, and more reactive that HNE (it reacts with protein nucleophiles through conjugate addition at a rate 6–31 times faster than does HNE) [Citation223]. As depicted in , ONE forms simple 4-ketoaldehyde Michael adducts by the reaction of C2 (11.1) and C3 (11.2) with His, Lys, and Cys that can exist as such or undergo ring-closure to furans (11.3 and 11.4). Additionally, 4-ketoaldehyde adducts can react with Lys forming 2,3- and 2,4-disubsitututed pyrrole cross-links (11.5 and 11.6) [Citation224]. ONE reacts also with the amino group of Lys forming the corresponding Schiff base (11.7), which then reacts with a second amine moiety at C3, followed by tautomerization and then a second two-electron autoxidation to give the fluorophore pyrrolium Lys–Lys cross link (11.8) which is also formed by HNE. More recently Zhu and Sayre [Citation225] reported that apparent long-lived Lys adducts with the mass expected of a Michael adduct are actually the isomeric 4-ketoamide (11.9) which together with Lys-derived pyrrolinone (11.10) and Lys–His imidazolyl pyrrole cross-links (11.5 or 11.6 with Nu = His) are all major products that form on proteins from ONE. Notably, Lys-derived pyrrolinone (11.10) which is in equilibrium with its corresponding hydroxy pyrrole tautomer, can dimerize generating the 2:2 ONE-Lys adduct (11.11). It should be noted that unlike HNE and other α,β-unsaturated aldehydes, ONE reacts also with Arg residues to give stable covalent adducts. Preference for the reaction of amino acid nucleophiles with ONE was determined to have the following order: Cys>> His> Lys> Arg [Citation226].
Figure 11. Reaction mechanisms of formation for the ONE-based adducts with nucleophilic sites. Adapted from [Citation22].
![Figure 11. Reaction mechanisms of formation for the ONE-based adducts with nucleophilic sites. Adapted from [Citation22].](/cms/asset/520d43be-3dca-4126-8eab-3c082ae28b30/ifra_a_815348_f0011_b.gif)
Another well-studied α,β-unsaturated aldehyde is represented by ACR (9.4) which has several sources other than peroxidation of PUFAs including the metabolism of amino acids, polyamines and drugs as well as the environment and food [Citation22]. Among the α,β-unsaturated aldehydes, ACR is by far the strongest electrophile and therefore shows the highest reactivity with protein nucleophiles, the order of reactivity (Cys > His > Lys) being the same as that established for HNE. As illustrated in and among these sites, the preferential formation of Michael-type adducts on cysteine residues (12.1) is believed to be the predominant way for ACR to exert its reactivity in biological systems. Besides a simple Michael adduct, ACR forms additional ALEs as studied by Uchida et al. In particular they identified an ACR–lysine adduct, Nϵ-(3-formyl-3,4-dehydropiperidino)lysine (FDP-lysine, 12.4). The mechanism proposed for FDP-Lys formation involves Michael addition of the Lys amino group to the ACR β-carbon atom to give a secondary amine with retention of the aldehyde group (12.3). This intermediate reacts with another ACR molecule via a Michael addition forming a derivative (12.5) that, after aldol condensation and dehydration, gives the FDP-Lys adduct [Citation227]. Later Uchida et al. identified another ACR-dependent ALE, the Nϵ-(3-methylpyridinium)-lysine (MP-lysine, 12.6), resulting from the initial Schiff base formation of ACR with the ϵ-amino group of lysine, which further reacts with a second ACR molecule via a Michael addition to generate an imine derivative. The subsequent conversion of this imine derivative to the final product (MP-lysine, 12.8) requires two oxidation steps and intramolecular cyclization, but its detailed mechanism has not yet been clarified [Citation228].
Figure 12. Reaction mechanisms of formation for the acrolein-based adducts with nucleophilic sites.
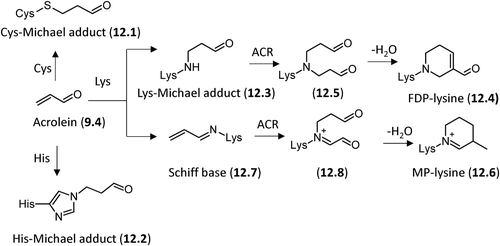
More recently the same research group, by using a reductive amination-based pyridylamination method, analyzed the ACR-specific adducts with a carbonyl function and found that ACR modification of the protein produced a number of carbonylated amino acids, including an ACR − histidine adduct. On the basis of the chemical and spectroscopic evidence, this adduct was identified as Nτ-(3-propanal)histidine (12.2) which appeared to be one of the major adducts generated in the oxidized LDL [Citation229].
Dicarbonyls
Exogenous and endogenous sources
The diverse sources of glyoxal and methylglyoxal have already been described in the previous sections and here attention is focused on those of malodialdeyde and isoketals. The former has endogenous sources almost identical to those of α,β-unsaturated carbonyls since its main in vivo source is represented by peroxidation of polyunsaturated fatty acids with two or more methylene-interrupted double bonds [Citation230]. Isoketals, also called LGs and isolevuglandins (isoLGs), are formed by the rearrangement of endoperoxide intermediates generated through the cyclooxygenase and free radical-induced oxidation of arachidonates [Citation231]. In detail, the endoperoxide intermediate undergoes a 1,2-hydride shift during the cleavage of three bonds in a concerted fashion with a polarized transition state. This reaction yields two γ-ketoaldehydes, namely levuglandin E2 (LGE2) and levuglandin D2 (LGD2) which are derivatives of levulinaldehyde with prostanoid side chains and which readily undergo dehydration leading to their anhydro analogs (AnLGE2 and AnLGD2) [Citation232,Citation233]. As mentioned before, the endoperoxide intermediate can be generated enzymatically through cyclooxygenase which adds two molecules of oxygen to a molecule of arachidonic acid to produce prostaglandin endoperoxide G2 (PGG2) as well as nonenzymatically by a free radical peroxidative mechanism as demonstrated by the observation that these isoprostanoids are generated in vivo, even after application of cyclooxygenase inhibitors [Citation234].
While isoketals are generated almost only endogenously, malondialdeyde can be present in foods arising from lipid peroxidation induced by heating. Indeed, it has been found in heated edible oils such as sunflower and palm oils and is contained at high levels in many rancid foods [Citation235].
ALEs formed by di-carbonyls
Different ALEs are generated by the reaction of di- carbonyls (di-aldehydes and cheto-aldehydes) with proteins. Some of these RCS, such as GO and MGO, besides being generated by a lipid oxidation/metabolism, are also formed by different enzymatic and nonenzymatic pathways involving sugars and their reactivity and formation of AGEs adducts have already been discussed under the AGEs section.
It should firstly be noted that the ability of the di- carbonyls to form stable and irreversible ALEs in relatively short periods is due to the presence of both the electron-withdrawing carbonyl groups, the chemical reactivity of which is further enhanced by the activation of one carbonyl group on the other. Indeed, the presence of only one carbonyl function can only give reversible Schiff bases that to be analytically detected require stabilization by a chemical reduction (reductive amination) using, for example, sodium borohydride.
