Abstract
Reactive derivatives of non-enzymatic glucose-protein condensation reactions integrate a heterogeneous group of irreversible adducts called advanced glycation end-products (AGEs). Numerous studies have investigated the role of the AGEs in cardiovascular system; however, its contribution to erectile dysfunction (ED) that is an early manifestation of cardiovascular disease has been less intensively investigated. This review summarizes the most recent advances concerning AGEs effects in the cavernous tissue of the penis and in ED onset, particularly on diabetes and aging, conditions that not only favor AGEs formation, but also increase risk of developing ED. The specific contribution of AGE on intra- and extracellular deposition of insoluble complexes, interference in activity of endothelial nitric oxide (NO) synthase, NO bioavailability, endothelial-dependent vasodilatation, as well as molecular pathways activated by receptor of AGEs are presented. Finally, the interventional actions that prevent AGEs formation, accumulation or activity in the cavernous tissue and that include nutritional pattern modulation, nutraceuticals, exercise, therapeutic strategies (statins, anti-diabetics, inhibitors of phosphodiesterase-5, anti-hypertensive drugs) and inhibitors of AGEs formation and crosslink breakers, are discussed. From this review, we conclude that despite the experiments conducted in animal models pointing to the AGE/RAGE axis as a potential interventional target with respect to ED associated with diabetes and aging, the clinical data have been very disappointing and, until now, did not provide evidence of benefits of treatments directed to AGE inactivation.
The corpus cavernosum
The penis is a high-vascularized organ. Two corpora cavernosa separated by an incomplete septum and constituted by erectile tissue extend along the length of the penis and fill with blood during erection which significantly increases their volume. Corpus cavernosum has a sponge-like texture, exhibiting a mesh of interconnected cavernous sinusoidal spaces, lined by endothelium and separated by trabeculae composed mainly of smooth muscle (SM) and connective tissue (collagen fibers and fibroblasts). Interestingly, and despite the anatomical and physiological homology, the rat cavernous tissue significantly differs from the human's counterpart in histological organization, particularly in the content of connective tissue. In fact, morphometric analysis after transmission electron microscopy observation demonstrated that cavernous tissue in young human presents 30% of connective tissue [Citation1], a level that ascends to more than 40% in the young adult rat [Citation2]. On the other hand, the percentage of smooth muscle cells (SMC) in human, at youth, in average doubles that observed in rat (50% vs 22%, respectively). Another important difference observed between rat's and human's corpus cavernosum is the organization of the fusiform SMCs, which are restricted to endothelium periphery in rodent, while in tissue of human origin are organized in long contractile fibers distributed within trabeculae ().
Figure 1. Hematoxylin and eosin staining of penile tissue from young rat (1) and human (2). Cavernous tissue has a sponge-like structure, with cavernous vascular spaces (VS) lined by endothelium (arrow) and separated by trabeculae composed mainly of connective tissue (CT), while smooth muscle fibers (SM) in the rat are restricted to endothelium periphery, in the human tissue organize in long contractile fibers distributed within trabeculae.
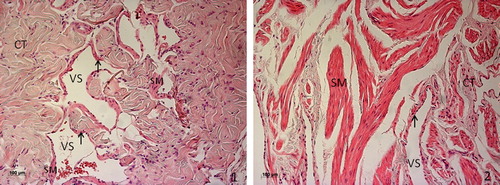
Regardless of these differences, the molecular mechanism of penile erection is common to both species. Indeed, rodents constitute a valuable model for the study of the factors that influence erectile function in the man, and most of the knowledge that now exists is supported by studies performed in rodent experimental models.
The erection mechanism
The development of an erection is a complex event involving integration of psychologic, neurologic, endocrine, vascular, and local anatomic systems. Sexual arousal is activated in higher cortical centers which then stimulate the medial pre-optic and paraventricular nuclei of the hypothalamus. These signals descend through a complex neural network involving the parasympathetic nervous system and eventually activate terminations in the sacral area. The neurovascular events that ultimately occur result in the inhibition of adrenergic tone and in the liberation of the non-adrenergic, non-cholinergic neurotransmitter gaseous molecule nitric oxide (NO) that is believed to be released from nerves and endothelial cells in penis erection, synthesized by neuronal NO synthase (nNOS) and endothelial NO synthase (eNOS), respectively. It has been thought that while NO produced by nNOS initiates penile erection, NO generated by eNOS participates in the sustained erection required for normal sexual performance [Citation3]. However, it was recently reported that nNOS also participate in NO synthesis along all the erectile process [Citation4]. NO is easily diffusible to other cells and, subsequently, stimulates the enzyme guanylate cyclase in penile SM that converts guanosine triphosphate (GTP) to cyclic guanosine monophosphate (cGMP). The increase in cGMP levels results in activation of protein kinase G and further decrease in intracellular calcium due to promotion of its efflux, which triggers the relaxation of arterial and trabecular SM. Vasodilatation enhances arterial blood flow into the lacunar spaces of the penis, which rises intracavernous pressure by a veno-oclusion mechanism that consists in the compression of the subtunical venules against the tunica albuginea and entrapment of the blood in corpora cavernosa. These coordinated actions establish the penis erection with adequate firmness for sexual activity [Citation5]. The molecular crosstalk between endothelium that produces most of the NO and SMCs that support vasodilatation presents an important role in erectile process. The sinusoidal small vessels of the penis are thus particularly sensitive to endothelial structural and functional changes [Citation6], and even minor modifications could lead to erectile dysfunction (ED). In fact, vasculogenic ED, the most prevalent form of ED, which directly results from vascular disease of the penile arteries, failure of the corporal veno-occlusive, or sinusoidal relax mechanisms [Citation7], is mainly provoked by an endothelium-dependent inability to vasodilatation [Citation8,Citation9], a condition named endothelial dysfunction that results from impaired bioavailability of NO, usually due to diminished synthesis or enhanced degradation in the perivascular SM. The contribution of endothelial dysfunction to ED onset is so relevant that these conditions were recently considered equivalent by some authors [Citation8,Citation9].
Epidemiology and risk factors for erectile dysfunction
Erectile dysfunction (ED), defined as the inability to develop and maintain an erection for satisfactory sexual intercourse or activity, is a highly prevalent disease affecting millions of men worldwide with a tendency for widespread increase [Citation10]. In 1994, the Massachusetts Male Aging Study [Citation11] reported a global prevalence of 52% considering any degree of ED, and demonstrated a significant age-related increase; while at 40 years, approximately 40% of men are affected, the rate increases to nearly 70% in men aged 70 years. Accordingly, the prevalence of complete ED increases from 5% to 15% as age increases from 40 to 70 years. Corroborating these data was recently reported that age is the independent variable most strongly associated with ED [Citation12]. Nevertheless, the direct causes of age-related ED are poorly understood and likely to be multifactorial in origin.
A European survey performed at 2005, demonstrated that besides aging, diabetes mellitus (DM), and hypertension (HTA) are the most important risk factors for the development of ED [Citation13]. Men with diabetes in average present a prevalence of ED higher than 50% [Citation14], and complaint of ED is 1.9–4 times more in diabetic than in non-diabetic men [Citation15]. Corroborating these findings, the Global Online Sexuality Survey (GOSS), an epidemiologic study based on validated questionnaires with applied age adjustment, reinforced that DM, HTA with and without anti-hypertensive treatment, coronary heart disease, obesity (defined by body mass index), and also psychological causes constitute serial risk factors for ED [Citation16]. Interestingly, neither smoking, nor alcohol consumption were associated with higher prevalence of ED, although duration of smoking was, in the group of smokers ED patients.
Analyzing together the conclusions of these important appraisals become clear that ED shares chief risk factors for cardiovascular disease (CVD), which support the common etiology of these diseases. In fact, this is consensually accepted, taking in account that endothelial dysfunction which underlies ED often precedes atherosclerosis formation [Citation17], a chronic inflammatory disease that develop over years and further evolves to CVD [Citation17]. Indeed, ED has recently become considered the earliest manifestation of atherosclerosis and a precursor of systemic vascular disease [Citation18], instead of an age-related complication of CVD. Furthermore, it becomes evident that the main risk factors for ED, in particular aging and DM, are those that more strongly associate with an increase in oxidative stress [Citation19,Citation20], characterized by an imbalance between the production of reactive oxygen species (ROS) and the endogenous antioxidant defenses at cellular level. This was not surprising, considering that increase in oxidative stress in cavernous tissue not only compromises NO bioavailability necessary to erectile function [Citation21], but also induces damage in the cavernous cells. Supporting that, it was demonstrated that aging and DM cause serious structural modifications in cavernous tissue, in both rat and human [Citation1,Citation2,Citation22,Citation23]. In addition, it is now clear that oxidative stress is involved in advanced glycation end-products (AGE) formation [Citation24], and that, on the other hand, AGEs induce oxidative stress [Citation25–28], constituting a deleterious vicious cycle that highly compromise normal erectile function. The increase in AGE formation in tissues and serum is strongly associated with DM [Citation29–31] and in lower extent to chronological aging [Citation24,Citation32,Citation33,Citation34], too.
AGE effect in cavernous tissue
Proteins could suffer reversible modifications by non- enzymatic glycosylation – glycation (often referred as the Maillard reaction) [Citation35] between free amino groups of amino acids such as lysine, arginine or protein terminal amino group and oxo group of sugars (glucose, fructose, ribose and others) and initially form Schiff bases, and further Amadori and Heyns rearrangements products [Citation36], that result from reaction with α-hydroxyl aldehydes (D-glucose) or α-ketone (D-fructose) groups in sugars, respectively. AGEs are formed ultimately, result of dehydration, condensation, fragmentation and crosslinking complex reactions ensuing initial glycation reactions (). Degradation of Schiff base and Amadori products generates highly reactive carbonyl compounds (glyoxal and methylglyoxal) [Citation37] that are also formed during glycolysis and by glucose autoxidation in the presence of catalytical metals, such as Cu2+, Fe2+, Mn2+ and Zn2+, and O2 [Citation38]. This class of carbonyl compounds could react directly with proteins to form glycoxidation products [Citation39,Citation40]. Methylglyoxal is the most reactive precursor of the formation of AGEs in aging [Citation41] and diabetes [Citation25]. Lipids and nucleic acids exposed to reducing sugars could also suffer glycation or glycoxidation reactions and form AGEs. Thus, AGEs constitute a chemically heterogenous group of compounds, conspiring that the pattern of AGE end products formed in any reaction is critically dependent in the precise conditions in which the reaction is occurring, and in contrast with Schiff bases their formation is non-reversible [Citation38]. AGEs were originally characterized as yellow/brown fluorescent compounds, but currently the term is used for a broad range of advanced end products, that show neither color, nor fluorescence. Further to stable end products of Maillard reaction, circulating AGEs of dietary origin [Citation42] and AGE peptides, result of activity of detoxifying enzymes, commonly called glycotoxins are very reactive, and can generate new AGE adducts after reaction with plasma or tissue components [Citation31,Citation42,Citation43]. Protein modifications may also lead to alteration of the interaction of target proteins, hence affecting the normal function of other non-modified proteins. Once formed, AGEs accelerate the processes of tissue deterioration.
Figure 2. Schematic presentation of the synthesis of AGEs.
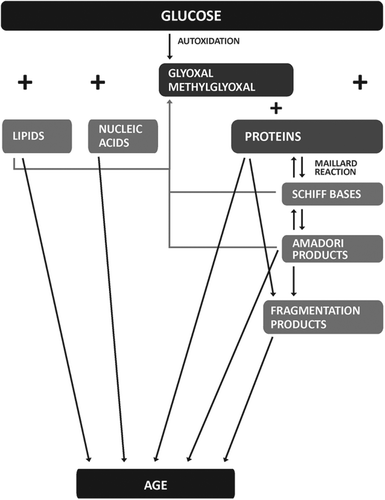
Diabetes is the condition that mostly favors AGE formation, considering that glucose in blood reacts with proteins leading to glycative, glycoxidative, and carbonyl modifications. Erythrocytes are freely permeable to glucose, and glycosylated hemoglobin (Hb) may be formed by reaction between Hb A and glucose, leading to the formation of an intermediary Amadori product HbA1c and accumulation of an irreversible end-product Hb-AGE [Citation44]. Similarly, endothelial cells present free non-insulin-dependent glucose transport which inside the cell provokes mitochondrial electron leakage and superoxide anion formation from oxygen reduction, which ultimately results in increased methylglyoxal-derived AGE formation [Citation45]. The formation of AGEs in DM correlates thus with glycemic control.
In contrast to DM, in chronological aging, AGE formation occurs more slowly and predominantly on long-lived proteins, such as extracellular matrix proteins that present a slow turnover. Over a person's lifespan, the amount of AGE-modified tissue progressively increases, partly due to the accumulation of AGE-modified proteins that resist to proteolytic digestion inside the cell. Interestingly, it is possible to estimate the “age” of any protein by its degree of AGE modification. Along aging, in parallel with glycation, proteins could suffer irreversible oxidation, due to the increased age-related oxidative stress. Direct oxidation of proteins result frequently in carbonyl group formation, which also exist in AGE adducts. Similar to AGEs, oxidized proteins present abnormal conformations that result in loss of native structure and catalytic activity and render them unable to exert their functions. Increased oxidative damage, accumulation of damaged proteins, and metabolic dysfunction normally considered independent age-related events could thus share a unique origin – modifications of proteins. In fact, carbonylated proteins and AGE formation are likely to be implicated in the molecular basis of aging and age-related diseases as suggested by in silico approaches [Citation24].
The mechanisms by which AGEs affect the cardiovascular system, and in particular the corpus cavernosum, are complex and include the following: (1) modification of extracellular matrix and intercellular adhesion, partly due to increase in collagen crosslinking, which favors its deposition in connective tissue (hallmark of dysfunctional heart and erectile tissue). AGEs form covalent bonds with collagen, which leads to vascular wall thickening and decreased elasticity that strongly contribute to dysfunction of cavernous tissue. Elastin could also be AGE-modified, but due to its low content of lysine residues compared to that of collagen, glycation occurs to a lesser extent. Despite that, Baumann [Citation46] speculated that glycative modifications of elastin lead to destruction of elastic fibers of arteries, strongly affecting its elasticity, as previously observed by transmission electron microscopy in human dermal tissue [Citation47]. AGE-modified proteins also affect intercellular interaction, namely in the heterotypic binding between cells and matrix, that once disturbed by glycation of matrix molecules, contribute to changes in cellular adhesion. On the other hand, modification of proteins involved in homotypic interactions required for epithelium assembly alters cell growth and induces loss of the epithelium phenotype [Citation48]. (2) Alteration in low-density lipoprotein (LDL) composition and trapping. Glycative modifications in extracellular matrix proteins contribute to the trapping of macromolecules such as lipoproteins in the vessel wall, which together with the impairment in degradation of AGE-modified proteins, lead to the accumulation of perivascular amorphous hyalinized material that characterizes diabetic vascular disease [Citation49] and atheroma formation [Citation50]. Indeed, AGEs have been identified in atherosclerotic plaques, even in the absence of diabetes [Citation51]. (3) It was also demonstrated that subendothelially deposited AGEs are chemotactic for monocytes, which allows its migration through the endothelial monolayer, and subsequent secretion of inflammatory molecules, attending that these cells are highly responsive to AGEs [Citation52]. (4) Increased expression of inflammatory cytokines induces proliferation of SMCs, as demonstrated in a co-culture system with monocytes, endothelial, and SMCs that revealed synergistic effects of AGEs on intercellular interactions implicated in atherosclerosis onset [Citation53]. Proliferation of SMCs is also promoted by insulin-like growth factor-1 (IGF-1) and platelet-derived growth factor (PDGF) isoform-BB, secreted by infiltrated monocytes triggered by AGE-modified proteins, in particular fibronectin [Citation52,Citation54,Citation55]. Platelet-derived growth factor engages PDGF receptor (PDGFR), inducing its dimerization and activation, and depending on the isoform of PDGF, PDGFR could be constituted by two chains α, two β, or otherwise one α and one β. Receptor chain β is a target to AGE binding in vivo, and AGE adducts formation alters mitogenic signaling in response to PDGF-BB. Indeed, circulating methylglyoxal and glyoxal contribute to induce PDGFR dysfunction and increase the vulnerability of atherosclerotic plaques [Citation56]. Interestingly, AGE-modified PDGF significantly loose its biological activity, too [Citation57]. Protein modifications due to AGE formation also contribute to transactivation of IGF-1 receptor as demonstrated in 3T3-L1 pre-adipocytes [Citation58], but as far as we know, no data on endothelial or SMCs are available. (5) Elevated serum AGEs contribute specifically to oxidative stress [Citation59], as AGEs upregulate the expression and activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, an important source of vascular oxidative stress [Citation60] and decrease expression of enzymes involved in antioxidant systems such as Mn-superoxide dismutase (SOD) observed in mice endothelial cells [Citation61], and (6) impairment of cellular NO bioavailability. The amount of NO has been demonstrated to be directly reduced by AGEs [Citation62], and levels of AGEs had been thus negatively associated with the extent of endothelium-dependent vasodilatation [Citation63]. In addition, it has been demonstrated in vitro that AGEs may diminish eNOS activity through receptor-mediated phosphorylation of serine residues in eNOS [Citation64], and not by competition with endogenous substrate (L-arginine) considering that neither L-arginine nor L-lysine-derived AGEs appear to impair NO biosynthesis by eNOS [Citation65]. AGEs also induce uncouple of eNOS that impedes NO synthesis, and reduce eNOS mRNA expression [Citation61]. Furthermore, AGEs are able to antagonize NO function by up regulating the expression of the vasoconstrictor endothelin-1 (ET-1) in endothelial cells [Citation66].
The production and accumulation of AGEs constitute thus a possible explanation for age- and diabetes-related increased ED risk. Supporting that, higher levels of AGE products, namely the fluorescent pentose-mediated protein crosslink pentosidine had been detected in corpus cavernosum of both aged and diabetic patients [Citation29,Citation32]. Interestingly, the levels of AGEs observed in cavernous tissue of diabetic patients were significantly higher than those observed in the serum, compared to those of controls, which clearly indicate that erectile tissue, specially cavernous collagen fibers, constitutes a preferential location for AGE accumulation [Citation29], with its consequent deleterious effect [Citation67]. In addition, it was reported that PDGF and PDGFR are constitutively expressed in the corpus cavernosum of the rat in endothelium and nerves, and that hypoxia, a direct consequence of advanced atherosclerosis, significantly up-regulates their expression [Citation68], suggesting that conditions that impedes the perfect blood perfusion in penis interfere in the PDGF system regulation. Considering that PDGF and its receptor could be the targets to AGE binding, it could not be excluded their interference on ED onset in aging and diabetes. Indeed, AGEs contribute specifically to ED by multiple mechanisms (1) physically decreases extensibility of the erectile tissue, (2) generates oxidative cell damage, and (3) reduces NO, which directly decreases cGMP bioavailability and impairs cavernosal SM relaxation [Citation30]. In addition, AGEs may also affect several different channels and receptors on the cavernosal SMCs, particularly potassium channels that mediate intracellular calcium release essential to SM relaxation [Citation14].
Mechanisms of AGE/RAGE in erectile dysfunction
Several AGE-affinity molecules, generally named AGE receptors (AGERs), have been identified. One of the functions attributed to AGE-binding molecules, expressed at cell surface mainly in monocytes and macrophages, is mediation of AGEs cell uptake from blood and tissues. This process is followed by AGE degradation in cells, improving its catabolism and turnover, constituting thus an anti-glycoxidant effect [Citation69]. The first cell surface receptor molecule found to be involved in endocytosis and that contributes to suppression of AGE-mediated mesangial cell inflammatory injury due to diabetes and aging is AGE receptor 1 (AGER1) [Citation70]. This 48-kDa transmembrane protein is found both in the endoplasmic reticulum and in the cytoplasmic membrane, exhibiting significant AGEbinding capacity and negative regulation of receptor of AGE (RAGE) effects, described below. It was also demonstrated that AGER1 provides protection against AGE-induced ROS generation via NADPH oxidase [Citation71]. This protective effect could, however, be lost under prolonged oral exposure to methylglyoxal-derived AGEs, considering that it induces depletion not only of AGER1 but also of SIRT1 [Citation72], a NAD+-dependent deacetylase with vascular protective effects, recently identified in the cavernous tissue of rodent and human [unpublished data, 73]. A 80- to 90-kDa protein was co-purified with AGER1 and named AGER2. An additional class of AGERs mainly expressed by macrophages but also found at endothelial cells is an β-galactoside high-affinity binding 32-kDa soluble lectin named galectin-3 or AGER3 [Citation74]. Galectin-3 abundant in the nucleus and cytoplasm is rapidly translocated to the cell surface upon exposure to AGEs, where it promotes endocytosis and degradation of AGE-moieties, apparently contributing to the maintenance of tissue integrity, supported by demonstration of increased inflammation in galectin-3 knockout mice [Citation75]. On the other hand, recent evidences such as upregulation of galectin-3 in the endothelium of Tie2-GFP transgenic mice under high-fat diet-induced diabetes [Citation76], galectin-3 detection in rat's SMC in rat aorta, apparently intervening in their AGE-induced proliferation [Citation77], demonstration that galectin-3 strongly increases vascular and heart fibrosis [Citation78], in line with the recent finding that galectin-3 inhibition in apoliprotein E-deficient mice avoids atherosclerosis formation [Citation79], do not agree with a protective role for galectin-3. Supporting that, a significant correlation between age, risk factors for CV disease, and all-cause mortality in the general population with galectin-3 levels was demonstrated in a cohort of 7968 subjects [Citation80]. Other AGE receptors such as the macrophages scavenger receptor (MSR) [Citation81] and the oxidized-LDL receptor CD36 [Citation82], both involved in the mechanisms of endocytosis and foam cells formation in atheromatous lesions, had been described.
Other well-studied class of AGERs triggers inflammatory processes in tissues, instead of being involved in AGE uptake and turnover. In fact, most of the effects normally attributed to AGEs occur after engagement and activation of RAGE (a member of the immunoglobulin superfamily). Receptor of AGE is the chief cellular receptor of AGEs and one of the best-characterized, and seems to amplify inflammatory signals initiated by AGEs by multiple mechanisms [Citation83]. The molecular cloning of RAGE predicted that it is constituted by an extracellular domain of three immunoglobulin (Ig)-like domains (one variable-type followed by two constant-type Ig domains), a single transmembrane domain and a very short highly charged cytoplasmic domain essential to RAGE-mediated signaling transduction [Citation84]. RAGE could engage non-AGE ligands too, such as proteins in the S100 family including S100A12, also called extracellular newly identified RAGE-binding protein (EN-RAGE) [Citation85], S100b, amyloid β peptide, the family of β sheet fibrils, amphoterin [Citation86], and high-mobility group box protein 1 (HMGB1), that mainly bind to its variable-type Ig domain [Citation83]. The exact role of RAGE in normal physiology is still unclear, considering that most of the RAGE is constitutively expressed at a low level in the absence of injury or inflammation in a limited number of cell types and tissues, such as endothelial cells, vascular SMCs, neurons, and leucocytes. However, RAGE expression is highly increased in response to AGE formation associated with specific conditions such as diabetes, dyslipidemia, and aging. In particular, an increased expression of RAGE was demonstrated in aged rodent vascular tissues, compared to that of young rats [Citation87]. Interestingly, RAGE might regulate levels of its ligands as AGE levels tend to reduce in the kidney of RAGE-deficient rats, compared to those of the controls, in equivalent conditions of glycemia [Citation88].
Despite recent evidences of RAGE intervention in regeneration of injured neurons and cardiac and skeletal muscles, especially at low-intensity RAGE signaling [89 for review], in a milieu of abundant AGEs as those existent in aged and diabetic individuals, that obligatory leads to excess RAGE stimulation, these protective effects do not probably occur. Moreover, endothelial and SMC respond to RAGE activation in the context of an inflammatory process, and so far, no evidences of any protective intervention of RAGE-activated signaling in these cells was reported. In fact, AGE/RAGE binding is likely involved in several signaling cascades, and in particular in those associated with atherosclerotic processes. Indeed, RAGE is expressed at atherosclerotic plaques in diabetic and in lower levels in non-diabetic individuals [Citation90]. In the atherosclerotic process, RAGE expression is particularly evident in endothelial cells, and in lower extent, in monocytes/macrophages and SMCs [Citation48]. Support for RAGE intervention in AGE-mediated atherosclerosis pathways come from demonstration that RAGE gene deletion resulted in marked reduction in atherosclerotic lesions and vascular inflammation in mice deficient in apolipoprotein E (apoE) or in low-density lipoproteins receptor [Citation48,Citation91] without modification in blood levels of glucose or lipids. These findings clearly evidenced that the mechanism of atherosclerosis formation strongly depends on RAGE-mediated pathways, and fundamentally that AGEs are not only markers but also mediators of chronic vascular complications. In line with this, it was verified that hypoxia is a strong activator of AGE formation, and supported by evidence that RAGE-deficient mice presented a diminished response to hypoxia compared to wild types, that downstream effects were mediated by RAGE [Citation83].
AGEs directly enhance the synthesis of free radicals of oxygen that apparently play an important role in oxidative stress RAGE-mediated signaling [Citation92]. Moreover, RAGE is involved in the generation of ROS partly due to NADPH oxidase activation [Citation93]. AGE downstream pathways through its receptor RAGE also (1) increases endothelial permeability to macromolecules, in part caused by loss of anionic sites which results from AGE-related impairment of heparin sulfate binding to extracellular matrix; (2) mediates migration of monocytes into the vessel wall, in a process that (3) increases intravascular fat accumulation and (4) promotes apoptosis and inhibits migration of endothelial progenitor cells [94 for review]. In addition, RAGE is a potent upregulator of NF-kappaB (NF-kB), a transcription factor responsible for many key biological processes, such as inflammation, apoptosis, and stress response. In fact, NF-kB modulates gene transcription of endothelin-1, vascular endothelial growth factor (VEGF), transforming growth factor β (TGF-β), pro-inflammatory cytokines, such as interleukins 1 α and 6 and tumor necrosis factor α (TNF-α), and of the adhesion molecules vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). Reduction or inhibition of NF-kB activation is the goal of many therapeutical interventions that successfully alleviate diseases of the myocardium [Citation95]. Indeed, AGE–RAGE strongly contributes to vascular injury, because further to facilitate the micro-environment conductive to AGE production (oxidative stress) also activates NF-kB, which strongly amplifies inflammatory response and leads to an evolving cycle of AGE production. Blockade of AGE/RAGE pathway is thus a potential target to control or avoid atherosclerosis progression.
Other studies suggest that AGE proteins are not exclusive drivers of RAGE-dependent pro-inflammatory cellular responses. In fact, EN-RAGE mediates inflammatory response in mice after RAGE engagement [Citation85], by a mechanism blocked by anti-RAGE/anti-EN-RAGE IgG treatments. Furthermore, Valencia et al. [Citation96] reported that high-affinity binding of AGE-modified albumin from bovine origin to RAGE is not sufficient to induce VCAM-1 and TNF-α secretion in endothelial cells grown in vitro, suggesting that only specific AGE modifications are capable of activating this cell type. In line with that, it was shown that methylglyoxal lysine- or carboxymethyl lysine-albumin neither bind nor induce activation of endothelial cells, evaluated by measurement of adhesion molecules expression in culture [Citation97]. In the same study, it was reported the existence of truncated splice variants of RAGE in cells, discussed below, which might explain the lack of interaction of intact RAGE with extracellular AGEs. Thus, the binding specificity and activation ability of the various AGE-modified molecules to RAGE remain to be elucidated, with all the evidences pointing to the existence of cell-specific mechanisms [Citation98].
As far as we know, expression of RAGE in corpus cavernosum was not yet demonstrated, neither in rodent, nor in human. Nevertheless, previous observation of foam cells of muscle origin in the corpus cavernosum of the aged rat [Citation99] supports the AGE/RAGE involvement in atherosclerosis progression in the penis vessels, considering that intracellular accumulation of AGEs promotes this phenotypic conversion within atherosclerotic plaques by RAGE activation [Citation100], further to the enhancement of SMCs proliferation. Similar to RAGE, no evidences of expression of the other classes of AGERs on cavernous tissue of either rodent or human origin were reported.