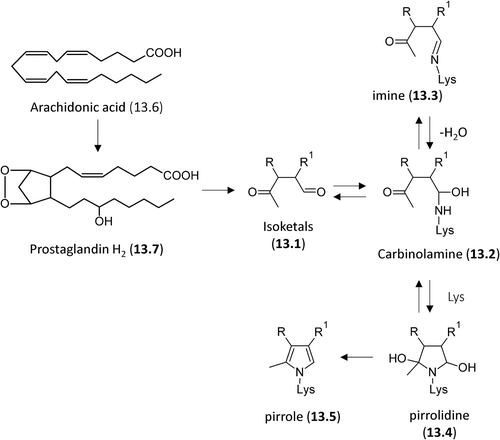
The reaction of the lipid-derived di-carbonyls usually involves the formation of a carbinolamine as a first step, followed by different rearrangement reactions leading to, in most cases, heterocylic cross-links as above reported in the cases of glyoxal and methylglyoxal. Isoketals is another example of di-carbonyls forming irreversible ALEs, which include heterocyclic cross-links. Isoketals are highly reactive γ-ketoaldehydes formed by free radical-mediated lipid peroxidation of H2-isoprostanes. Unlike the F2-isoprostanes, the biological effect of these products is due to their ability to modify proteins rather than through activation of specific receptors [Citation234]. reports the reaction of isoketals (13.1) with the amino group of Lys which is based on the formation of a carbinolamine intermediate (13.2) that is in equilibrium with the corresponding reversible Schiff base (13.3) or the remaining carbonyl of hemiaminal can undergo an intramolecular nucleophilic attack by the ϵ-amino group of a second Lys residue to yield a highly unstable pyrrolidine adduct (12.4) which undergoes a rapid dehydration to form a pyrrole adduct (12.5) [Citation232].
Figure 13. Reaction mechanisms of formation for the isoketal-based adducts with nucleophilic sites.
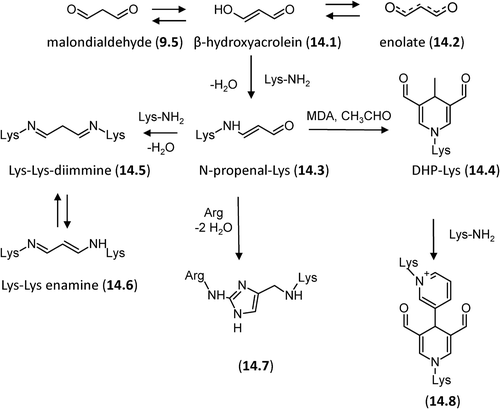
Malondialdehyde (9.5, MDA) is often the most abundant individual aldehyde resulting from lipid peroxidation and occurs in biological materials in various covalently bound forms [Citation218]. As shown in and compared with other RCS, MDA is relatively less reactive at neutral pH and this is explained by considering that it enolizes rapidly and then loses a proton at neutral pH forming the enolate salt (14.2) which is stabilized by a conjugated π-bond system. The reactivity of MDA is due to the nondissociated enolate (the β-hydroxyacrolein, 14.1) which is able to react with the ϵ-amino group of Lys forming the semistable Nϵ-(2-propenal)lysine (N-propenal-Lys, 14.3). N-propenal-Lys can further react with another molecule of MDA and a molecule of acetaldehyde (derived from MDA breakdown) to form a stable fluorescent product containing a dihydropyridine moiety (DHP-Lys, 14.4) or with an additional Lys residue to form a Lys–Lys-diimine (14.5) which would exist as its resonance-stabilized enamine (14.6, ) [Citation236]. The aldehyde side chains on the stable dihydropyridine ring can further react with Lys residues to form a reversible cross-link [Citation237]. MDA protein adducts and, in particular, N-propenal-Lys and the dihydropyridine cross-links have been more recently characterized by MS approaches and using insulin and albumin as target proteins [Citation238,Citation239]. Beside Lys, MDA reacts with other nucleophilic residues such as Arg forming the stable Nδ-(2-pirimidyl)-L- ornitine (14.7) [Citation240]. Cross-linking of MDA involving Lys and Arg residues, 2-ornithinyl-4-methyl(1ϵ-lysyl) 1,3-imidazole (14.8), has been elucidated by NMR and MS and identified in diabetic patients [Citation241].
Figure 14. Reaction mechanisms of formation for the MDA-based adducts with nucleophilic sites.
Acknowledgment
This work was supported by funds from the Italian Ministry of University and Research (Project PRIN 2009) and by the European Project COST CM1001.
Declaration of interest
The authors report no declarations of interest. The authors alone are responsible for the content and writing of the paper.
References
- Baynes JW. Chemical modification of proteins by lipids in diabetes. Clin Chem Lab Med 2003;41:1159–1165.
- Yamagishi S, Maeda S, Matsui T, Ueda S, Fukami K, Okuda S. Role of advanced glycation end products (AGEs) and oxidative stress in vascular complications in diabetes. Biochim Biophys Acta 2012;1820:663–671.
- Yap FYT, Kantharidis P, Coughlan MT, Slattery R, Forbes JM. Advanced Glycation End Products as Environmental Risk Factors for the Development of Type 1 Diabetes. Curr Drug Targets 2012;13:526–540.
- Sugiyama S, Miyata T, Inagi R, Kurokawa K. Implication of the glycoxidation and lipoxidation reactions in the pathogenesis of dialysis-related amyloidosis (Review). Int J Mol Med 1998;2:561–565.
- Iacobini C, Menini S, Ricci C, Scipioni A, Sansoni V, Mazzitelli G, et al. Advanced lipoxidation end-products mediate lipid-induced glomerular injury: role of receptor-mediated mechanisms. J Pathol 2009;218:360–369.
- Del Turco S, Basta G. An update on advanced glycation endproducts and atherosclerosis. Biofactors 2012;38: 266–274.
- Prasad A, Bekker P, Tsimikas S. Advanced Glycation End Products and Diabetic Cardiovascular Disease. Cardiol Rev 2012;20:177–183.
- Li JL, Liu DN, Sun L, Lu Y, Zhang Z. Advanced glycation end products and neurodegenerative diseases: Mechanisms and perspective. J Neurol Sci 2012;317:1–5.
- Butterfield DA, Reed T, Sultana R. Roles of 3-nitroty rosine- and 4-hydroxynonenal-modified brain proteins in the progression and pathogenesis of Alzheimer‘s disease. Free Radic Res 2011;45:59–72.
- Fu MX, Requena JR, Jenkins AJ, Lyons TJ, Baynes JW, Thorpe SR. The advanced glycation end product, N-(epsilon)(carboxymethyl)lysine, is a product of both lipid peroxidation and glycoxidation reactions. J Biol Chem 1996;271: 9982–9986.
- Ciulla MM, Paliotti R, Carini M, Aldini G. Fibrosis, enzymatic and non-enzymatic cross-links in hypertensive heart disease. Cardiovasc Hematol Disord Drug Targets 2011.
- Kurien BT, Hensley K, Bachmann M, Scofield RH. Oxidatively modified autoantigens in autoimmune diseases. Free Radic Biol Med 2006;41:549–556.
- Ramasamy R, Vannucci SJ, Yan SS, Herold K, Yan SF, Schmidt AM. Advanced glycation end products and RAGE: a common thread in aging, diabetes, neurodegeneration, and inflammation. Glycobiology 2005;15:16R–28R.
- Thornalley PJ. Cell activation by glycated proteins. AGE receptors, receptor recognition factors and functional classification of AGEs Cell Mol Biol 1998;44:1013–1023.
- LoPachin RM, Gavin T, DeCaprio A, Barber DS. Application of the Hard and Soft, Acids and Bases (HSAB) Theory to Toxicant-Target Interactions. Chem Res Toxicol 2012;25:239–251.
- Baumgart T, Hammond AT, Sengupta P, Hess ST, Holowka DA, Baird BA, Webb WW. Large-scale fluid/fluid phase separation of proteins and lipids in giant plasma membrane vesicles. Proc Natl Acad Sci U S A 2007;104: 3165–3170.