Soluble RAGEs
Besides RAGE expression in multiple cell types, soluble forms of RAGE generated by alternative splicing of C- terminal circulate in blood [Citation101]. The soluble forms of RAGE (sRAGE) do not contain neither the transmembrane nor the effector cytoplasmatic domains, but retain the ability to bind to AGEs. At least two main types of sRAGE forms can be distinguished, considering that further to sRAGE which encompasses the material formed by proteolytic cleavage from cell surface receptor, a novel splice variant expressed and secreted in human vascular cells, that is named endogenous secretory RAGE (esRAGE), was recently found. This peptide possesses a different sequence of amino acids in one of the constant Ig-like domains, compared with the other sRAGE, which allows its specific detection in serum. Endogenous secretory RAGE neutralizes AGE effects in endothelial cells and is present in human serum [Citation102]. In healthy individuals, sRAGE is present in blood and in secretions and, similar to esRAGE, seems to be involved in targeting senescent molecules to degradation. A significantly lower level of sRAGE was observed in diabetic individuals which was associated with an age-independent increase in arterial stiffness [Citation103]. Interestingly, soluble forms of RAGE seem to protect against atherosclerotic plaque formation, considering that sRAGE administration to diabetic mice deficient in apoE led to a significant reduction in lesion area when compared with the controls that received the vehicle [Citation104]. Administration for 6 weeks reduced inflammation due to inactivation of NF-kB and, consequently decreased expression of TGFβ, VCAM-1, fibronectin, and type IV collagen in aorta [Citation105]. Moreover, administration of sRAGE to aged rats prevented aging-associated impairment of endothelial function, a condition that often precedes atherosclerosis [Citation87]. Despite the promissory effect of sRAGE administration in AGEs scavenging that apparently prevents its interaction with cell-surface pro-inflammatory receptors [Citation94], the role of endogenous sRAGE is still a matter of discussion, considering that high levels were associated with an increased risk of coronary heart disease in DM type 2 patients [Citation106]. This finding suggested that proteolytic cleavage of RAGE could mediate inflammatory processes in these conditions. On the other hand, a potential protective effect of esRAGE for atherosclerosis was observed in patients [Citation107], supported by the strong negative correlation observed between esRAGE blood levels and subclinical atherosclerosis and CVD risk factors [Citation107] that in turn associate with ED. This evidences prompted Emanuele and Bretonato to hypothesize that esRAGE blood levels could act as a biomarker for ED [Citation108]. However, further studies will be necessary to clarify this aspect.
As the accumulation of AGEs is likely involved in ED risk, strategies that intervene in its formation, accumulation, and RAGE-mediated pathways have been investigated. These approaches were classified as the following: (1) nutritional pattern modulation, (2) nutraceuticals (products derived from food sources that besides nutritional value provide extra health benefits), (3) exercise, and (4) therapeutic strategies that include drugs currently employed in the treatment of other diseases ().
Figure 3. Schematic presentation of the nutritional and therapeutic interventions on the formation of AGEs and ED onset. ACEI, angiotensin-converting enzyme inhibitors; ARB, angiotensin II receptor-1 blocker; cGMP, cyclic guanosine monophosphate; ER, energy restriction; LDL, low-density lipoprotein; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; PDE5, phosphodiesterase-5; ROS, reactive oxygen species; SMC, smooth muscle cells.
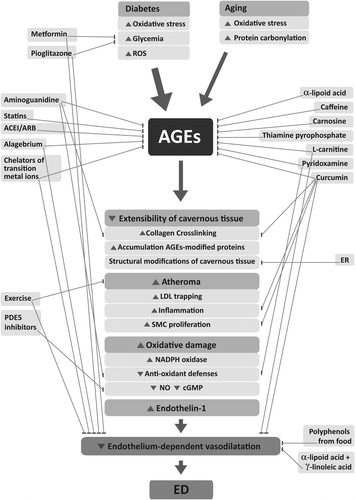
Prevention of AGE formation or AGE-related damage
Nutritional pattern modulation
AGEs are heterogeneous substances, and diet represents an important source especially if the preparation of food was carried out at high temperature that is demonstrated to facilitate AGE formation. Browned or caramelized foodstuffs, such as coffee or cola drinks, tend to have the highest levels of AGEs that are often incorporated into foods for intensifying the natural flavors. In fact, the AGE content of Western diet significantly increased in the last 50 years, together with sugars and lipids. This could be deleterious to the population, considering that exogenous AGEs, result of reactions between sugars and proteins from diet, are believed to be absorbed after ingestion, and passed to the circulation. In this setting, a strong correlation between AGE consumption and serum levels was demonstrated [Citation109]. In diabetic patients, as renal impairment progresses, an increase in blood AGEs, positively related to creatinine levels, also occurs. Circulating AGEs from diet have been correlated with inflammatory marker high-sensitivity C-reactive protein (hsCRP) blood levels [Citation110] and vascular compliance in humans [Citation111], and may therefore contribute to ED onset. Thus, a low AGE intake could represent a strategy to avoid vascular disturbances, and despite the fact that it may be difficult to maintain for a long term in humans, the benefits in the prevention of atherosclerosis and ED, even for healthy individuals, could be valuable. Supporting that, previous studies evidenced that energy restriction (ER), a dietary pattern that reduced the energy ingestion normally to 60–75% of the ad libitum ingested amount and that significantly decreased serum AGEs in obese humans after a short-term intervention [Citation112], induced profound modifications in the structural organization of the corpus cavernosum of the rat, both in short and in long-term treatments, being able to reverse the connective tissue deposition induced by high-fat diet regular consumption [Citation113,Citation114]. The decrease in cavernous connective tissue content manifests as ameliorated erectile function in the elderly [Citation115]. Thus, we hypothesize that this improvement can be partly due to reduction in cavernous AGE formation. Decreasing the glycation burden through ER will minimize the formation of Amadori products and prevent AGE accumulation in corpus cavernosum. In the absence of these compounds, that normally co-localize and crosslink with collagen fibers [Citation116,Citation117], the percentage of cavernous fibrous collagen deposed in connective tissue tends to reduce. In line with this, others reported a significant reduction in skin collagen AGE levels in rhesus monkeys submitted to long-term ER [Citation118]. However, the exact contribution of the glycation-modified collagen in the cavernous connective tissue organization remains elusive, and further studies will be necessary to clarify its function.
Beyond the ER or dietary AGE reduction, some natural compounds bioavailable in food, most of them with antioxidant capability, could exert protective effects in the cardiovascular system. The main advantage is the accessibility and safety of its consumption. Considering that some studies employed entire foodstuffs or fruit juices, and not isolated compounds, the global effect could be due to multiple mechanisms. Polyphenols constitute a class of compounds that possess potent free radical scavenging capacities. Indeed, the consumption of food naturally rich in polyphenols by mice and humans significantly inhibits oxidative stress, atherogenesis, and atherosclerotic lesion development. In what concerns cavernous tissue, it was demonstrated that polyphenols-rich food improve erectile function in both artherogenic ED and non-pathogenic conditions, and prevent ischemia-induced fibrosis [Citation119]. In fact, it has been demonstrated that chronic administration of pomegranate whole juice, or polyphenols extract to rabbits or rats, caused significant increases in intracavernous blood flow and SMCs relaxation, possibly via increasing NO bioavailability [Citation119–121]. Corroborating these findings, it was reported that pomegranate juice consumption improved erections in a cohort of 53 men with mild to moderate erectile dysfunction. However, the results in this study did not achieve statistical significance [Citation122]. Several evidences support that consumption of polyphenols of plant origin could not only induce NO formation in the endothelial cells, but also suppress the activation of inducible NOS (iNOS), a isoform of NOS activated in inflammation and infection [Citation123]. The inhibition of iNOS activity is particularly beneficial to erectile function, considering that iNOS-derived NO strongly contributes to nitration of proteins that causes damage in tissues and premature aging [Citation124]. In line with these findings, it was demonstrated that 6-month consumption of red wine or green tea, beverages also naturally rich in polyphenols, induces an apparent diminished atherosclerotic progression in cavernous tissue in aged rats and activates selected mechanisms for the maintenance of cavernous tissue vascularization [Citation99,Citation125].
Nutraceuticals
Polyphenols
Several studies elucidate the effects of polyphenols at cellular level. Most of these experiments use specific compounds, such as curcumin found in Indian spice turmeric, epigallocatechin-3-gallate (EGCG), the more abundant polyphenol of green tea, resveratrol available in grapes and red wine, and quercetin present in capers, lovage, apples, onions, and grapes.
Curcumin has been extensively studied and recently recognized as an antiaging nutrient [Citation126,Citation127]. Accumulating evidence suggests that it is a highly pleiotropic molecule that modulates abundant targets in cells, which influence numerous biochemical and molecular functions. Curcumin exhibits an inhibitory effect in AGE effects, apparently by direct trapping of methylglyoxal, which intracellular levels were demonstrated to decrease in curcumin-treated endothelial cell lines in culture [Citation128]. In hepatic stellate cells isolated from rodents, curcumin was also shown to eliminate the effects of AGEs by attenuation of oxidative stress, inhibition of expression of RAGE gene, increase in the activity of the nuclear receptor peroxisome proliferator-activated receptor (PPARγ) [Citation129], and by promoting AGER1 protective role [Citation130]. An equivalent mechanism was also observed in cultured rabbit chondrocytes, considering that it significantly decreased AGE-stimulated TNF-α expression and suppressed the NF-kB activation [Citation131]. In fact, curcumin presents important anti-inflammatory properties, as shown in endothelial cells where it further to inhibit activation of inflammatory transcription factors AP-1 and NF-kB [Citation132,Citation133], downregulate expression of intracellular adhesion molecule-1 (ICAM-1) secondary to increase in anti-inflammatory haem-oxygenase-1 (HO-1), VCAM-1, and monocyte chemoattractant protein-1 [Citation134–136], which compromise monocyte adhesion to endothelium as demonstrated in co-cultures of these cell types [Citation137]. Curcumin inhibits response to hypoxia, considering that it downregulates expression of the major transcription factor hypoxia- inducible factor 1 (HIF-1) [Citation138], and through the inhibition of c-Jun N-terminal kinase (JNK) signaling pathway [Citation139], impedes methylglyoxal-induced PARP degradation in endothelial cells [Citation140]. Curcumin downregulates HMGB1 release under lipopolysaccharide triggering, and ensuing pro-inflammatory response in endothelial cultured cells [Citation141]. A recent report demonstrated that this cell survival under H2O2-induced oxidative stress was increased by curcumin that promotes autophagy mechanisms, through inhibition of the phosphatidylinositol 3-kinase-AKT-mechanistic target of rapamycin (MTOR) signaling pathway and nuclear translocation of forkhead box protein O1 (FOXO1) [Citation142]. Conversely, induces an increment of its cytoplasmic acetylated form, and despite the fact that FOXO1 is a putative target to SIRT1-mediated deacetylation, the activity of this enzyme is not affected by curcumin [Citation143]. An additional mechanism apparently involved in endothelial cells oxidative damage is the Notch signaling pathway, that curcumin also inhibits [Citation144].
In what concerns SMC, curcumin presents an inhibitory effect in migration and proliferation while promoting apoptosis [Citation145,Citation146], in part by upregulation of expression of HO-1 after binding to the promotor region antioxidant responsive element (ARE) [Citation147]. It also inhibits the IGF-1- and PDGF-induced SMC proliferation, collagen synthesis, and intracellular ROS generation [Citation148–150]. In addition, curcumin upregulates expression of aldose reductase, an enzyme that strongly contributes to detoxification of methylglyoxal, via phosphatidylinositol 3-kinase/ Akt, p38 mitogen-activated protein kinase (MAPK), and nuclear factor-erythroid 2-related factor 2 [Citation151]. It is also able to suppress lipopolysaccharide-induced overexpression of inflammatory mediators and iNOS-derived NO via inhibiting the Toll-like receptor 4 (TLR4)-MAPK/NF-kB pathway and ET-1-induced downstream proliferative signaling events, in a concentration-dependent fashion [Citation152,Citation153].
Curcumin dietary supplementation lowers oxidative stress and improves vascular dysfunction both in young and in aged experimental rodent models [Citation154–157]. As recently shown in apo-E-deficient mice treated with (0,2%) curcumin-supplemented food, gene expression of molecules that intervene in leukocyte adhesion and transendothelial migration via NF-kB-dependent pathways is repressed which resulted in reduction in atherosclerotic lesions, which agrees with results obtained in a microarray analysis in a endothelial cell line [Citation158]. Fleenor and collaborators demonstrated that an equivalent curcumin treatment for 4 weeks induced a significant decrease in aorta AGE formation of old mice, while did not affect the levels in the younger ones [Citation154]. Their collagen I expression was also reduced compared with that of the old controls. Moreover, 8-week orally administrated curcumin to STZ-induced diabetic rats reduced AGE formation and crosslinking of collagen [Citation159]. In line with that, it was recently described that regular administration of water-soluble curcumin improved the erectile function in a rat model of diabetes, coupled to increase in expression of eNOS and nNOS, and repression of inflammatory genes and iNOS [Citation160]. These findings do not completely agree with the data reported by Farhangkhoee and collaborators, considering that they found a concomitant curcumin-induced upregulation of eNOS, iNOS, and ET-1 expression, coupled with a decrease in protein and DNA oxidative damage in the heart of diabetic rats [Citation161]. Interestingly, curcumin, EGCG, and quercetin share action mechanisms, considering that all of them interact with the membranes of endothelial cells, restoring transmembrane potential and fluidity in the presence of AGEs, apparently repressing the formation of atherosclerotic lesions [Citation162]. Supporting these findings that reveal important intervening of polyphenols in cellular functions, it was also demonstrated that resveratrol inhibits AGE-induced proliferation and collagen synthesis activity in vascular SMCs [Citation163] and that EGCG inhibits crosslink of collagen in the heart of diabetic rats [Citation164].
L-Carnitine
L-Carnitine is the biologically active form of a small peptide constituted by lysine and methionine, required for the transport of fatty acids from the cytosol into the mitochondria during the breakdown of lipids. Demonstration of anti-glycating effect of L-carnitine, in line with reduction in the levels of methylglyoxal and glycated Hb in blood, and collagen crosslink in skin and tail tendon observed in high-fructose diet-fed rats [Citation165] indicated this compound mostly available in red meat and dairy products, as a possible nutraceutical with applications in the control of diabetes and age-related diseases. In what concerns ED, administration of L-carnitine to patients seems promissory, considering reports that demonstrate that oral administration of derivatives of L-carnitine significantly improved nocturnal penile tumescence and International Index of Erectile Function score, as well as other symptoms associated with male aging [Citation166]. Treatment with an association of propionyl-L-carnitine with niacin and L-arginine was also demonstrated to have measurable effects on erectile function and sexual life satisfaction in a single-blind trial that includes 54 ED patients [Citation167]. Derivatives of L-carnitine have also been employed as adjuvants of inhibitors of phosphodiesterase-5 (PDE5, first-line treatment for ED, discussed below), with very promising results for ED ensuing bilateral nerve-sparing radical retropubic prostatectomy [Citation168] and DM-related ED [Citation169,Citation170]. Association of L-carnitine with the treatment was particularly beneficial for patients with ED refractory to PDE5 inhibitors monotherapy, considering that it frankly improved the endothelial function [Citation171]. Despite the amelioration in markers of inflammatory processes and vascular damage, an unexpected maintenance in circulating AGEs was observed in diabetes-related ED patients treated with L-carnitine derivatives.