- Guéraud F, Atalay M, Bresgen N, Cipak A, Eckl PM, Huc L, et al. Chemistry and biochemistry of lipid peroxidation products. Free Radic Res 2010;44:1098–1124.
- Huber J, Vales A, Mitulovic G, Blumer M, Schmid R, Witztum JL, et al. Oxidized membrane vesicles and blebs from apoptotic cells contain biologically active oxidized phospholipids that induce monocyte-endothelial interactions. Arterioscler Thromb Vasc Biol 2002;22:101–107.
- Breyer V, Frischmann M, Bidmon C, Schemm A, Schiebel K, Pischetsrieder M. Analysis and biological relevance of advanced glycation end-products of DNA in eukaryotic cells. FEBS J 2008;275:914–925.
- Pamplona R. Membrane phospholipids, lipoxidative damage and molecular integrity: a causal role in aging and longevity. Biochim Biophys Acta 2008;1777:1249–1262.
- Takeuchi M, Takino J, Yamagishi S. Involvement of the toxic AGEs (TAGE)-RAGE system in the pathogenesis of diabetic vascular complications: a novel therapeutic strategy. Curr Drug Targets 2010;11:1468–1482.
- Aldini G, Dalle-Donne I, Facino RM, Milzani A, Carini M. Intervention strategies to inhibit protein carbonylation by lipoxidation-derived reactive carbonyls. Med Res Rev 2007; 27:817–868.
- Negre-Salvayre A, Coatrieux C, Ingueneau C, Salvayre R. Advanced lipid peroxidation end products in oxidative damage to proteins. Potential role in diseases and therapeutic prospects for the inhibitors Br J Pharmacol 2008;153: 6–20.
- Maillard L. Action des acides aminés sur les sucres: formation des mélanoȉdines par voie méthodique CR Acad Sci 1912;154:66–68.
- Hodge J. Chemistry of browning reactions in model systems. J Agric Food Chem 1953;1:928–943.
- Kunkel HG, Wallenius G. New hemoglobin in normal adult blood. Science 1955;122:288.
- Monnier VM, Cerami A. Nonenzymatic browning in vivo: possible process for aging of long-lived proteins. Science 1981;211:491–493.
- Tessier FJ. The Maillard reaction in the human body. The main discoveries and factors that affect glycation Pathol Biol 2010;58:214–219.
- Sell DR, Monnier VM. Molecular basis of arterial stiffening: role of glycation- A mini-review. Gerontology 2012;58: 227–237.
- Goh S-Y, Cooper ME. The role of advanced glycation end products in progression and complications of diabetes. J Clin Endocrinol Metab 2008;93:1143–1152.
- Nagaraj RH, Linetsky M, Stitt AW. The pathogenic role of Maillard reaction in the aging eye. Amino Acids 2012;42:1205–1220.
- Schalkwijk CG, Brouwers O, Stehouwer CDA. Modulation of insulin action by advanced glycation endproducts: a new player in the field. Horm Metab Res 2008;40:614–619.
- Amadori M. The product of the condensation of glucose and p-phenetidine. Atti Reale Accad Nazl Lincei 1929;9: 68–73.
- Isbell H, Frush H. Mutarotation, hydrolysis, and rearrangement reactions of glycosylamines. J Org Chem 1958;23: 1309–1319.
- Morales FJ, Somoza V, Fogliano V. Physiological relevance of dietary melanoidins. Amino Acids 2012;42:1097–1109.
- Heyns K, Beilfuss W. Ketosylamine rearrangement of D-threo-pentulose (D-xylulose) with alpha-amino acids. Chem Ber 1970;103:2873–2876.
- Zeitsch K. The chemistry and technology of furfural and its many by-products. Amsterdam: Elsevier; 2000.
- Abraham K, Gurtler R, Berg K, Heinemeyer G, Lampen A, Appel KE. Toxicology and risk assessment of 5-Hydroxymethylfurfural in food. Mol Nutr Food Res 2011; 55:667–678.
- Nursten H. Maillard reaction: chemistry, biochemistry and implications. London: Royal Society of Chemistry; 2005.
- Kurata T, Otsuka Y. Amino-reductones. Formation mechanisms and structural characteristics. Adv Exp Med Biol 1998;434:269–276.
- Slaughter JC. The naturally occurring furanones: formation and function from pheromone to food. Biol Rev Camb Philos Soc 1999;74:259–276.
- Davidek T, Robert F, Devaud S, Vera FA, Blank I. Sugar fragmentation in the Maillard reaction cascade: Formation of short-chain carboxylic acids by a new oxidative alpha-dicarbonyl cleavage pathway. J Agric Food Chem 2006;54:6677–6684.
- Cammerer B, Wedzicha BL, Kroh LW. Nonenzymatic browning reactions of retro-aldol degradation products of carbohydrates. Eur Food Res Technol 1999;209:261–265.
- Namiki M, Hayashi T. A new mechanism of the Maillard reaction involving sugar fragmentation and free radical formation. Washington, DC: American Chemical Society; 1983.
- Hayase F, Usui T, Ono Y, Shirahashi Y, Machida T, Ito T, et al. Formation mechanisms of melanoidins and fluorescent pyridinium compounds as advanced glycation end products. Ann N Y Acad Sci 2008;1126:53–58.
- Wang HY, Qian H, Yao WR. Melanoidins produced by the Mail lard reaction: structure and biological activity. Food Chem 2011;128:573–584.
- Bunn HF, Higgins PJ. Reaction of monosaccharides with proteins: possible evolutionary significance. Science 1981; 213:222–224.
- Laroque D, Inisan C, Berger C, Vouland E, Dufosse L, Guerard F. Kinetic study on the Maillard reaction. Consideration of sugar reactivity Food Chem 2008;111:1032–1042.
- Suárez G, Rajaram R, Oronsky AL, Gawinowicz MA. Nonenzymatic glycation of bovine serum-albumin by fructose (fructation) - comparison with the maillard reaction initiated by glucose. J Biol Chem 1989;264:3674–3679.
- Dills WL. Protein fructosylation – fructose and the maillard reaction. Am J Clin Nutr 1993;58:S779–S787.
- Schalkwijk CG, Stehouwer CDA, van Hinsbergh VWM. Fructose-mediated non-enzymatic glycation: sweet coupling or bad modification. Diabetes Metab Res Rev 2004;20: 369–382.
- Rippe JM, Angelopoulos TJ. Sucrose, high-fructose corn syrup, and fructose, their metabolism and potential health effects: what do we really know?Adv Nutr 2013;4:236–245.
- Wang Y, Ho CT. Formation of 2,5-dimethyl-4-hydroxy-3(2H)-furanone through methylglyoxal: A Maillard reaction intermediate. J Agric Food Chem 2008;56:7405–7409.
- Noda Y, Peterson DG. Structure-reactivity relationships of flavan-3-ols on product generation in aqueous glucose/glycine model systems. J Agric Food Chem 2007;55:3686–3691.
- Nishibori S, Berhnard RA, Osawa T, Kawakishi S. Volatile components formed from reaction of sugar and beta-alanine as a model system of cookie processing. Adv Exp Med Biol 1998;434:255–267.
- Ota M, Kohmura M, Kawaguchi H. Characterization of a new Maillard type reaction product generated by heating 1-deoxymaltulosyl-glycine in the presence of cysteine. J Agric Food Chem 2006;54:5127–5131.
- Choudhary V, Mushrif SH, Ho C, Anderko A, Nikolakis V, Marinkovic NS, et al. Insights into the interplay of Lewis and Brønsted acid catalysts in glucose and fructose conversion to 5-(hydroxymethyl)furfural and levulinic acid in aqueous media. J Am Chem Soc 2013;135:3997–4006.