Thiamine pyrophosphate and pyridoxamine
Thiamine pyrophosphate and pyridoxamine are derivatives of vitamins B1 and B6, respectively, compounds that naturally exist in food. Thiamin is naturally present in cereal grains, liver, and eggs, itself however, particularly when supplied in supplement formulations is poorly absorbed and taken into the cells. The most bioavailable thiamine-derived compounds are those found in crushed roasted garlic, also called allithiamines, that could result in a 5- to 120-fold increase in intracellular thiamine levels when compared with an equivalent amount of isolated thiamine [Citation49]. On the other hand, pyridoxamine exists in fish, chicken, walnuts, and carrots. Indeed, the toxicity of their ingestion is very low, and the tolerance for most of the individuals is very good. Both thiamine pyrophosphate and pyridoxamine are able to inhibit the formation of AGEs [Citation172]. Until now, no study has been carried out concerning the effect of supplementation of vitamins B1 or B6 in erectile function; however, several evidences support that thiamine could result in inhibition of human arterial muscle cell proliferation and of endothelial cells apoptosis induced by high glucose and insulin in cell culture [Citation173,Citation174]. A recently performed clinical trial revealed disappointing, considering that it does not demonstrate the effects of benfotiamine, a lipid-soluble thiamine derivative on intermediate pathways of hyperglycemia-induced endothelial dysfunction [Citation175].
In what concerns pyridoxamine, it was demonstrated that it could exert a protective effect on vascular system; it inhibits glycation of collagen, decreases plasma triglycerides and cholesterol, and reduces HTA and thickening of the vascular wall in obese, diabetic, and aged rats [Citation176–179]. It was also demonstrated that pyridoxamine rather than limiting the formation of AGEs also functions as an antioxidant that protect cells against oxidative stress [Citation180]. In line with these evidences, pyridoxamine treatment strongly attenuated the progression of established diabetes-associated atherosclerosis in diabetic apolipoprotein E knockout mice [Citation179]. In what concerns clinical trials, carried out exclusively in patients with type 2 diabetic nephropathy, no effect of pyridoxamine on the progression of serum creatinine at 1 year was demonstrated [Citation181,Citation182]. Nevertheless, in another study, significant reduction in the rise of serum creatinine and urinary albumin excretion was observed in the treated group [Citation183]. No studies in cavernous tissue have been performed.
Other nutrients that could interfere in AGE formation in corpus cavernosum
Alpha-lipoid acid could be considered a promissory strategy for ED treatment, particularly diabetes-related ED, considering that it prevents formation of AGE-modified products in the presence of glucose, counteracts oxidative stress, and blocks inflammatory responses induced by AGEs in endothelial cells [Citation184]. In addition, an interesting study demonstrated that α-lipoic and γ-linolenic acids interact synergistically to improve NO-mediated neural and endothelium-dependent relaxation of corpus cavernosum in streptozotocin-induced diabetes in rat, despite the absence of effect observed when each was employed alone [Citation185].
Other nutrients such as carnosine and caffeine, available in meat and coffee, respectively, likely possess AGE scavenge activity, regardless no evidence of benefic effects in vascular or erectile function exists. Carnosine, a dipeptide of the amino acids β-alanine and histidine, highly concentrated in muscle and brain, not only suppresses AGE formation, but also inhibits its reactivity toward normal peptides [Citation186]. Indeed, serum AGE levels in diabetic vegetarians are significantly higher than those observed in diabetic omnivores, apparently justified by reduced intake of carnosine [Citation187]. In what concerns caffeine, it is an activator of lysozyme activity. Lysozyme contains a domain with high-affinity binding for AGEs [Citation188], and is likely to function as a scavenger of circulating AGEs in serum and mucosal secretions. On the other hand, coffee, the main source of caffeine, is also rich in AGEs, in particular methylglyoxal [Citation189], and despite the recognized benefits of its ingestion, its final effect in terms of glycative action remains to be clarified. Other AGE-rich foodstuffs, further to coffee, seem to favorably affect health, as the bread crust extracts. Despite induce increase in ROS and inflammatory signaling pathways that include NF-kB in cultured heart fibroblasts of murine origin, it is able to reduce the expression of miofibroblast markers and secretion of extracellular matrix molecules, apparently contributing to the prevention of heart fibrosis [Citation190]. In addition, bread crust improves antioxidant defenses in heart fibroblasts cell lines, apparently in a RAGE-dependent fashion [Citation191], a result supported by in vivo experiments, which suggest cardioprotective effects in the case of severe oxidative stress [Citation192]. Another study conducted in mice demonstrated that melanoidins, high molecular weight heterogeneous polymers formed by Maillard reaction, in high heated pellets, significantly reduced inflammation in experimental colitis [Citation193]. No evidences of beneficial effects of AGE-rich food in cavernous tissue were reported so far.
Exercise
Studies in animal models indicate that regular practice of exercise favors eNOS activity, NO synthesis, endothelium-dependent vasodilatation, and even reversion of atherogenesis as shown in hypercholesterolemic apolipoprotein E-deficient mice [Citation194]. In addition, exercise was recently recognized as an important non-pharmacological intervention in the avoidance of ED [Citation195]. Despite the fact that the exact mechanisms are still elusive, recently reported data suggest that AGE reduction could be involved in vascular protection exerted by exercise. In fact, it was recently demonstrated that aerobic exercise decreases plasma AGE-associated fluorescence in obese Zucker rats [Citation196] and increases sRAGE levels with improvement of vascular function in DM type 2 [Citation197] and aged [Citation198] patients. These findings strongly suggest that exercise practice affects multiple mechanisms intracellularly that interfere in glycative and oxidative modifications of proteins.
Further to nutritional modulation, nutraceuticals, or exercise, several drugs or medicinal products had been tested, mainly in animal models, tentatively to control AGE-induced damage. The most relevant data are further discussed with a particular focus to possible applications to erectile dysfunction amelioration.
Therapeutic strategies
Chelators of transition metal ions
Intermediates of AGE formation is highly favored in the presence of transition metals, and chelators that impede their binding to proteins had been proposed as inhibitors of formation of AGEs. In fact, this class of agents is able to inhibit AGE formation in vitro and to improve cardiovascular function, independently of plasma glucose or lipid concentrations in rodent experimental models of diabetes [Citation199]. In line with those findings, it was found that transition metal chelators, such as penicillamine, reverse the inhibitory effect of hyperhomocysteinemia on endothelium-dependent relaxation in the corpus cavernosum in the rabbit [Citation200] and that trientene improved both aorta and corpus cavernosum endothelium-dependent relaxation [Citation201]. These evidences strongly support the concept that metal- catalyzed oxidation is an important contributor to AGE formation in vivo and to AGE-mediated damage, too [Citation202]. Interestingly, other AGE inhibitors whose main activity is to trap reactive carbonyl or dicarbonyl intermediates in AGE generation also present potent chelating activity, making it difficult to dissociate the antioxidative and chelating activity from the carbonyl trapping action. The most important example of a multifunctional drug is aminoguanidine, described below.
Aminoguanidine
Aminoguanidine (Pimagedine), a nucleophilic hydrazine-like compound, has been introduced as a drug that is able to block the Maillard reaction in vivo, considering that it reacts with early glycosylation products creating unreactive substitutes that do not go on to form AGEs [Citation203]. Additional protection against the formation of AGEs could be related to direct antioxidant effects, inhibition of catalase activity [Citation204], scavenging of peroxynitrite radicals [Citation205], and transition metal-chelating activity [Citation206]. Indeed, aminoguanidine prevents AGE formation [Citation207] and crosslinking of collagen in the arterial wall in diabetic rats [Citation208], an effect that was not so evident in normal aged rats [Citation209]. Aminoguanidine constitutes also a selective and strong inhibitor of iNOS activity [Citation210,Citation211] while activates n-NOS and eNOS-mediated NO synthesis [Citation30,Citation61]. It was demonstrated that these isoenzymes are differentially regulated and that gene transfer of iNOS to endothelial cells downregulates expression of eNOS in cavernous tissue [Citation212], which in the long-term could compromise erectile function. Aminoguanidine has no direct effect on cavernosal SM relaxation as demonstrated by Cartledge and collaborators in tissue strips of aminoguanidine-treated non-diabetic control animals. In fact, the protective effect of aminoguanidine on erectile function is due to reduction in cavernosal AGE formation as shown in STZ-induced diabetes rat models [Citation30,Citation213,Citation214]. Moreover, treatment with aminoguanidine significantly diminishes expression of the AGER galectin-3 and restores erectile function, independently of the timing of treatment, despite the time-dependency observed for cavernosal AGE formation in diabetic rats [Citation214]. Unfortunately, many studies reported high rates of pancreatic and renal tumors in aminoguanidine-treated animals. In addition, aminoguanidine traps pyridoxal phosphate, a derivative of vitamin B6, so long-term administration leads to serial deficiency of it. Thus, taking these evidences in account and considering the adverse effects, particularly the toxicity observed during the human trials in patients with type 1 (ACTION 1) and type 2 diabetes (ACTION 2), it is unlikely that the benefits of aminoguanidine treatments observed in rats could be extended to humans.
Alagebrium
Another class of drugs that apparently decrease AGE products in tissues are the breakers of glycation crosslinks or, crosslink breakers of AGEs, being the most extensively studied the alagebrium (phenyl-4,5-dimethylthiazolium bromide). Initially thought that this class of drugs could disrupt in vivo crosslinks of advanced Maillard reaction, no evidence to date exists [Citation38], and recent reports strongly suggested that alagebrium, similar to aminoguanidine, could interfere with AGE formation in tissues [Citation49]. Independently of the mechanism of inhibition of AGE products, alagebrium is able to reduce vascular AGE accumulation in experimental diabetes [Citation49] and to attenuate the development of atherosclerosis in diabetic ApoE-deficient mice [Citation215]. The vascular beneficial effects of alagebrium were also observed in aged animals, where this compound is able to prevent vascular and myocardial stiffness [Citation216]. In what concerns erectile tissue, it was reported that treatment for 6 weeks reversed diabetes-induced erectile dysfunction in rodents [Citation217], concomitantly with a decrease in serum and cavernous levels of AGEs. More recently, it was also assayed the combined action of alagebrium with sildenafil, an PDE5 inhibitor in an equivalent rodent model [Citation218], which induced increase in intracavernous pressure coupled to reduction in AGEs, iNOS, inflammatory markers, and apoptosis levels. Despite the promissory results observed in rodents, no study concerning erectile function has been performed in human and the recently trial conducted in The Netherlands did not show beneficial effect of alagebrium in patients with heart failure and systolic dysfunction [Citation219].
Anti-diabetics: metformin and pioglitazone
Metformin is an oral biguanide insulin-sensitizing agent that inhibits hepatic glucose production and increases sensitivity to insulin while reduces glucotoxicity in type 2 DM [Citation220]. One of the side effects of metformin treatment is decrease in body weight, particularly in non-diabetic patients [Citation221,Citation222]. This phenomenon is particularly valuable to obese man with complaints of ED, considering that weight loss in obese man is strongly related to improvement in erectile function [Citation195]. By reducing glucose level in blood, metformin induces decrease in AGE formation reducing methylglyoxal levels [Citation223,Citation224], and decreases oxidative and nitrosative stress in type 2 DM patients [Citation225]. Thus, metformin emerges as a putative therapeutic to erectile dysfunction, which is strongly supported by demonstration that 4-week treatment restored cavernous levels of expression of eNOS and nNOS in an experimental rodent model of high-fat diet induced obesity. Moreover, it was demonstrated that metformin´s effects in NOS levels seem to be associated with activation of 5′ adenosine monophosphate-activated protein kinase (AMPK) [Citation226]. Recently, a randomized pilot study was performed in eugonadal non-diabetic men with insulin resistance that investigated the effect of association of metformin to sildenafil treatment, and first results of this study demonstrated that metformin not only reduced insulin resistance but improved erectile function, too [Citation227].
Pioglitazone is a member of the family of thiazolidinedione compounds, an inhibitor of glycation and AGE formation [Citation228], and similar to metformin, it is used in the treatment of diabetes considering that it sensitizes peripheral tissues to insulin. Pioglitazone has been demonstrated to ameliorate ED in rat models of type 2 DM and aging [Citation229,Citation230], and to increase response to sildenafil in poor-responsive patients [Citation231].
PDE5 inhibitors
PDE5 is the predominant form of the PDE isoenzymes in the cavernous tissue. This catabolic enzyme naturally involved in detumescence of the penis after erection is highly selective to cGMP, catalyzing its breakdown and significantly reducing its accumulation due to NO signaling. In line with that, PDE5 inhibitors are currently considered by most physicians the first-line treatment for ED. Interestingly, this class of drugs does not have relaxant effect on isolated human corpus cavernosum, but when sexual stimulation arose, it causes local release of NO that coupled to inhibition of PDE5 leads to increased levels of cGMP in cavernous tissue. The prolonged level of cGMP improves SM relaxation favoring erection maintenance. Vardenafil, sildenafil, and taladafil are drugs that belong to the family of PDE5 inhibitors, and largely used in the pharmacological treatment of erectile dysfunction. Interestingly, besides cGMP protective effect, it was demonstrated that vardenafil is able to block AGE/RAGE-activated molecular responses in cultured endothelial cells due to suppression of AGE-induced ROS generation coupled to RAGE expression [Citation232]. This evidence indicates that diabetes and age-related ED could be partially induced by AGE/RAGE axis and that it constitutes a potential target for developing novel anti-ED therapeutics.
Statins
Statins belong to a vastly employed class of drugs, mainly used to lower cholesterol levels in blood by inhibiting the enzyme HMG-CoA reductase, which plays a central role in the production of cholesterol in the liver. Several statins are currently available in market, produced by different pharmaceutical laboratories, and are administrated to patients with a high grade of security. In fact, the direct beneficial effect of statin in cholesterol blood levels and reducing the risk of stroke and heart attach [Citation233] strongly superimposes the adverse effects that treatment provoke particularly in diabetics [Citation234] and elderly patients [Citation235]. Besides the direct cholesterol-lowering effect, statins also present pleiotropic activity, exerting endothelium protection [Citation236–238]. For instance, cerivastatin is able to interfere with RAGE signaling and to inhibit AGE-induced angiogenesis in vitro [Citation239]. Atorvastatin, one of the most widely used statins, can increase circulating soluble RAGE levels in type 2 DM patients [Citation240,Citation241], while decreases the AGE levels [Citation242]. In vitro and in vivo studies demonstrated that atorvastatin inhibits CRP-induced expression and activation of RAGE and downstream signaling pathways [Citation243]. It was also demonstrated that simvastatin (an additional member of the family) not only reduced AGE formation, but also significantly decreased vascular SMCs proliferation [Citation244,Citation245], strongly supporting that statins could counteract atherosclerosis progression. This finding is particularly relevant, considering that erectile dysfunction is often a manifestation of atherosclerosis. In line with this, it was demonstrated that statin treatments can alleviate ED [Citation246], despite the fact that the effect of this family of drugs on erectile function is far from being consensual [Citation247,Citation248]. On the other hand, statins are also employed with success in association with PDE5 inhibitors, considering their apparent synergetic effects [Citation249–253]. Since ED is associated with impaired vascular function and statin treatment improves vascular function, its administration to PDE5 inhibitors non-responsive ED patients, strongly increases the efficacy of the treatment.
Anti-hypertensive drugs
Although no direct effect of the AGE/RAGE axis is observed on essential HTA, augmented AGEs have been evidenced in the vasculature of these individuals, linked with an increased cardiovascular risk [Citation46]. In line with that, most of the drugs employed in the treatment of HTA (angiotensin-converting enzyme inhibitors – ACEI, and angiotensin II receptor-1 blocker – ARB) possess anti-AGE activity, mainly by chelating transition metals, but also by blocking oxidative steps in the pre-Amadori, and in lower extent in post-Amadori, stages of AGE formation () [Citation45]. In addition, it was reported that perindopril (an ACEI) reduces AGE accumulation and increases sRAGE levels in human [Citation254]. Indeed, anti-hypertensive drugs of the family of ACEIs and ARBs likely exert favorable effect on AGE decoy, and in the control of ED. Two drugs of these families (ramipril – ACEI, and telmisartan – ARB) were tested in a murine model of atherosclerosis ApoE(-/-), and significantly improved endothelial function of aortic and cavernosal tissues, via reduction of oxidative stress [Citation255]. On the other hand, a pre-specified substudy (ONTARGET/TRANSCEND) trial, where 1549 patients underwent double-blind randomization, and the same anti-hypertensive drugs were tested isolated and, in association, lead the investigators conclude that these treatment did not significantly improve or worsen ED [Citation256]. Thus, further studies with an enlarged range of anti-hypertensive compounds will be necessary to clarify its potential application in the treatment or co-adjuvation of treatment of ED.
Other drugs that affect AGE formation
Further to agents that mitigate AGE effects, it was observed that treatment with nitrates, drugs vastly used in the treatment of coronary artery disease (CAD) significantly increase AGE formation in the pericardial fluid, as verified in a cohort of 75 patients undergoing surgical coronary artery revascularization [Citation257]. The concomitant use of nitrates and PDE5 inhibitors for patients that present CAD and ED is contraindicated, and unfortunately contributed to several cardiac deaths due to severe hypotension [Citation258]. A novel drug ranozolyne could represent an alternative to nitrates in the treatment of CAD for patients that intent to initiate PDE5 inhibitors therapy; however, more studies are needed to examine this possibility [Citation259]. Interestingly, ranozolyne seems to counteract AGE formation, considering that it strongly decreases HbA1c levels in serum of diabetic patients [Citation260].
Concluding remarks
The biology of diabetes and aging owns in common the formation and accumulation of AGEs and the activation of AGE/RAGE axis. Several evidences indicate that ED, a surrogate symptom of systemic cardiovascular disease, particularly prevalent in the elderly and DM patients, could be partly caused by AGE-dependent erectile tissue damage coupled to NO decrease. AGE/RAGE axis emerges thus as a potential interventional target for treatment of ED.
The strict control of blood glucose levels in DM patients and the reduction of AGE ingestion along life are the first therapies for reducing AGE formation and damage. However, novel treatments, such as those targeting AGEs, seem promising in combating endothelial dysfunction and ED. This review discusses possible strategies, including nutritional modulation, nutraceuticals, and drugs, which may be useful in the management of age and diabetes-related ED. These substances have been successfully used in animal models, but the clinical data have been very disappointing. Indeed, the best strategy to intervene in the avoidance of AGE accumulation and activity in the cavernous tissue remains to be elucidated. In the future, it is likely that more potent and specific therapies will emerge to counteract the action of AGEs in erectile function, but until the best agent becomes a reality, optimal metabolic and blood pressure control, achieved and sustained from early age and from DM onset, remains the best resource to prevent AGE-related damage.
Acknowledgment
The author thanks Sérgio Evangelista from Laboratório de Iconografia, Faculdade de Medicina da Universidade do Porto, for his contribution in the preparation of the illustrations.
Declaration of interest
The author reports no declaration of interest. The author alone is responsible for the content and writing of the paper.
References
- Tomada N, Oliveira R, Tomada I, Vendeira P, Neves D. Comparative ultrastructural study of human corpus cavernosum during ageing. Microsc Microanal 2008;14:S3152–S3155.
- Cordeiro AL, Figueiredo A, Godinho F, Martins I, Vendeira P, Almeida H, Neves D. Ultrastructural characterization of corpus cavernosum of ageing, orchidectomy and diabetes rat experimental models. Microsc Microanal 2008;14:S397–S398.
- Burnett AL, Nelson RJ, Calvin DC, Liu JX, Demas GE, Klein SL, et al. Nitric oxide-dependent penile erection in mice lacking neuronal nitric oxide synthase. Mol Med 1996; 2:288–296.
- Hurt KJ, Sezen SF, Lagoda GF, Musicki B, Rameau GA, Snyder SH, Burnett AL. Cyclic AMP-dependent phosphorylation of neuronal nitric oxide synthase mediates penile erection. Proc Natl Acad Sci USA 2012;109:16624–16629.
- Ignarro LJ, Bush PA, Buga GM, Wood KS, Fukuto JM, Rajfer J. Nitric oxide and cyclic GMP formation upon electrical field stimulation cause relaxation of corpus cavernosum smooth muscle. Biochem Biophys Res Commun 1990;31: 843–850.
- Kaya C, Uslu Z, Karaman I. Is endothelial function impaired in erectile dysfunction patients?Int J Impot Res 2006;18: 55–60.
- Shamloul R, Ghanem H. Erectile dysfunction. 2013;381: 153–165.
- Goldstein I. The association of ED (erectile dysfunction) with ED (endothelium dysfunction) in the international journal of impotence research: The journal of sexual medicine. Int J Impot Res 2003;15:229–230.
- Guay AT. ED2: erectile dysfunction = endothelial dysfunction. Endocrinol Metabol Clin N Am 2007;36:453–463.
- Ayta IA, Mckinlay JB, Krane RJ. The likely worldwide increase in erectile dysfunction between 1995 and 2025 and some possible policy consequences. BJU Int 1999;84: 50–56.
- Feldman HA, Goldstein I, Hatzichristou DG, Krane RJ, McKinlay JB. Impotence and its medical and psychosocial correlates: Results of the Massachusetts Male Aging Study. J Urol 1994;151:54–61.
- Ghalayini IF, Al-Ghazo MA, Al-Azab R, Bani-Hani I, Matani YS, Barham AE, et al. Erectile dysfunction in a Mediterranean country: Results of an epidemiological survey of a representative sample of men. Int J Impot Res 2010; 22:196–203.
- Ponholzer A, Temml C, Mock K, Marzlaekm M, Obermayr R, Maderbacher S. Prevalence and risk factors for erectile dysfunction in 2869 men using a validated questionnaire. Eur Urol 2005;47:80–86.
- Thorve VS, Kshirsagar AD, Vyawahare NS, Joshi VS, Ingale KG, Mohite RJ. Diabetes-induced erectile dysfunction: epidemiology, pathophysiology and management. J Diabetes Complications 2011;25:129–136.
- Lewis RW. Epidemiology of erectile dysfunction. Urol Clin North Am 1996;28:209–216.
- Shaeer O, Shaeer K. The Global Online Sexuality Survey (GOSS): the United States of America in 2011. Chapter I: Erectile Dysfunction Among English-Speakers. J Sex Med. 2012 [Epub ahead of print].
- Lusis AJ. Atherosclerosis. Nature 2000;407:233–241.
- Cheitlin CMD. Erectile dysfunction: the earliest sign of generalized vascular disease?J Am Coll Cardiol 2004;43: 185–186.
- Stadtman ER. Protein oxidation and aging. Science 1992;257:1220–1224.
- Bonnefont-Rousselot D, Bastard JP, Jaudon MC, Delattre J. Consequences of the diabetic status on the oxidant/antioxidant balance. Diabetes Metab 2000;26:163–176.
- Silva FH, Mónica FZ, Báu FR, Brugnerotto AF, Priviero FB, Toque HA, Antunes E. Superoxide anion production by NADPH oxidase plays a major role in erectile dysfunction in middle-gged rats: prevention by antioxidant therapy. J Sex Med2013 [Epub ahead of print].
- Figueiredo A, Cordeiro AL, Tomada N, Tomada I, Rodrigues A, Gouveia A, Neves D. Real-time PCR study of Ang1, Ang2, Tie-2, VEGF and KDR expression in human erectile tissue during aging. J Sex Med 2011;8:1341–1351.
- Tomada N, Tomada I, Vendeira P, Cruz F, Neves D. Characterization of VEGF and Angiopoietins expression in human corpus cavernosum during aging. J Sex Med 2011; 7:1410–1418.
- Baraibar MA, Liu L, Ahmed EK, Friguet B. Protein oxidative damage at the crossroads of cellular senescence, aging and aging-related diseases. Oxid Med Cell Longev2012 [Epub ahead of print].
- Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001;414:813–820.
- Vlassara H, Uribarri J. Glycoxidation and diabetic complications: modern lessons and a warning?Rev Endocr Metab Disord 2004;5:181–188.
- Mohamed AK, Bierhaus A, Schiekofer S, Tritschler H, Ziegler R, Nawroth PP. The role of oxidative stress and NF-kappaB activation in late diabetic complications. Biofactors 1999;10:157–167.
- Higashi Y, Noma K, Yoshizumi M, Kihara Y. Endothelial function and oxidative stress in cardiovascular diseases. Circ J 2009;73:411–418.
- Seftel AD, Vaziri ND, Ni Z, Razmjouei K, Fogarty J, Hampel N, et al. Advanced glycation end products in human penis: elevation in diabetic tissue, site of deposition, and possible effect through iNOS or eNOS. Urology 1997; 50:1016–1026.
- Cartledge JJ, Eardley I, Morrison JF. Advanced glycation end-products are responsible for the impairment of corpus cavernosal smooth muscle relaxation seen in diabetes. BJU Int 2001;87:402–407.
- Jakus V, Rietbrock N. Advanced glycation end-products and the progress of diabetic vascular complications. Physiol Res 2004;53:131–142.
- Jiaan DB, Seftel AD, Fogarty J, Hampel N, Cruz W, Pomerantz J, et al. Age-related increase in an advanced glycation end product in penile tissue. World J Urol 1995;13:369–375.
- Hipkiss AR. Accumulation of altered proteins and ageing: causes and effects. Expt Gerontolol 2006;41:464–473.
- Schöneich C. Protein modification in aging: an update. Exp Gerontol 2006;41807–41812.
- Maillard LC. Action des acides amines sur les sucres: formation des mélanoides par voie méthodique. C R Acad Sci 1912;154:66–68.
- Ahem N, Thornally PJ. Advanced glycation end-products: what is their relevance to diabetic complications?Diabetes Obes Metab 2007;9:233–245.
- Rabbani N, Thornalley PJ. Glycation research in aminoacids: a place to call home. Aminoacids 2012;42:1087–1096.
- Sell DR, Monnier VM. Molecular basis of arterial stiffening: role of glycation – A mini-review. Gerontology 2012;58: 227–237.
- Lyons T, Jenkins AJ. Glycation, oxidation and lipoxidation in the development of the complications of diabetes mellitus: a carbonyl stress hypothesis. Diabetes Rev 1997;5: 365–391.
- Degenhardt TP, Thorpe SR, Baynes JW. Chemical modification of proteins by methylglyoxal. Cell Mol Biol 1998; 44:1139–1145.
- Xue M, Rabbani N, Thornalley PJ. Glyoxalase in ageing. Sem Cell Dev Biol 2011;22:293–301.
- He C, Sabol J, Mitsuhashi T, Vlassara H. Dietary glycotoxins: inhibition of reactive products by aminoguanidine facilitates renal clearance and reduces tissue sequestration. Diabetes 1999;48:1308–1315.
- Bucala R, Makita Z, Vega G, Grundy S, Koschinsky T, Cerami A, Vlassara H. Modification of low density lipoprotein by advanced glycation end products contributes to the dyslipidemia of diabetes and renal insufficiency. Proc Natl Acad Sci USA 1994;91:9441–9445.
- Makita Z, Vlassara H, Rayfield E, Cartwright K, Friedman E, Rodby R, et al. Hemoglobin-AGE: a circulating marker of advanced glycosylation. Science 1992;258:651–653.
- Peyroux J, Sternberg M. Advanced glycation endproducts (AGEs): pharmacological inhibition in diabetes. Pathologie Biologie 2006;54:405–419.
- Baumann M. Role of advanced glycation end products in hypertension and cardiovascular risk: human studies. J Am Soc Hypertens 2012;6:427–435.
- Mizutari K, Ono T, Ikeda K, Kayashima K, Horiuchi S. Photo-enhanced modification of human skin elastin in actinic elastosis by N(epsilon)-(carboxymethyl)lysine, one of the glycoxidation products of the Maillard reaction. J Invest Dermatol 1997;108:797–802.
- Haitoglou CS, Tsilibary EC, Brownlee M, Charonis AS. Altered cellular interactions between endothelial cells and nonenzymatically glucosylated laminin/type IV collagen. J Biol Chem 1992;267:12404–12407.
- Thomas MC, Baynes JW, Thorpe SR, Cooper ME. The role of AGEs and AGE inhibitors in diabetic cardiovascular disease. Curr Drug Targets 2005;6:453–474.
- Knott HM, Brown BE, Davies MJ, Dean RT. Glycation and glycoxidation of low-density lipoproteins by glucose and low-molecular mass aldehydes. Formation of modified and oxidized particles. Eur J Biochem 2003;270: 3572–3582.
- Harja E, Bu DX, Hudson BI, Chang JS, Shen X, Hallam K, et al. Vascular and inflammatory stresses mediate atherosclerosis via RAGE and its ligands in apoE-/- mice. J Clin Invest 2008;118:183–194.
- Kirstein M, Brett J, Radoff S, Ogawa S, Stern D, Vlassara H. Advanced protein glycosylation induces transendothelial human monocyte chemotaxis and secretion of platelet-derived growth factor: role in vascular disease of diabetes and aging. Proc Natl Acad Sci USA 1990;87:9010–9014.
- Nam MH, Lee HS, Seomun Y, Lee Y, Lee KW. Monocyte-endothelium-smooth muscle cell interaction in co-culture: proliferation and cytokine productions in response to advanced glycation end products. Biochim Biophys Acta 2011;1810:907–912.
- Kirstein M, Aston C, Hintz R, Vlassara H. Receptor-specific induction of insulin-like growth factor I in human monocytes by advanced glycosylation end product-modified proteins. J Clin Invest 1992;90:439–446.
- Corrêa-Giannella ML, de Azevedo MR, Leroith D, Giannella-Neto D. Fibronectin glycation increases IGF-I induced proliferation of human aortic smooth muscle cells. Diabetol Metab Syndr 2012;4:19.