- Rabbani N, Thornalley PJ. Glycation research in amino acids: a place to call home. Amino Acids 2012;42:1087–1096.
- Cho SJ, Roman G, Yeboah F, Konishi Y. The road to advanced glycation end products: a mechanistic perspective. Curr Med Chem 2007;14:1653–1671.
- Luevano-Contreras C, Chapman-Novakofski K. Dietary advanced glycation end products and aging. Nutrients 2010; 2:1247–1265.
- Van Lancker F, Adams A, De Kimpe N. Chemical modifications of peptides and their impact on food properties. Chem Rev 2011;111:7876–7903.
- Henle T. AGEs in foods: do they play a role in uremia?Kidney Int 2003;63:S145–S147.
- Sebekova K, Somoza V. Dietary advanced glycation endproducts (AGEs) and their health effects – PRO. Mol Nutr Food Res 2007;51:1079–1084.
- Henle T, Schwarzenbolz U, Klostermeyer H. Detection and quantification of pentosidine in foods. Z Lebensm Unters Forsch 1997;204:95–98.
- Biemel KM, Buhler HP, Reihl O, Lederer MO. Identification and quantitative evaluation of the lysine-arginine crosslinks GODIC, MODIC, DODIC, and glucosepan in foods. Nahrung 2001;45:210–214.
- Erbersdobler HF, Somoza V. Forty years of furosine – Forty years of using Maillard reaction products as indicators of the nutritional quality of foods. Mol Nutr Food Res 2007;51: 423–430.
- Wang Y, Ho CT. Flavour chemistry of methylglyoxal and glyoxal. Chem Soc Rev 2012;41:4140–4149.
- Avzianova E, Brooks SD. Raman spectroscopy of glyoxal oligomers in aqueous solutions. Spectrochim Acta A Mol Biomol Spectrosc 2013;101:40–48.
- Kielhorn J, Pohlenz-Michel C, Schmidt S. Glyoxal. Concise international chemical assessment. Geneva, Switzerland: World Health Organization; 2004.
- Degen J, Hellwig M, Henle T. 1,2-dicarbonyl compounds in commonly consumed foods. J Agric Food Chem 2012; 60:7071–7079.
- Fu TM, Jacob DJ, Wittrock F, Burrows JP, Vrekoussis M, Henze DK. Global budgets of atmospheric glyoxal and methylglyoxal, and implications for formation of secondary organic aerosols. J Geophys Res Atmos 2008;113.
- O’Brien PJ, Siraki AG, Shangari N. Aldehyde sources, metabolism, molecular toxicity mechanisms, and possible effects on human health. Crit Rev Toxicol 2005;35:609–662.
- Manini P, La Pietra P, Panzella L, Napolitano A, d’Ischia M. Glyoxal formation by Fenton-induced degradation of carbohydrates and related compounds. Carbohydr Res 2006;341: 1828–1833.
- Wellsknecht KJ, Zyzak DV, Litchfield JE, Thorpe SR, Baynes JW. Mechanism of autoxidative glycosylation – identification of glyoxal and arabinose as intermediates in the autoxidative modification of proteins by glucose. Biochemistry 1995;34:3702–3709.
- Thornalley PJ, Langborg A, Minhas HS. Formation of glyoxal, methylglyoxal and 3-deoxyglucosone in the glycation of proteins by glucose. Biochem J 1999;344:109–116.
- Yin HY, Porter NA. New insights regarding the autoxidation of polyunsaturated fatty acids. Antioxid Redox Signal 2005;7:170–184.
- Shangari N, O’Brien PJ. The cytotoxic mechanism of glyoxal involves oxidative stress. Biochem Pharmacol 2004;68: 1433–1442.
- Mannervik B. Molecular enzymology of the glyoxalase system. Drug Metabol Drug Interact 2008;23:13–27.
- Shangari N, Bruce WR, Poon R, O’Brien PJ. Toxicity of glyoxals – role of oxidative stress, metabolic detoxification and thiamine deficiency. Biochem Soc Trans 2003;31: 1390–1393.
- Eisner BH, Porten SP, Bechis SK, Stoller ML. Diabetic kidney stone formers excrete more oxalate and have lower urine pH than nondiabetic stone formers. J Urol 2010;183:2244–2248.
- Lange JN, Wood KD, Knight J, Assimos DG, Holmes RP. Glyoxal formation and its role in endogenous oxalate synthesis. Adv Urol 2012;2012:819202.
- Thornalley PJ. Advances in glyoxalase research. Glyoxalase expression in malignancy, anti-proliferative effects of methylglyoxal, glyoxalase I inhibitor diesters and S-D- lactoylglutathione, and methylglyoxal-modified protein binding and endocytosis by the advanced glycation endproduct receptorCrit Rev Oncol Hematol 1995;20:99–128.
- Schwarzenbolz U, Henle T, Haebner R, Klostermeyer A. On the reaction of glyoxal with proteins. Z Lebensm Unters Forsch 1997;205:121–124.
- Zeng JM, Davies MJ. Evidence for the formation of adducts and S-(carboxymethyl)cysteine on reaction of alpha-dicarbonyl compounds with thiol groups on amino acids, peptides, and proteins. Chem Res Toxicol 2005;18:1232–1241.
- Mitchell SC, Steventon GB. S-Carboxymethyl-L-cysteine. Drug Metab Rev 2012;44:129–147.
- Glomb MA, Lang G. Isolation and characterization of glyoxal-arginine modifications. J Agric Food Chem 2001; 49:1493–1501.
- Iijima K, Murata M, Takahara H, Irie S, Fujimoto D. Identification of N(omega)-carboxymethylarginine as a novel acid-labileadvanced glycation end product in collagen. Biochem J 2000;347:23–27.
- Cotham WE, Metz TO, Ferguson PL, Brock JWC, Hinton DJS, Thorpe SR, et al. Proteomic analysis of arginine adducts on glyoxal-modified ribonuclease. Mol Cell Proteomics 2004;3:1145–1153.
- Schwarzenbolz U, Mende S, Henle T. Model studies on protein glycation: influence of cysteine on the reactivity of arginine and lysine residues toward glyoxal. Ann N Y Acad Sci 2008;1126:248–252.
- Sell DR, Monnier VM. Conversion of arginine into ornithine by advanced glycation in senescent human collagen and lens crystallins. J Biol Chem 2004;279:54173–54184.
- Lederer MO, Klaiber RG. Cross-linking of proteins by Maillard processes: characterization and detection of lysine-arginine cross-links derived from glyoxal and methylglyoxal. Bioorg Med Chem 1999;7:2499–2507.
- Nasiri R, Field MJ, Zahedi M, Moosavi-Movahedi AA. Cross-linking mechanisms of arginine and lysine with α,β-dicarbonyl compounds in aqueous solution. J Phys Chem A 2011;115:13542–13555.
- Adamiec J, Rossner J, Velisek J, Cejpek K, Savel J. Minor Strecker degradation products of phenylalanine and phenylglycine. Eur Food Res Technol 2001;212:135–140.
- Glomb MA, Pfahler C. Amides are novel protein modifications formed by physiological sugars. J Biol Chem 2001; 276:41638–41647.
- Pfeifer YV, Haase PT, Kroh LW. Reactivity of thermally treated α-dicarbonyl compounds. J Agric Food Chem 2013;61:3090–3096.
- Nemet I, Varga-Defterdarovic L. Methylglyoxal-derived beta-carbolines formed from tryptophan and its derivates in the Maillard reaction. Amino Acids 2007;32:291–293.
- HomokiFarkas P, Orsi F, Kroh LW. Methylglyoxal determination from different carbohydrates during heat processing. Food Chem 1997;59:157–163.