- Cantero AV, Portero-Otin M, Ayala V, Auge N, Sanson M, Elbaz M, et al. Methylglyoxal induces advanced glycation end product (AGEs) formation and dysfunction of PDGF receptor-beta: implications for diabetic atherosclerosis. FASEB J 2007;21:3096–3106.
- Nass N, Vogel K, Hofmann B, Presek P, Silber RE, Simm A. Glycation of PDGF results in decreased biological activity. Int J Biochem Cell Biol 2010;42:749–754.
- Yang SJ, Chen CY, Chang GD, Wen HC, Chen CY, Chang SC, et al. Activation of Akt by advanced glycation end products (AGEs): involvement of IGF-1 receptor and caveolin-1. PLoS One 2013;8:e58100.
- Forbes JM, Soulis T, Thallas V, Panagiotopoulos S, Long DM, Vasan S, et al. Renoprotective effects of a novel inhibitor of advanced glycation. Diabetologia 2001;44: 108–114.
- Christ M, Bauersachs J, Liebetrau C, Heck M, Günther A, Wehling M. Glucose increases endothelial-dependent superoxide formation in coronary arteries by NAD(P)H oxidase activation: attenuation by the 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitor atorvastatin. Diabetes 2002;51:2648–2652.
- Su J, Lucchesi PA, Gonzalez-Villalobos RA, Palen DI, Rezk BM, Suzuki Y, et al. Role of advanced glycation end products with oxidative stress in resistance artery dysfunction in type 2 diabetic mice. Arterioscler Thromb Vasc Biol 2008;28:1432–1438.
- Bucala R, Tracey KJ, Cerami A. Advanced glycosylation products quench nitric oxide and mediate defective endothelium-dependent vasodilatation in experimental diabetes. J Clin Invest 1991;87:432–438.
- Sena CM, Matafome P, Crisóstomo J, Rodrigues L, Fernandes R, Pereira P, Seiça RM. Methylglyoxal promotes oxidative stress and endothelial dysfunction. Pharmacol Res 2012;65:497–506.
- Xu B, Ji Y, Yao K, Cao YX, Ferro A. Inhibition of human endothelial cell nitric oxide synthesis by advanced glycation end-products but not glucose: relevance to diabetes. Clin Sci (Lond) 2005;10:439–446.
- Lai YL, Aoyama S, Nagai R, Miyoshi N, Ohshima H. Inhibition of L-arginine metabolizing enzymes by L-arginine-derived advanced glycation end products. J Clin Biochem Nutr 2010;46:177–185.
- Quehenberger P, Bierhaus A, Fasching P, Muellner C, Klevesath M, Hong M, et al. Endothelin 1 transcription is controlled by nuclear factor-kappaB in AGE-stimulated cultured endothelial cells. Diabetes 2000;49:1561–1570.
- Giuseppe C, Ferdinando F, Ciro I, Vincenzo M. Pharmacology of erectile dysfunction in man. Pharmacol Ther 2006;111:400–423.
- Aversa A, Basciani S, Visca P, Arizzi M, Gnessi L, Frajese G, Fabbri A. Platelet-derived growth factor (PDGF) and PDGF receptors in rat corpus cavernosum: changes in expression after transient in vivo hypoxia. J Endocrinol 2001;170:395–402.
- Vlassara H. The AGE-receptor in the in the pathogenesis of diabetic complications. Diabetes Metab Res Rev 2001;17: 436–443.
- Lu C, He JC, Cai W, Liu H, Zhu L, Vlassara H. Advanced glycation endproduct (AGE) receptor 1 is a negative regulator of the inflammatory response to AGE in mesangial cells. Proc Nat Acad Sci 2004;101:11767–11772.
- Cai W, Torreggiani M, Zhu L, Chen X, He JC, Striker GE, Vlassara H. AGER1 regulates endothelial cell NADPH oxidase-dependent oxidant stress via PKC-delta: implications for vascular disease. Am J Physiol Cell Physiol 2010; 298:C624–C634.
- Cai W, Ramdas M, Zhu L, Chen X, Striker GE, Vlassara H. Oral advanced glycation endproducts (AGEs) promote insulin resistance and diabetes by depleting the antioxidant defenses AGE receptor-1 and sirtuin 1. Proc Natl Acad Sci U S A. 2012;109:15888–15893.
- Tomada I, Tomada N, Almeida H, Neves D. Androgen depletion in humans leads to cavernous tissue reorganization and upregulation of Sirt1-eNOS axis. AGE 2013;35:35–47.
- Vlassara H, Li YM, Imani F, Wojciechowicz D, Yang Z, Liu FT, Cerami A. Identification of galectin-3 as a high- affinity binding protein for advanced glycation end products (AGE): a new member of the AGE-receptor complex. Mol Med 1995;1:634–646.
- Pang J, Rhodes DH, Pini M, Akasheh RT, Castellanos KJ, Cabay RJ, et al. Increased adiposity, dysregulated glucose metabolism and systemic inflammation in Galectin-3 KO mice. PLoS One 2013;8:e57915.
- Darrow AL, Shohet RV, Maresh JG. Transcriptional analysis of the endothelial response to diabetes reveals a role for galectin-3. Physiol Genomics 2011;43:1144–1152.
- Seki N, Hashimoto N, Sano H, Horiuchi S, Yagui K, Makino H, Saito Y. Mechanisms involved in the stimulatory effect of advanced glycation end products on growth of rat aortic smooth muscle cells. Metabolism 2003;52:1558–1563.
- Calvier L, Miana M, Reboul P, Cachofeiro V, Martinez-Martinez E, de Boer RA, et al. Galectin-3 mediates aldosterone-induced vascular fibrosis. Arterioscler Thromb Vasc Biol 2013;33:67–75.
- Mackinnon AC, Liu X, Hadoke PW, Miller MR, Newby DE, Sethi T. Inhibition of galectin-3 reduces atherosclerosis in apolipoprotein E-deficient mice. Glycobiology 2013;23: 654–663.
- de Boer RA, van Veldhuisen DJ, Gansevoort RT, Muller Kobold AC, van Gilst WH, Hillege HL, et al. The fibrosis marker galectin-3 and outcome in the general population. J Intern Med 2012;272:55–64.
- Ohgami N, Nagai R, Ikemoto M, Arai H, Kuniyasu A, Horiuchi S, Nakayama H. Cd36, a member of the class b scavenger receptor family, as a receptor for advanced glycation end products. J Biol Chem 2001;276:3195–3202.
- Araki N, Higashi T, Mori T, Shibayama R, Kawabe Y, Kodama T, et al. Macrophage scavenger receptor mediates the endocytic uptake and degradation of advanced glycation end products of the Maillard reaction. Eur J Biochem 1995;230:408–415.
- Ramasamy R, Yan SF, Schmidt AM. Advanced glycation endproducts: from precursors to RAGE: round and round we go. Amino Acids 2012;42:1151–1161.
- Neeper M, Schmidt AM, Brett J, Yan SD, Wang F, Pan YC, et al. Cloning and expression of a cell surface receptor for advanced glycosylation end products of proteins. J Biol Chem 1992;267:14998–15004.
- Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell 1999;97:889–901.
- Bucciarelli LG, Wendt T, Rong L, Lalla E, Hofmann MA, Goova MT, et al. RAGE is a multiligand receptor of the immunoglobulin superfamily: implications for homeostasis and chronic disease. Cell Mol Life Sci 2002;59:1117–1128.
- Hallam KM, Li Q, Ananthakrishnan R, Kalea A, Zou YS, Vedantham S, et al. Aldose reductase and AGE-RAGE pathways: central roles in the pathogenesis of vascular dysfunction in aging rats. Aging Cell 2010;5:776–784.
- Myint KM, Yamamoto Y, Doi T, Kato I, Harashima A, Yonekura H, et al. RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes 2006; 55:2510–2522.
- Sorci G, Riuzzi, Giambanco I, Donato R. RAGE in tissue homeostasis, repair and regeneration. Biochim Biophys Acta 2013;1833:101–109.
- Mahajan N, Malik N, Bahl A, Dhawan V. Receptor for advanced glycation end products (RAGE) and its inflammatory ligand EN-RAGE in non-diabetic subjects with pre-mature coronary artery disease. Atherosclerosis 2009; 207:597–602.
- Sun L, Ishida T, Yasuda T, Kojima Y, Honjo T, Yamamoto Y, et al. RAGE mediates oxidized LDL-induced pro-inflammatory effects and atherosclerosis in non-diabetic LDL receptor-deficient mice. Cardiovasc Res 2009;82: 371–381.
- Lal MA, Brismar H, Eklöf AC, Aperia A. Role of oxidative stress in advanced glycation end product-induced mesangial cell activation. Kidney Int 2002;61:2006–2014.
- Wautier MP, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier JL. Activation of NADPH oxidase by AGE links oxidant stress to altered gene expression via RAGE. Am J Physiol Endocrinol Metab 2001;280:E685–E694.
- Barlovic DP, Soro-Paavonen A, Jandeleit-Dahm KA. RAGE biology, atherosclerosis and diabetes. Clin Sci (Lond) 2011;121:43–55.
- Hall G, Hasday JD, Rogers TB. Regulating the regulator: NF-kappaB signaling in heart. J Mol Cell Cardiol 2006;41:580–591.
- Valencia JV, Mone M, Koehne C, Rediske J, Hughes TE. Binding of receptor for advanced glycation end products (RAGE) ligands is not sufficient to induce inflammatory signals: lack of activity of endotoxin-free albumin-derived advanced glycation end products. Diabetologia 2004;47: 844–852.
- Lieuw-a-Fa ML, Schalkwijk CG, Engelse M, van Hinsbergh VW. Interaction of Nepsilon(carboxymethyl) lysine- and methylglyoxal-modified albumin with endothelial cells and macrophages. Splice variants of RAGE may limit the responsiveness of human endothelial cells to AGEs. Thromb Haemost 2006;95:320–328.
- Pötzsch S, Blankenhorn A, Navarrete Santos A, Silber RE, Somoza V, Simm A. The effect of an AGE-rich dietary extract on the activation of NF-κB depends on the cell model used. Food Funct 2013 [Epub ahead of print].
- Neves D, Assunção M, Marques F, Andrade JP, Almeida H. Does regular consumption of green tea influence VEGF and its receptors expression in aged rat erectile tissue?Possible implications in vasculogenic erectile dysfunction progression. AGE 2008;30:217–228.
- Higashi T, Sano H, Saishoji T, Ikeda K, Jinnouchi Y, Kanzaki T, et al. The receptor for advanced glycation end products mediates the chemotaxis of rabbit smooth muscle cells. Diabetes 1997;46:463–472.
- Kalea AZ, Reiniger N, Yang H, Arriero M, Schmidt AM, Hudson BI. Alternative splicing of the murine receptor for advanced glycation end-products (RAGE) gene. FASEB J 2009;23:1766–1774.
- Yonekura H, Yamamoto Y, Sakurai S, Petrova RG, Abedin MJ, Li H, et al. Novel splice variants of the receptor for advanced glycation end-products expressed in human vascular endothelial cells and pericytes, and their putative roles in diabetes-induced vascular injury. Biochem J 2003; 370:1097–1109.
- Choi KM, Yoo HJ, Kim HY, Lee KW, Seo JA, Kim SG, et al. Association between endogenous secretory RAGE, inflammatory markers and arterial stiffness. Int J Cardiol 2009, 132:96–101.
- Bucciarelli LG, Wendt T, Qu W, Lu Y, Lalla E, Rong LL, et al. RAGE blockade stabilizes established atherosclerosis in diabetic apolipoprotein E-null mice. Circulation 2002;106: 2827–2835.
- Kislinger T, Tanji N, Wendt T, Qu W, Lu Y, Ferran LJ Jr, et al. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol 2001;21:905–910.
- Colhoun HM, Betteridge DJ, Durrington P, Hitman G, Neil A, Livingstone S, et al. Total soluble and endogenous secretory receptor for advanced glycation end products as predictive biomarkers of coronary heart disease risk in patients with type 2 diabetes: an analysis from the CARDS trial. Diabetes 2011;60:2379–2385.
- Koyama H, Shoji T, Yokoyama H, Motoyama K, Mori K, Fukumoto S, et al. Plasma level of endogenous secretory RAGE is associated with components of the metabolic syndrome and atherosclerosis. Arterioscler Thromb Vasc Biol 2005;25:2587–2593.
- Emanuele E, Bertona M. Endogenous secretory RAGE as a potential biochemical screening tool for erectile dysfunction. Med Hypotheses 2006;67:668–669.
- Semba RD, Ang A, Talegawkar S, Crasto C, Dalal M, Jardack P, et al. Dietary intake associated with serum versus urinary carboxymethyl-lysine, a major advanced glycation end product, in adults: the Energetics Study. Eur J Clin Nutr 2012;66:3–9.
- Uribarri J, Cai W, Peppa M, Goodman S, Ferrucci L, Striker G, Vlassara H. Circulating glycotoxins and dietary advanced glycation endproducts: two links to inflammatory response, oxidative stress, and aging. J Gerontol A Biol Sci Med Sci 2007;62:427–433.
- Uribarri J, Cai W, Sandu O, Peppa M, Goldberg T, Vlassara H. Diet-derived advanced glycation end products are major contributors to the body's AGE pool and induce inflammation in healthy subjects. Ann N Y Acad Sci 2005;1043:461–466.
- Gugliucci A, Kotani K, Taing J, Matsuoka Y, Sano Y, Yoshimura M, et al. Short-term low calorie diet intervention reduces serum advanced glycation end products in healthy overweight or obese adults. Ann Nutr Metab 2009;54: 197–201.
- Tomada I, Tomada N, Almeida H, Neves D. Energy restriction and exercise modulate Angiopoietins and Vascular Endothelial Growth Factor expression in the cavernous tissue of high-fat diet-fed rats. Asian J Androl 2012;14:635–642.
- Tomada I, Fernandes D, Guimarães JT, Almeida H, Neves D. Energy restriction ameliorates Metabolic Syndrome-induced cavernous tissue structural modifications in aged rats. Age (Dordr)2012 [Epub ahead of print].
- Maio MT, Hannan JL, Komolova M, Adams MA. Caloric restriction prevents visceral adipose tissue accumulation and maintains erectile function in aging rats. J Sex Med 2012; 9:2273–2283.
- Sakata N, Imanaga Y, Meng J, Tachikawa Y, Takebayashi S, Nagai R, et al. Immunohistochemical localization of different epitopes of advanced glycation end products in human atherosclerotic lesions. Atherosclerosis 1998;141:61–75.
- Woods TM, Kamalov M, Harris PW, Cooper GJ, Brimble M. Synthesis of monolysyl advanced glycation endproducts and their incorporation into collagen model peptides. Org Lett 2012;14:5740–5743.
- Sell DR, Lane MA, Obrenovich ME, Mattison JA, Handy A, Ingram DK, et al. The effect of caloric restriction on glycation and glycoxidation in skin collagen of nonhuman primates. J Gerontol Biol Sci 2003;58A:508–516.
- Azadzoi KM, Schulman RN, Aviram M, Siroky MB. Oxidative stress in arteriogenic erectile dysfunction: prophylactic role of antioxidants. J Urol 2005;174:386–393.
- Zhang Q, Radisavljevic ZM, Siroky MB, Azadzoi KM. Dietary antioxidants improve arteriogenic erectile dysfunction. Int J Androl 2011;34:225–235.
- Ha US, Koh JS, Kim HS, Woo JC, Kim SJ, Jang H, et al. Cyanidin-3-O-β-D-glucopyranoside concentrated materials from mulberry fruit have a potency to protect erectile function by minimizing oxidative stress in a rat model of diabetic erectile dysfunction. Urol Int 2012;88:470–476.
- Forest CP, Padma-Nathan H, Liker HR. Efficacy and safety of pomegranate juice on improvement of erectile dysfunction in male patients with mild to moderate erectile dysfunction: a randomized, placebo-controlled, double-blind, crossover study. Int J Impot Res 2007;19:564–567.
- Achike FI, Kwan CY. Nitric oxide, human diseases and the herbal products that affect the nitric oxide signalling pathway. Clin Exp Pharmacol Physiol 2003;30:605–615.
- McCann SM, Licinio J, Wong ML, Yu WH, Karanth S, Rettorri V. The nitric oxide hypothesis of aging. Exp Gerontol 1998;33:813–826.
- Neves D, Tomada I, Assunção M, Marques F, Almeida H, Andrade JP. Effects of chronic red wine consumption on the expression of vascular endothelial growth factor, angiopoietin 1, angiopoietin 2 and its receptors in rat erectile tissue. J Food Science 2010;75:H79–H86.
- Lima CF, Pereira-Wilson C, Rattan SI. Curcumin induces heme oxygenase-1 in normal human skin fibroblasts through redox signaling: relevance for anti-aging intervention. Mol Nutr Food Res 2011;55:430–442.
- Shen LR, Xiao F, Yuan P, Chen Y, Gao QK, Parnell LD et al. Curcumin-supplemented diets increase superoxide dismutase activity and mean lifespan in Drosophila. Age (Dordr)2012 [Epub ahead of print].
- Hu TY, Liu CL, Chyau CC, Hu ML. Trapping of methylglyoxal by curcumin in cell-free systems and in human umbilical vein endothelial cells. J Agric Food Chem 2012; 60:8190–8196.
- Lin J, Tang Y, Kang Q, Feng Y, Chen A. Curcumin inhibits gene expression of receptor for advanced glycation end-products (RAGE) in hepatic stellate cells in vitro by elevating PPARγ activity and attenuating oxidative stress. Br J Pharmacol 2012;166:2212–2227.
- Lin J, Tang Y, Kang Q, Chen A. Curcumin eliminates the inhibitory effect of advanced glycation end-products (AGEs) on gene expression of AGE receptor-1 in hepatic stellate cells in vitro. Lab Invest 2012;92:827–841.
- Yang Q, Wu S, Mao X, Wang W, Tai H. Inhibition effect of curcumin on TNF-α and MMP-13 expression induced by advanced glycation end products in chondrocytes. Pharmacology 2013;91:77–85.
- Pendurthi UR, Williams JT, Rao LV. Inhibition of tissue factor gene activation in cultured endothelial cells by curcumin. Suppression of activation of transcription factors Egr-1, AP-1, and NF-kappa B.Arterioscler Thromb Vasc Biol. 1997;17:3406–3413.
- Bierhaus A, Zhang Y, Quehenberger P, Luther T, Haase M, Müller M et al. The dietary pigment curcumin reduces endothelial tissue factor gene expression by inhibiting binding of AP-1 to the DNA and activation of NF-kappa B. Thromb Haemost 1997;77:772–782.
- Olszanecki R, Gebska A, Korbut R. The role of haem oxygenase-1 in the decrease of endothelial intercellular adhesion molecule-1 expression by curcumin. Basic Clin Pharmacol Toxicol. 2007;101:411–415.
- Binion DG, Heidemann J, Li MS, Nelson VM, Otterson MF, Rafiee P. Vascular cell adhesion molecule-1 expression in human intestinal microvascular endothelial cells is regulated by PI 3-kinase/Akt/MAPK/NF-kappaB: inhibitory role of curcumin. Am J Physiol Gastrointest Liver Physiol. 2009; 297:G259–268.
- Panicker SR, Kartha CC. Curcumin attenuates glucose- induced monocyte chemoattractant protein-1 synthesis in aortic endothelial cells by modulating the nuclear factor-kappaB pathway. Pharmacology 2010;85:18–26.
- Kumar A, Dhawan S, Hardegen NJ, Aggarwal BB. Curcumin (Diferuloylmethane) inhibition of tumor necrosis factor (TNF)-mediated adhesion of monocytes to endothelial cells by suppression of cell surface expression of adhesion molecules and of nuclear factor-kappaB activation. Biochem Pharmacol. 1998;55:775–783.
- Bae MK, Kim SH, Jeong JW, Lee YM, Kim HS, Kim SR et al. Curcumin inhibits hypoxia-induced angiogenesis via down-regulation of HIF-1. Oncol Rep. 2006;15:1557–1562.
- Chen YR, Tan TH. Inhibition of the c-Jun N-terminal kinase (JNK) signaling pathway by curcumin. Oncogene 1998; 17:173–178.
- Akhand AA, Hossain K, Mitsui H, Kato M, Miyata T, Inagi R et al. Glyoxal and methylglyoxal trigger distinct signals for map family kinases and caspase activation in human endothelial cells. Free Radic Biol Med 2001;31:20–30.
- Kim DC, Lee W, Bae JS. Vascular anti-inflammatory effects of curcumin on HMGB1-mediated responses in vitro. Inflamm Res 2011;60:1161–1168.
- Han J, Pan XY, Xu Y, Xiao Y, An Y, Tie L et al. Curcumin induces autophagy to protect vascular endothelial cell survival from oxidative stress damage. Autophagy 2012;8: 812–825.
- Bradford PG. Curcumin and obesity. Biofactors 2013;39: 78–87.
- Yang Y, Duan W, Liang Z, Yi W, Yan J, Wang N et al. Curcumin attenuates endothelial cell oxidative stress injury through Notch signaling inhibition. Cell Signal 2013;25: 615–629.
- Yu YM, Lin HC. Curcumin prevents human aortic smooth muscle cells migration by inhibiting of MMP-9 expression. Nutr Metab Cardiovasc Dis 2010;20:125–132.
- Chen HW, Huang HC. Effect of curcumin on cell cycle progression and apoptosis in vascular smooth muscle cells. Br J Pharmacol 1998;124:1029–1040.
- Pae HO, Jeong GS, Jeong SO, Kim HS, Kim SA, Kim YC, et al. Roles of heme oxygenase-1 in curcumin-induced growth inhibition in rat smooth muscle cells. Exp Mol Med 2007;39:267–277.
- Youreva V, Kapakos G, Srivastava AK. Insulin-like growth-factor-1-induced PKB signaling and Egr-1 expression is inhibited by curcumin in A-10 vascular smooth muscle cells. Can J Physiol Pharmacol 2013;91:241–247.
- Yang X, Thomas DP, Zhang X, Culver BW, Alexander BM, Murdoch WJ, et al. Curcumin inhibits platelet-derived growth factor-stimulated vascular smooth muscle cell function and injury-induced neointima formation. Arterioscler Thromb Vasc Biol 2006;26:85–90.
- Hua Y, Dolence J, Ramanan S, Ren J, Nair S. Bisdemethoxycurcumin inhibits PDGF-induced vascular smooth muscle cell motility and proliferation. Mol Nutr Food Res 2013 [Epub ahead of print].
- Kang ES, Woo IS, Kim HJ, Eun SY, Paek KS, Kim HJ, et al. Up-regulation of aldose reductase expression mediated by phosphatidylinositol 3-kinase/Akt and Nrf2 is involved in the protective effect of curcumin against oxidative damage. Free Radic Biol Med 2007;43:535–545.
- Meng Z, Yan C, Deng Q, Gao DF, Niu XL. Curcumin inhibits LPS-induced inflammation in rat vascular smooth muscle cells in vitro via ROS-relative TLR4-MAPK/NF-κB pathways. Acta Pharmacol Sin 2013 [Epub ahead of print].
- Kapakos G, Youreva V, Srivastava AK. Attenuation of endothelin-1-induced PKB and ERK1/2 signaling, as well as Egr-1 expression, by curcumin in A-10 vascular smooth muscle cells. Can J Physiol Pharmacol 2012;90:1277–1285.
- Fleenor BS, Sindler AL, Marvi NK, Howell KL, Zigler ML, Yoshizawa M, Seals DR. Curcumin ameliorates arterial dysfunction and oxidative stress with aging. Exp Gerontol2012 [Epub ahead of print].
- Majithiya JB, Balaraman R. Time-dependent changes in antioxidant enzymes and vascular reactivity of aorta in streptozotocin-induced diabetic rats treated with curcumin. J Cardiovasc Pharmacol 2005;46:697–705.
- Nakmareong S, Kukongviriyapan U, Pakdeechote P, Kukongviriyapan V, Kongyingyoes B, Donpunha W, et al. Tetrahydrocurcumin alleviates hypertension, aortic stiffening and oxidative stress in rats with nitric oxide deficiency. Hypertens Res 2012;35:418–425.
- Rungseesantivanon S, Thenchaisri N, Ruangvejvorachai P, Patumraj S. Curcumin supplementation could improve diabetes-induced endothelial dysfunction associated with decreased vascular superoxide production and PKC inhibition. BMC Complement Altern Med 2010;10:57.
- Coban D, Milenkovic D, Chanet A, Khallou-Laschet J, Sabbe L, Palagani A, et al. Dietary curcumin inhibits atherosclerosis by affecting the expression of genes involved in leukocyte adhesion and transendothelial migration. Mol Nutr Food Res 2012;56:1270–1281.
- Sajithlal GB, Chithra P, Chandrakasan G. Effect of curcumin on the advanced glycation and cross-linking of collagen in diabetic rats. Biochem Pharmacol 1998;56:1607–1614.
- Abdel Aziz MT, Motawi T, Rezq A, Mostafa T, Fouad HH, Ahmed HH, et al. Effects of a water-soluble curcumin protein conjugate vs. pure curcumin in a diabetic model of erectile dysfunction. J Sex Med 2012;9:1815–1833.
- Farhangkhoee H, Khan ZA, Chen S, Chakrabarti S. Differential effects of curcumin on vasoactive factors in the diabetic rat heart. Nutr Metab 2006;183:27.
- Margina D, Gradinaru D, Manda G, Neagoe I, Ilie M. Membranar effects exerted in vitro by polyphenols – quercetin, epigallocatechin gallate and curcumin – on HUVEC and Jurkat cells, relevant for diabetes mellitus. Food Chem Toxicol 2013 [Epub ahead of print].
- Mizutani K, Ikeda K, Yamori Y. Resveratrol inhibits AGEs-induced proliferation and collagen synthesis activity in vascular smooth muscle cells from stroke-prone spontaneously hypertensive rats. Biochem Biophys Res Commun 2000;274:61–67.
- Babu PV, Sabitha KE, Srinivasan P, Shyamaladevi CS. Green tea attenuates diabetes induced Maillard-type fluorescence and collagen cross-linking in the heart of streptozotocin diabetic rats. Pharmacol Res 2007;55:433–40.
- Rajasekar P, Anuradha CV. L-Carnitine inhibits protein glycation in vitro and in vivo: evidence for a role in diabetic management. Acta Diabetol 2007;44:83–90.
- Cavallini G, Caracciolo S, Vitali G, Modenini F, Biagiotti G. Carnitine versus androgen administration in the treatment of sexual dysfunction, depressed mood, and fatigue associated with male aging. Urology 2004;63:641–646.
- Gianfrilli D, Lauretta R, Di Dato C, Graziadio C, Pozza C, De Larichaudy J, et al. Propionyl-L-carnitine, L-arginine and niacin in sexual medicine: a nutraceutical approach to erectile dysfunction. Andrologia 2012;44:600–604.
- Cavallini G, Modenini F, Vitali G, Koverech A. Acetyl-L-carnitine plus propionyl-L-carnitine improve efficacy of sildenafil in treatment of erectile dysfunction after bilateral nerve-sparing radical retropubic prostatectomy. Urology 2005;66:1080–1085.
- Gentile V, Vicini P, Prigiotti G, Koverech A, Di Silverio F. Preliminary observations on the use of propionyl-L-carnitine in combination with sildenafil in patients with erectile dysfunction and diabetes. Curr Med Res Opin 2004;20: 1377–1384.
- Gentile V, Antonini G, Antonella Bertozzi M, Dinelli N, Rizzo C, Ashraf Virmani M, Koverech A. Effect of propionyl-L-carnitine, L-arginine and nicotinic acid on the efficacy of vardenafil in the treatment of erectile dysfunction in diabetes. Curr Med Res Opin 2009;25:2223–2228.
- Morano S, Mandosi E, Fallarino M, Gatti A, Tiberti C, Sensi M, et al. Antioxidant treatment associated with sildenafil reduces monocyte activation and markers of endothelial damage in patients with diabetic erectile dysfunction: a double-blind, placebo-controlled study. Eur Urol 2007; 52:1768–1774.
- Booth AA, Khalifah RG, Hudson BG. Thiamine pyrophosphate and pyridoxamine inhibit the formation of antigenic advanced glycation end-products: comparison with aminoguanidine. Biochem Biophys Res Commun 1996;220: 113–119.
- Avena R, Arora S, Carmody BJ, Cosby K, Sidawy NA. Thiamine (Vitamin B1) protects against glucose- and insulin-mediated proliferation of human infragenicular arterial smooth muscle cells. Ann Vasc Surg 2000;14:37–43.
- Beltramo E, Berrone E, Buttiglieri S, Porta M. Thiamine and benfotiamine prevent increased apoptosis in endothelial cells and pericytes cultured in high glucose. Diabetes Metab Res Rev 2004;20:330–336.
- Alkhalaf A, Kleefstra N, Groenier KH, Bilo HJ, Gans RO, Heeringa P, et al. Effect of benfotiamine on advanced glycation endproducts and markers of endothelial dysfunction and inflammation in diabetic nephropathy. PLoS One 2012;7:e40427.
- Alderson NL, Chachich ME, Youssef NN, Beattie RJ, Nachtigal M, Thorpe SR, Baynes JW. The AGE inhibitor pyridoxamine inhibits lipemia and development of renal and vascular disease in Zucker obese rats. Kidney Int 2003; 63:2123–2133.