- Hollnagel A, Kroh L. Formation of α-dicarbonyl fragments from mono- and disaccharides under caramelization and Maillard reaction conditions. Z Lebensm Unters Forsch 1998;207:50–54.
- Kuntz S, Rudloff S, Ehl J, Bretzel RG, Kunz C. Food derived carbonyl compounds affect basal and stimulated secretion of interleukin-6 and-8 in Caco-2 cells. Eur J Nutr 2009;48:499–503.
- Casazza JP, Felver ME, Veech RL. The metabolism of acetone in rat. J Biol Chem 1984;259:231–236.
- Cooper R. Metabolism of methylglyoxal in microorganisms. Annu Rev Microbiol 1984;38:49–68.
- Fujioka K, Shibamoto T. Determination of toxic carbonyl compounds in cigarette smoke. Environ Toxicol 2006;21: 47–54.
- Camel V, Bermond A. The use of ozone and associated oxidation processes in drinking water treatment. Water Res 1998;32:3208–3222.
- Beisswenger PJ, Howell SK, Nelson RG, Mauer M, Szwergold BS. alpha-oxoaldehyde metabolism and diabetic complications. Biochem Soc Trans 2003;31:1358–1363.
- Kalapos MP. Methylglyoxal in living organisms – Chemistry, biochemistry, toxicology and biological implications. Toxicol Lett 1999;110:145–175.
- Kalapos M. Methylglyoxal and glucose metabolism: a historical perspective and future avenues for research. Drug Metabol Drug Interact 2008;23:69–91.
- Kalapos MP. The tandem of free radicals and methylglyoxal. Chem Biol Interact 2008;171:251–271.
- Cui Q, Karplus M. Catalysis and specificity in enzymes: a study of triosephosphate isomerase and comparison with methyl glyoxal synthase. Adv Protein Chem 2003;66: 315–372.
- Richard JP. Mechanism for the formation of methylglyoxal from triosephosphates. Biochem Soc Trans 1993;21:549–553.
- Cooper RA. Methylglyoxal synthase. Methods Enzymol 1975;41:502–508.
- Dhar A, Desai K, Kazachmov M, Yu P, Wu LY. Methylglyoxal production in vascular smooth muscle cells from different metabolic precursors. Metabolism 2008;57: 1211–1220.
- Bondoc FY, Bao ZP, Hu WY, Gonzalez FJ, Wang YY, Yang CS, Hong JY. Acetone catabolism by cytochrome P450 2E1: studies with CYP2E1-null mice. Biochem Pharmacol 1999;58:461–463.
- Turk Z, Nemet I, Varga-Defteardarovic L, Car N. Elevated level of methylglyoxal during diabetic ketoacidosis and its recovery phase. Diabetes Metab 2006;32:176–180.
- Jung JY, Yun HS, Lee J, Oh MK. Production of 1,2- Propanediol from Glycerol in Saccharomyces cerevisiae. J Microbiol Biotechnol 2011;21:846–853.
- Bechara EJH, Dutra F, Cardoso VES, Sartori A, Olympio KPK, Penatti CAA, et al. The dual face of endogenous alpha-aminoketones: Pro-oxidizing metabolic weapons. Comp Biochem Physiol C Toxicol Pharmacol 2007;146:88–110.
- Callingham BA, Crosbie AE, Rous BA. Some aspects of the pathophysiology of semicarbazide-sensitive amine oxidase enzymes. Prog Brain Res 1995;106:305–321.
- Lyles GA. Mammalian plasma and tissue-bound semicarbazide-sensitive amine oxidases: Biochemical, pharmacological and toxicological aspects. Int J Biochem Cell Biol 1996;28:259–274.
- Vander Jagt DL, Hunsaker LA. Methylglyoxal metabolism and diabetic complications: roles of aldose reductase, glyoxalase-I, betaine aldehyde dehydrogenase and 2-oxoaldehyde dehydrogenase. Chem Biol Interact 2003;143–144:341–351.
- Vander Jagt DL. Methylglyoxal, diabetes mellitus and diabetic complications. Drug Metabol Drug Interact 2008; 23:93–124.
- Yang K, Qiang D, Delaney S, Mehta R, Bruce WR, O’Brien PJ. Differences in glyoxal and methylglyoxal metabolism determine cellular susceptibility to protein carbonylation and cytotoxicity. Chem Biol Interact 2011;191:322–329.
- Vedantham S, Ananthakrishnan R, Schmidt AM, Ramasamy R. Aldose reductase, oxidative stress and diabetic cardiovascular complications. Cardiovasc Hematol Agents Med Chem 2012;10:234–240.
- Choi CH, Park SJ, Jeong SY, Yim HS, Kang SO. Methylglyoxal accumulation by glutathione depletion leads to cell cycle arrest in Dictyostelium. Mol Microbiol 2008; 70:1293–1304.
- Marchitti SA, Brocker C, Stagos D, Vasiliou V. Non-P450 aldehyde oxidizing enzymes: the aldehyde dehydrogenase superfamily. Expert Opin Drug Metab Toxicol 2008;4:697–720.
- Munoz-Clares RA, Diaz-Sanchez AG, Gonzalez-Segura L, Montiel C. Kinetic and structural features of betaine aldehyde dehydrogenases: Mechanistic and regulatory implications. Arch Biochem Biophys 2010;493:71–81.
- Dunkerton J, James SP. Purification of 2-oxoaldehyde dehydrogenase and its dependence on unusual amines. Biochem J 1975;149:609–617.
- Degenhardt TP, Thorpe SR, Baynes JW. Chemical modification of proteins by methylglyoxal. Cell Mol Biol (Noisy- le-grand) 1998;44:1139–1145.
- Westwood ME, Thornalley PJ. Molecular characteristics of methylglyoxal-modified bovine and human serum albumins – comparison with glucose-derived advanced glycation endproduct-modified serum albumins. J Protein Chem 1995; 14:359–372.
- Thornalley PJ. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification – a role in pathogenesis and antiproliferative chemotherapy. Gen Pharmacol 1996;27:565–573.
- Henle T, Walter AW, Haessner R, Klostermeyer H. Detection and identification of a protein-bound imidazolone resulting from the reaction of arginine residues and methylglyoxal. Z Lebensm Unters Forsch 1994;199:55–58.
- Lo TWC, Westwood ME, McLellan AC, Selwood T, Thornalley PJ. Binding and modification of proteins by methylglyoxal under physiological conditions – a kinetic and mechanistic study with n-alpha-acetylarginine, n-alpha- acetylcysteine, and n-alpha-acetyllysine, and bovine serum-albumin. J Biol Chem 1994;269:32299–32305.
- Wang TN, Kartika R, Spiegel DA. Exploring Post- translational Arginine Modification Using Chemically Synthesized Methylglyoxal Hydroimidazolones. J Am Chem Soc 2012;134:8958–8967.
- Klopfer A, Spanneberg R, Glomb MA. Formation of arginine modifications in a model system of N-alpha-tert- Butoxycarbonyl (Boc)-Arginine with methylglyoxal. J Agric Food Chem 2011;59:394–401.
- Oya T, Hattori N, Mizuno Y, Miyata S, Maeda S, Osawa T, Uchida K. Methylglyoxal modification of protein – Chemical and immunochemical characterization of methylglyoxal- arginine adducts. J Biol Chem 1999;274:18492–18502.
- Shipanova IN, Glomb MA, Nagaraj RH. Protein modification by methylglyoxal: Chemical nature and synthetic mechanism of a major fluorescent adduct. Arch Biochem Biophys 1997;344:29–36.
- Ahmed MU, Brinkmann Frye E, Degenhardt TP, Thorpe SR, Baynes JW. N-epsilon-(carboxyethyl)lysine, a product of the chemical modification of proteins by methylglyoxal, increases with age in human lens proteins. Biochem J 1997;324:565–570.
- Sell DR, Strauch CM, Shen W, Monnier VM. 2-Aminoadipic acid is a marker of protein carbonyl oxidation in the aging human skin: effects of diabetes, renal failure and sepsis. Biochem J 2007;404:269–277.
- Nagaraj RH, Shipanova IN, Faust FM. Protein cross-linking by the Maillard reaction – Isolation, characterization, and in vivo detection of a lysine-lysine cross-link derived from methylglyoxal. J Biol Chem 1996;271:19338–19345.
- Meade SJ, Miller AG, Gerrard JA. The role of dicarbonyl compounds in non-enzymatic crosslinking: a structure- activity study. Bioorg Med Chem 2003;11:853–862.
- Nemet I, Varga-Defterdarovic L. The role of methylglyoxal in the non-enzymatic conversion of tryptophan, its methyl ester and tryptamine to 1-acetyl-beta-carbolines. Bioorg Med Chem 2008;16:4551–4562.
- Niwa T. 3-deoxyglucosone: metabolism, analysis, biological activity, and clinical implication. J Chromatogr B 1999; 731:23–36.
- Usui T, Yanagisawa S, Ohguchi M, Yoshino M, Kawabata R, Kishimoto J, et al. Identification and determination of alpha-dicarbonyl compounds formed in the degradation of sugars. Biosci Biotechnol Biochem 2007;71:2465–2472.
- Gobert J, Glomb MA. Degradation of glucose: reinvestigation of reactive alpha-dicarbonyl compounds. J Agric Food Chem 2009;57:8591–8597.
- Andrewes P. Changes in mail lard reaction products in ghee during storage. Food Chem 2012;135:921–928.
- Gensberger S, Mittelmaier S, Glomb MA, Pischetsrieder M. Identification and quantification of six major α-dicarbonyl process contaminants in high-fructose corn syrup. Anal Bioanal Chem 2012;403:2923–2931.
- Jadhav H, Pedersen CM, Soiling T, Bols M. 3-Deoxy- glucosone is an intermediate in the formation of furfurals from D-glucose. Chemsuschem 2011;4:1049–1051.
- Tsukushi S, Katsuzaki T, Aoyama I, Takayama F, Miyazaki T, Shimokata K, Niwa T. Increased erythrocyte 3-DG and AGEs in diabetic hemodialysis patients: role of the polyol pathway. Kidney Int 1999;55:1970–1976.
- Sakai M, Oimomi M, Kasuga M. Experimental studies on the role of fructose in the development of diabetic complications. Kobe J Med Sci 2002;48:125–136.
- Hamada Y, Araki N, Koh N, Nakamura J, Horiuchi S, Hotta N. Rapid formation of advanced glycation end products by intermediate metabolites of glycolytic pathway and polyol pathway. Biochem Biophys Res Commun 1996;228:539–543.
- Delpierre G, Collard F, Fortpied J, Van Schaftingen E. Fructosamine 3-kinase is involved in an intracellular deglycation pathway in human erythrocytes. Biochem J 2002;365:801–808.
- Lal S, Randall WC, Taylor AH, Kappler F, Walker M, Brown TR, Szwergold BS. Fructose-3-phosphate production and polyol pathway metabolism in diabetic rat hearts. Metabolism 1997;46:1333–1338.
- Wellsknecht KJ, Lyons TJ, McCance DR, Thorpe SR, Feather MS, Baynes JW. 3-deoxyfructose concentrations are increased in human plasma and urine in diabetes. Diabetes 1994;43:1152–1156.
- Collard F, Vertommen D, Fortpied J, Duester G, Van Schaftingen E. Identification of 3-deoxyglucosone dehydrogenase as aldehyde dehydrogenase 1A1 (retinaldehyde dehydrogenase 1). Biochimie 2007;89:369–373.
- Linden T, Cohen A, Deppisch R, Kjellstrand P, Wieslander A. 3,4-Dideoxyglucosone-3-ene (3,4-DGE): A cytotoxic glucose degradation product in fluids for peritoneal dialysis. Kidney Int 2002;62:697–703.
- Ortiz A, Wieslander A, Linden T, Santamaria B, Sanz A, Justo P, et al. 3,4-DGE is important for side effects in peritoneal dialysis what about its role in diabetes. Curr Med Chem 2006;13:2695–2702.
- Mittelmaier S, Niwa T, Pischetsrieder M. Chemical and physiological relevance of glucose degradation products in peritoneal dialysis. J Ren Nutr 2012;22:181–185.
- Jono T, Nagai R, Lin X, Ahmed N, Thornalley PJ, Takeya M, Horiuchi S. N-epsilon-(carboxymethyl)lysine and 3-DG-imidazolone are major AGE structures in protein modification by 3-deoxyglucosone. J Biochem 2004;136: 351–358.
- Hayase F, Konishi Y, Kato H. Identification of the modified structure of arginine residues in proteins with 3-deoxyglucosone, a maillard reaction intermediate. Biosci Biotechnol Biochem 1995;59:1407–1411.
- Niwa T, Katsuzaki T, Ishizaki Y, Hayase F, Miyazaki T, Uematsu T, et al. Imidazolone, a novel advanced glycation end product, is present at high levels in kidneys of rats with streptozotocin-induced diabetes. Febs Lett 1997;407:297–302.
- Franke S, Niwa T, Deuther-Conrad W, Sommer M, Hein G, Stein G. Immunochemical detection of imidazolone in uremia and rheumatoid arthritis. Clin Chim Acta 2000;300: 29–41.
- Kroh LW, Fiedler T, Wagner J. alpha-dicarbonyl compounds – key intermediates for the formation of carbohydrate-based melanoidins. Ann N Y Acad Sci 2008;1126:210–215.
- Miyata S, Liu BP, Shoda H, Ohara T, Yamada H, Suzuki K, Kasuga M. Accumulation of pyrraline-modified albumin in phagocytes due to reduced degradation by lysosomal enzymes. J Biol Chem 1997;272:4037–4042.
- Monnier VM, Nagaraj RH, PorteroOtin M, Glomb M, Elgawish AH, Sell DR, Friedlander MA. Structure of advanced Maillard reaction products and their pathological role. Nephrol Dial Transplant 1996;11:20–26.
- Hellwig M, Henle T. Formyline, a new glycation compound from the reaction of lysine and 3-deoxypentosone. Eur Food Res Technol 2010;230:903–914.
- Hidalgo FJ, Zamora R. In-vitro production of long-chain pyrrole fatty esters from carbonyl-amine reactions. J Lipid Res 1995;36:725–735.
- Nagaraj RH, PorteroOtin M, Monnier VM. Pyrraline ether crosslinks as a basis for protein crosslinking by the advanced Maillard reaction in aging and diabetes. Arch Biochem Biophys 1996;325:152–158.
- Geissler S, Hellwig M, Zwarg M, Markwardt F, Henle T, Brandsch M. Transport of the advanced glycation end products alanylpyrraline and pyrralylalanine by the human proton-coupled peptide transporter hPEPT1. J Agric Food Chem 2010;58:2543–2547.
- Niwa H, Takeda A, Wakai M, Miyata T, Yasuda Y, Mitsuma T, et al. Accelerated formation of N-epsilon-(carboxymethyl) lysine, an advanced glycation end product, by glyoxal and 3-deoxyglucosone in cultured rat sensory neurons. Biochem Biophys Res Commun 1998;248:93–97.
- Skovsted IC, Christensen M, Breinholt J, Mortensen SB. Characterisation of a novel AGE-compound derived from lysine and 3-deoxyglucosone. Cell Mol Biol 1998;44:1159–1163.
- Dai ZY, Wang BL, Sun G, Fan XJ, Anderson VE, Monnier VM. Identification of glucose-derived cross-linking sites in ribonuclease A. J Proteome Res 2008;7:2756–2768.
- Biemel KM, Friedl DA, Lederer MO. Identification and quantification of major Maillard cross-links in human serum albumin and lens protein – evidence for glucosepane as the dominant compound. J Biol Chem 2002;277:24907–24915.
- Reihl O, Biemel KM, Eipper W, Lederer MO, Schwack W. Spiro cross-links: Representatives of a new class of glycoxidation products. J Agric Food Chem 2003;51:4810–4818.
- Karachalias N, Babaei-Jadidi R, Ahmed N, Thornalley PJ. Accumulation of fructosyl-lysine and advanced glycation end products in the kidney, retina and peripheral nerve of streptozotocin-induced diabetic rats. Biochem Soc Trans 2003;31:1423–1425.
- Krause R, Schlegel K, Schwarzer E, Henle T. Formation of peptide-bound Heyns compounds. J Agric Food Chem 2008;56:2522–2527.
- Reihl O, Rothenbacher TM, Lederer MO, Schwack W. Carbohydrate carbonyl mobility – the key process in the formation of alpha-dicarbonyl intermediates. Carbohydr Res 2004;339:1609–1618.
- Hartkopf J, Erbersdobler HF. Model experiments on the formation of N-epsilon-carboxymethyllysine in food-products. Z Lebensm Unters Forsch 1994;198:15–19.
- Sjoberg JS, Bulterijs S. Characteristics, formation, and pathophysiology of glucosepane: a major protein cross-link. Rejuvenation Res 2009;12:137–148.
- Nasiri R, Field MJ, Zahedi M, Moosavi-Movahedi AA. Comparative DFT study to determine if α-oxoaldehydes are precursors for pentosidine formation. J Phys Chem A 2012; 116:2986–2996.
- Sell DR, Biemel KM, Reihl O, Lederer MO, Strauch CM, Monnier VM. Glucosepane is a major protein cross-link of the senescent human extracellular matrix – relationship with diabetes. J Biol Chem 2005;280:12310–12315.
- Fan XJ, Sell DR, Zhang JY, Nemet I, Theves M, Lu J, et al. Anaerobic vs aerobic pathways of carbonyl and oxidant stress in human lens and skin during aging and in diabetes: A comparative analysis. Free Radic Biol Med 2010;49:847–856.
- Grandhee SK, Monnier VM. Mechanism of formation of the maillard protein cross-link pentosidine – glucose, fructose, and ascorbate as pentosidine precursors. J Biol Chem 1991;266:11649–11653.
- Nakayama T, Hayase F, Kato H. Formation of ϵ-(2-formyl- 5-hydroxymethyl-pyrrol-1-yl)-L-norleucine in the Maillard reaction between D-glucose and L-lysine. Agric Biol Chem 1980;44:1201–1202.
- Requena JR, Price DL, Thorpe SR, Baynes JW. Measurement of pentosidine in biological samples. Methods Mol Med 2000;38:209–217.
- Biemel KM, Reihl O, Conrad J, Lederer MO. Formation pathways for lysine-arginine cross-links derived from hexoses and pentoses by Maillard processes – Unraveling the structure of a pentosidine precursor. J Biol Chem 2001; 276:23405–23412.
- Dyer DG, Blackledge JA, Thorpe SR, Baynes JW. Formation of pentosidine during nonenzymatic browning of proteins by glucose - identification of glucose and other carbohydrates as possible precursors of pentosidine invivo. J Biol Chem 1991;266:11654–11660.
- Nasiri R, Zahedi M, Jamet H, Moosavi-Movahedi AA. Theoretical studies on models of lysine-arginine cross-links derived from α-oxoaldehydes: a new mechanism for glucosepane formation. J Mol Model 2012;18:1645–1659.
- Nakamura K, Nakazawa Y, Ienaga K. Acid-stable fluorescent advanced glycation end products: Vesperlysines A, B, and C are formed as crosslinked products in the maillard reaction between lysine or proteins with glucose. Biochem Biophys Res Commun 1997;232:227–230.
- Tessier F, Obrenovich M, Monnier VM. Structure and mechanism of formation of human lens fluorophore LM-1 – relationship to vesperlysine A and the advanced Maillard reaction in aging, diabetes, and cataractogenesis. J Biol Chem 1999;274:20796–20804.
- Nemet I, Strauch CM, Monnier VM. Favored and disfavored pathways of protein crosslinking by glucose: glucose lysine dimer (GLUCOLD) and crossline versus glucosepane. Amino Acids 2011;40:167–181.
- Obayashi H, Nakano K, Shigeta H, Yamaguchi M, Yoshimori K, Fukui M, et al. Formation of crossline as a fluorescent advanced glycation end product in vitro and in vivo. Biochem Biophys Res Commun 1996;226:37–41.
- Pamplona R. Advanced lipoxidation end-products. Chem Biol Interact 2011;192:14–20.
- Hermetter A, Kinnunen P, Spickett C. Oxidized phospholipids-Their properties and interactions with proteins. Biochim Biophys Acta 2012;1818:2373–2373.
- Bochkov VN, Oskolkova OV, Birukov KG, Levonen AL, Binder CJ, Stockl J. Generation and biological activities of oxidized phospholipids. Antioxid Redox Signal 2010;12:1009–1059.
- Hoff HF, O’Neil J, Wu ZP, Hoppe G, Salomon RL. Phospholipid hydroxyalkenals - Biological and chemical properties of specific oxidized lipids present in atherosclerotic lesions. Arterioscler Thromb Vasc Biol 2003;23: 275–282.
- Silva AMN, Borralho AC, Pinho SA, Domingues MRM, Domingues P. Cross-oxidation of angiotensin II by glycerophosphatidylcholine oxidation products. Rapid Commun Mass Spectrom 2011;25:1413–1421.
- Shibata T, Kondo M, Osawa T, Shibata N, Kobayashi M, Uchida K. 15-deoxy-Delta(12,14)-prostaglandin J(2) – a prostaglandin D-2 metabolite generated during inflammatory processes. J Biol Chem 2002;277:10459–10466.
- Garzon B, Oeste CL, Diez-Dacal B, Perez-Sala D. Proteomic studies on protein modification by cyclopentenone prostaglandins: Expanding our view on electrophile actions. J Proteomics 2011;74:2243–2263.
- Schneider C, Porter NA, Brash AR. Routes to 4-hydroxynonenal: fundamental issues in the mechanisms of lipid peroxidation. J Biol Chem 2008;283:15539–15543.
- Long EK, Picklo MJ, Sr. Trans-4-hydroxy-2-hexenal, a product of n-3 fatty acid peroxidation: Make some room HNE. Free Radic Biol Med 2010;49:1–8.
- Niki E. Lipid peroxidation: Physiological levels and dual biological effects. Free Radic Biol Med 2009;47:469–484.
- Szori M, Abou-Abdo T, Fittschen C, Csizmadia IG, Viskolcz B. Allylic H-abstraction hydrogen abstraction from 1,4 type polyalkenes as a model for free radical trapping by polyunsaturated fatty acids (PUFAs). Phys Chem Chem Phys 2007;9:1931–1940.
- Catala A. A synopsis of the process of lipid peroxidation since the discovery of the essential fatty acids. Biochem Biophys Res Commun 2010;399:318–323.
- Stevens JF, Maier CS. Acrolein: sources, metabolism, and biomolecular interactions relevant to human health and disease. Mol Nutr Food Res 2008;52:7–25.
- Seiler N. Catabolism of polyamines. Amino Acids 2004; 26:217–233.
- Balogh LM, Atkins WM. Interactions of glutathione transferases with 4-hydroxynonenal. Drug Metab Rev 2011;43:165–178.
- Alary J, Gueraud F, Cravedi JP. Fate of 4-hydroxynonenal in vivo: disposition and metabolic pathways. Mol Aspects Med 2003;24:177–187.
- Kuiper HC, Miranda CL, Sowell JD, Stevens JF. Mercapturic acid conjugates of 4-hydroxy-2-nonenal and 4-oxo-2- nonenal metabolites are in vivo markers of oxidative stress. J Biol Chem 2008;283:17131–17138.
- Zarkovic N. 4-hydroxynonenal as a bioactive marker of pathophysiological processes. Mol Aspects Med 2003;24: 281–291.
- Jaganjac M, Prah IO, Cipak A, Cindric M, Mrakovcic L, Tatzber F, et al. Effects of bioreactive acrolein from automotive exhaust gases on human cells in vitro. Environ Toxicol 2012;27:644–652.
- Abraham K, Andres S, Palavinskas R, Berg K, Appel KE, Lampen A. Toxicology and risk assessment of acrolein in food. Mol Nutr Food Res 2011;55:1277–1290.
- Liu L, Ye XP, Bozell JJ. A comparative review of petroleum-based and bio-based acrolein production. Chemsuschem 2012;5:1162–1180.
- Arntz D, Fischer A, Höpp M, Jacobi S, Sauer J, Ohara T, et al. Acrolein and Methacrolein. Ullmann’s Encyclopedia of Industrial Chemistry. New York: Wiley; 2007.
- Bein K, Leikauf GD. Acrolein – a pulmonary hazard. Mol Nutr Food Res 2011;55:1342–1360.
- Talhout R, Opperhuizen A, van Amsterdam JGC. Sugars as tobacco ingredient: Effects on mainstream smoke composition. Food Chem Toxicol 2006;44:1789–1798.
- Carmella SG, Chen M, Han S, Briggs A, Jensen J, Hatsukami DK, Hecht SS. Effects of smoking cessation on eight urinary tobacco carcinogen and toxicant biomarkers. Chem Res Toxicol 2009;22:734–741.
- LoPachin RM, Gavin T, Petersen DR, Barber DS. Molecular mechanisms of 4-hydroxy-2-nonenal and Acrolein Toxicity: Nucleophilic Targets and Adduct Formation. Chem Res Toxicol 2009;22:1499–1508.
- Nadkarni DV, Sayre LM. Structural definition of early lysine and histidine adduction chemistry of 4-hydroxynonenal. Chem Res Toxicol 1995;8:284–291.
- Lin D, Lee HG, Liu Q, Perry G, Smith MA, Sayre LM. 4-oxo-2-nonenal is both more neurotoxic and more protein reactive than 4-hydroxy-2-nonenal. Chem Res Toxicol 2005;18:1219–1231.
- Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med 1991;11:81–128.
- Sayre LM, Lin D, Yuan Q, Zhu XC, Tang XX. Protein adducts generated from products of lipid oxidation: Focus on HNE and ONE. Drug Metab Rev 2006;38: 651–675.
- Uchida K, Stadtman ER. Covalent attachment of 4-hydroxynonenal to glyceraldehyde-3-phosphate dehydrogenase - a possible involvement of intramolecular and intermole- cular cross-linking reaction. J Biol Chem 1993;268: 6388–6393.
- Lee SH, Blair IA. Characterization of 4-oxo-2-nonenal as a novel product of lipid peroxidation. Chem Res Toxicol 2000;13:698–702.
- Spiteller P, Kern W, Reiner J, Spiteller G. Aldehydic lipid peroxidation products derived from linoleic acid. Biochim Biophys Acta 2001;1531:188–208.
- Liu ZF, Minkler PE, Sayre LA. Mass spectroscopic characterization of protein modification by 4-Hydroxy-2-(E)- nonenal and 4-Oxo-2-(E)-nonenal. Chem Res Toxicol 2003; 16:901–911.
- Zhang WH, Liu JY, Xu GZ, Yuan Q, Sayre LM. Model studies on protein side chain modification by 4-oxo-2-nonenal. Chem Res Toxicol 2003;16:512–523.
- Zhu XC, Sayre LM. Mass spectrometric evidence for long-lived protein adducts of 4-oxo-2-nonenal. Redox Rep 2007; 12:45–49.
- Doorn JA, Petersen DR. Covalent adduction of nucleophilic amino acids by 4-hydroxynonenal and 4-oxononenal. Chem Biol Interact 2003;143:93–100.
- Uchida K. Current status of acrolein as a lipid peroxidation product. Trends Cardiovasc Med 1999;9:109–113.
- Furuhata A, Ishii T, Kumazawa S, Yamada T, Nakayama T, Uchida K. N-epsilon-(3-methylpyridinium) lysine, a major antigenic adduct generated in acrolein-modified protein. J Biol Chem 2003;278:48658–48665.
- Maeshima T, Honda K, Chikazawa M, Shibata T, Kawai Y, Akagawa M, Uchida K. Quantitative analysis of acrolein-specific adducts generated during lipid peroxidation-modification of proteins in vitro: identification of N-tau-(3-Propanal)histidine as the major adduct. Chem Res Toxicol 2012;25:1384–1392.
- Del Rio D, Stewart AJ, Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis 2005;15:316–328.
- Zhang M, Li W, Li T. Generation and detection of levuglandins and isolevuglandins in vitro and in vivo. Molecules 2011;16:5333–5348.
- Davies SS, Amarnath V, Roberts LJ. Isoketals: highly reactive gamma-ketoaldehydes formed from the H-2-isoprostane pathway. Chem Phys Lipids 2004;128:85–99.
- Salomon RG. Distinguishing levuglandins produced through the cyclooxygenase and isoprostane pathways. Chem Phys Lipids 2005;134:1–20.
- Montuschi P, Barnes P, Jackson Roberts L. Insights into oxidative stress: the isoprostanes. Curr Med Chem 2007; 14:703–717.
- Doureradjou P, Koner BC. Effect of different cooking vessels on heat induced lipid peroxidation of different edible oils. J Food Biochem 2008;32:740–751.
- Slatter DA, Bolton CH, Bailey AJ. The importance of lipid-derived malondialdehyde in diabetes mellitus. Diabetologia 2000;43:550–557.
- Slatter DA, Murray M, Bailey AJ. Formation of a dihydropyridine derivative as a potential cross-link derived from malondialdehyde in physiological systems. Febs Lett 1998; 421:180–184.
- Ishii T, Ito S, Kumazawa S, Sakurai T, Yamaguchi S, Mori T, et al. Site-specific modification of positively-charged surfaces on human serum albumin by malondialdehyde. Biochem Biophys Res Commun 2008;371:28–32.
- Ishii T, Kumazawa S, Sakurai T, Nakayama T, Uchida K. Mass spectroscopic characterization of protein modification by malondialdehyde. Chem Res Toxicol 2006;19:122–129.
- Slatter DA, Paul RG, Murray M, Bailey AJ. Reactions of lipid-derived malondialdehyde with collagen. J Biol Chem 1999;274:19661–19669.
- Slatter DA, Avery NC, Bailey AJ. Identification of a new cross-link and unique histidine adduct from bovine serum albumin incubated with malondialdehyde. J Biol Chem 2004;279:61–69.