- Chang KC, Liang JT, Tsai PS, Wu MS, Hsu KL. Prevention of arterial stiffening by pyridoxamine in diabetes is associated with inhibition of the pathogenic glycation on aortic collagen. Br J Pharmacol 2009;157:1419–1426.
- Wu ET, Liang JT, Wu MS, Chang KC. Pyridoxamine prevents age-related aortic stiffening and vascular resistance in association with reduced collagen glycation. Exp Gerontol 2011;46:482–488.
- Watson AM, Soro-Paavonen A, Sheehy K, Li J, Calkin AC, Koitka A, et al. Delayed intervention with AGE inhibitors attenuates the progression of diabetes-accelerated atherosclerosis in diabetic apolipoprotein E knockout mice. Diabetologia 2011;54:681–689.
- Jain SK, Lim G. Pyridoxine and pyridoxamine inhibits superoxide radicals and prevents lipid peroxidation, protein glycosylation, and (Na+ + K+)-ATPase activity reduction in high glucose-treated human erythrocytes. Free Radic Biol Med 2001;30:232–237.
- Giannoukakis N. Pyridoxamine (BioStratum). Curr Opin Investig Drugs 2005;6:410–418.
- Lewis EJ, Greene T, Spitalewiz S, Blumenthal S, Berl T, Hunsicker LG, et al. Pyridorin in type 2 diabetic nephropathy. J Am Soc Nephrol 2012;23:131–136.
- Williams ME, Bolton WK, Khalifah RG, Degenhardt TP, Schotzinger RJ, McGill JB. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am J Nephrol 2007;27: 605–614.
- Bierhaus A, Chevion S, Chevion M, Hofmann M, Quehenberger P, Illmer T, et al. Advanced glycation end product-induced activation of NF-kappaB is suppressed by alpha-lipoic acid in cultured endothelial cells. Diabetes 1997;46:1481–1490.
- Keegan A, Cotter MA, Cameron NE. Corpus cavernosum dysfunction in diabetic rats: effects of combined alpha-lipoic acid and gamma-linolenic acid treatment. Diabetes Metab Res Rev 2001;17:380–386.
- Hipkiss AR. Carnosine and its possible roles in nutrition and health. Adv Food Nutr Res 2009;57:87–154.
- Krajcovicová-Kudlácková M, Sebeková K, Schinzel R, Klvanová J. Advanced glycation end products and nutrition. Physiol Res 2002;51:313–316.
- Li YM, Tan AX, Vlassara H. Antibacterial activity of lysozyme and lactoferrin is inhibited by binding of advanced glycation-modified proteins to a conserved motif. Nat Med 1995;1:1057–1061.
- Tan D, Wang Y, Lo CY, Sang S, Ho CT. Methylglyoxal: its presence in beverages and potential scavengers. Ann N Y Acad Sci 2008;1126:72–75.
- Ruhs S, Nass N, Somoza V, Friess U, Schinzel R, Silber RE, Simm A. Maillard reaction products enriched food extract reduce the expression of myofibroblast phenotype markers. Mol Nutr Food Res 2007;51:488–495.
- Leuner B, Ruhs S, Brömme HJ, Bierhaus A, Sel S, Silber RE, et al. RAGE-dependent activation of gene expression of superoxide dismutase and vanins by AGE-rich extracts in mice cardiac tissue and murine cardiac fibroblasts. Food Funct 2012;3:1091–1098.
- Ruhs S, Nass N, Bartling B, Brömme HJ, Leuner B, Somoza V, et al. Preconditioning with Maillard reaction products improves antioxidant defence leading to increased stress tolerance in cardiac cells. Exp Gerontol 2010;45: 752–762.
- Anton PM, Craus A, Niquet-Léridon C, Tessier FJ. Highly heated food rich in Maillard reaction products limit an experimental colitis in mice. Food Funct 2012;3:941–949
- Niebauer J, Maxwell AJ, Lin PS, Wang D, Tsao PS, Cooke JP. NOS inhibition accelerates atherogenesis: reversal by exercise. Am J Physiol Heart Circ Physiol 2003; 285:H535–H540.
- Esposito K, Giugliano F, Di Palo C, Giugliano G, Marfella R, D’Andrea F, et al. Effect of lifestyle changes on erectile dysfunction in obese men: a randomized controlled trial. JAMA 2004;291:2978–2984.
- Boor P, Celec P, Behuliak M, Grancic P, Kebis A, Kukan M, et al. Regular moderate exercise reduces advanced glycation and ameliorates early diabetic nephropathy in obese Zucker rats. Metabolism 2009;58:1669–1677.
- Choi KM, Han KA, Ahn HJ, Hwang SY, Hong HC, Choi HY, et al. Effects of exercise on sRAGE levels and cardiometabolic risk factors in patients with type 2 diabetes: a randomized controlled trial. J Clin Endocrinol Metab 2012;97:3751–3758.
- Kotani K, Caccavello R, Sakane N, Yamada T, Taniguchi N, Gugliucci A. Influence of physical activity intervention on circulating soluble receptor for advanced glycation end products in elderly subjects. J Clin Med Res 2011;3:252–257.
- Lu J, Gong D, Choong SY, Xu H, Chan YK, Chen X, et al. Copper(II)-selective chelation improves function and antioxidant defences in cardiovascular tissues of rats as a model of diabetes: comparisons between triethylenetetramine and three less copper-selective transition-metal-targeted treatments. Diabetologia 2010;53:1217–1226.
- Koupparis AJ, Jeremy J, Angelini G, Persad R, Shukla N. Penicillamine administration reverses the inhibitory effect of hyperhomocysteinaemia on endothelium-dependent relaxation in the corpus cavernosum in the rabbit. BJU Int 2006;98:440–444.
- Keegan A, Cotter MA, Cameron NE. Effects of chelator treatment on aorta and corpus cavernosum from diabetic rats. Free Radic Biol Med 1999;27:536–543.
- Gur S, Kadowitz PJ, Hellstrom WJ. A critical appraisal of erectile function in animal models of diabetes mellitus. Int J Androl 2009;32:93–114.
- Vlassara H, Bucala R, Striker L. Pathogenic effects of advanced glycosylation: biochemical, biologic, and clinical implications for diabetes and aging. Lab Invest 1994;70: 138–151.
- Ihm SH, Yoo HJ, Park SW, Ihm J. Effect of aminoguanidine on lipid peroxidation in streptozotocin-induced diabetic rats. Metabolism 1999;48:1141–1145.
- Szabó C, Ferrer-Sueta G, Zingarelli B, Southan GJ, Salzman AL, Radi R. Mercaptoethylguanidine and guanidine inhibitors of nitric-oxide synthase react with peroxynitrite and protect against peroxynitrite-induced oxidative damage. J Biol Chem 1997;272:9030–9036.
- Price DL, Rhett PM, Thorpe SR, Baynes JW. Chelating activity of advanced glycation end-product inhibitors. J Biol Chem 2001;276:48967–48972.
- Thornalley PJ. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch Biochem Biophys 2003;419:31–40.
- Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Aminoguanidine prevents diabetes-induced arterial wall protein cross-linking. Science 1986;232:1629–1632.
- Sell DR, Nelson JF, Monnier VM. Effect of chronic aminoguanidine treatment on age-related glycation, glycoxidation, and collagen cross-linking in the Fischer 344 rat. J Gerontol A Biol Sci Med Sci 2001;56:B405–B411.
- Griffiths MJ, Messent M, MacAllister RJ, Evans TW. Aminoguanidine selectively inhibits inducible nitric oxide synthase. Br J Pharmacol 1993;110:963–968.
- Misko TP, Moore WM, Kasten TP, Nickols GA, Corbett JA, Tilton RG, et al. Selective inhibition of the inducible nitric oxide synthase by aminoguanidine. Eur J Pharmacol 1993;233:119–125.
- Wessells H, Teal TH, Luttrell IP, Sullivan CJ. Effect of endothelial cell-based iNOS gene transfer on cavernosal eNOS expression and mouse erectile responses. Int J Impot Res 2006;18:438–445.
- Usta MF, Bivalacqua TJ, Yang DY, Ramanitharan A, Sell DR, Viswanathan A, et al. The protective effect of aminoguanidine on erectile function in streptozotocin diabetic rats. J Urol 2003;170:1437–1442.
- Usta MF, Bivalacqua TJ, Koksal IT, Toptas B, Surmen S, Hellstrom WJ. The protective effect of aminoguanidine on erectile function in diabetic rats is not related to the timing of treatment. BJU Int 2004;94:429–432.
- Forbes JM, Yee LT, Thallas V, Lassila M, Candido R, Jandeleit-Dahm KA, et al. Advanced glycation end product interventions reduce diabetes-accelerated atherosclerosis. Diabetes 2004;53:1813–1823.
- Asif M, Egan J, Vasan S, Jyothirmayi GN, Masurekar MR, Lopez S, et al. An advanced glycation end product cross-link breaker can reverse age-related increases in myocardial stiffness. Proc Natl Acad Sci U S A 2000;97:2809–2813.
- Usta MF, Kendirci M, Gur S, Foxwell NA, Bivalacqua TJ, Cellek S, Hellstrom WJ. The breakdown of preformed advanced glycation end products reverses erectile dysfunction in streptozotocin-induced diabetic rats: preventive versus curative treatment. J Sex Med 2006;3:242–250.
- Gurbuz N, Sagdic G, Sanli A, Ciftcioglu A, Bassorgun I, Baykal A, Usta MF. Therapeutic effect of combination of alagebrium (ALT-711) and sildenafil on erectile function in diabetic rats. Int J Impot Res 2012;24:114–121.
- Hartog JW, Willemsen S, van Veldhuisen DJ, Posma JL, van Wijk LM, Hummel YM, et al. Effects of alagebrium, an advanced glycation endproduct breaker, on exercise tolerance and cardiac function in patients with chronic heart failure. Eur J Heart Fail 2011;13:899–908.
- Wiernsperger NF, Bailey CJ. The antihyperglycaemic effect of metformin: therapeutic and cellular mechanisms. Drugs 1999;58:31–39.
- Paolisso G, Amato L, Eccellente R, Gambardella A, Tagliamonte MR, Varricchio G, et al. Effect of metformin on food intake in obese subjects. Eur J Clin Invest 1998;28:441–446.
- Seifarth C, Schehler B, Schneider HJ. Effectiveness of metformin on weight loss in non-diabetic individuals with obesity. Exp Clin Endocrinol Diabetes 2012 [Epub ahead of print].
- Beisswenger PA, Howell SK, Touchette AD, Lal S, Szwergold BS. Metformin reduces systematic methylglyoxal levels in type 2 diabetes. Diabetes 1999;48:198–202.
- Beisswenger P, Ruggiero-Lopez D. Metformin inhibition of glycation processes. Diabetes Metab 2003;29:6S95–103.
- Chakraborty A, Chowdhury S, Bhattacharyya M. Effect of metformin on oxidative stress, nitrosative stress and inflammatory biomarkers in type 2 diabetes patients. Diabetes Res Clin Pract 2011;93:56–62.
- Kim YW, Park SY, Kim JY, Huh JY, Jeon WS, Yoon CJ, et al. Metformin restores the penile expression of nitric oxide synthase in high-fat-fed obese rats. J Androl 2007;28: 555–560.
- Rey-Valzacchi GJ, Costanzo PR, Finger LA, Layus AO, Gueglio GM, Litwak LE, Knoblovits P. Addition of metformin to sildenafil treatment for erectile dysfunction in eugonadal nondiabetic men with insulin resistance. A prospective, randomized, double-blind pilot study. J Androl 2012;33:608–614.
- Rahbar S, Natarajan R, Yerneni K, Scott S, Gonzales N, Nadler JL. Evidence that pioglitazone, metformin and pentoxifylline are inhibitors of glycation. Clin Chim Acta 2000;301:65–77.
- Kovanecz I, Ferrini MG, Vernet D, Nolazco G, Rajfer J, Gonzalez-Cadavid NF. Pioglitazone prevents corporal veno-occlusive dysfunction in a rat model of type 2 diabetes mellitus. BJU Int 2006;98:116–124.
- Kovanecz I, Ferrini MG, Vernet D, Nolazco G, Rajfer J, Gonzalez-Cadavid NF. Ageing-related corpora veno-occlusive dysfunction in the rat is ameliorated by pioglitazone. BJU Int 2007;100:867–874.
- Gholamine B, Shafiei M, Motevallian M, Mahmoudian M. Effects of pioglitazone on erectile dysfunction in sildenafil poor-responders: a randomized, controlled study. J Pharm Pharm Sci 2008;11:22–31.
- Ishibashi Y, Matsui T, Takeuchi M, Yamagishi S. Vardenafil, an inhibitor of phosphodiesterase-5, blocks advanced glycation end product (AGE)-induced up- regulation of monocyte chemoattractant protein-1 mRNA levels in endothelial cells by suppressing AGE receptor (RAGE) expression via elevation of cGMP. Clin Exp Med 2011;11:131–135.
- Ridker PM, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, et al. Rosuvastatin to prevent vascular events in men and women with elevated C-reactive protein. N Engl J Med 2008;359:2195–2207.
- de Vries FM, Denig P, Pouwels KB, Postma MJ, Hak E. Primary prevention of major cardiovascular and cerebrovascular events with statins in diabetic patients: a meta-analysis. Drugs 2012;72:2365–2373.
- Long SB, Blaha MJ, Blumenthal RS, Michos ED. Clinical utility of rosuvastatin and other statins for cardiovascular risk reduction among the elderly. Clin Interv Aging 2011;6:27–35.
- Antonopoulos AS, Margaritis M, Shirodaria C, Antoniades C. Translating the effects of statins: From redox regulation to suppression of vascular wall inflammation. Thromb Haemost 2012;108:840–848.
- Balakumar P, Kathuria S, Taneja G, Kalra S, Mahadevan N. Is targeting eNOS a key mechanistic insight of cardiovascular defensive potentials of statins?J Mol Cell Cardiol 2012;52:83–92.
- Ishida K, Matsumoto T, Taguchi K, Kamata K, Kobayashi T. Pravastatin normalizes endothelium-derived contracting factor-mediated response via suppression of Rho-kinase signalling in mesenteric artery from aged type 2 diabetic rat. Acta Physiol 2012;205:255–265.
- Okamoto T, Yamagishi S, Inagaki Y, Amano S, Koga K, Abe R, et al. Angiogenesis induced by advanced glycation end products and its prevention by cerivastatin. FASEB J 2002;16:1928–1930.
- Santilli F, Bucciarelli L, Noto D, Cefalù AB, Davì V, Ferrante E, et al. Decreased plasma soluble RAGE in patients with hypercholesterolemia: effects of statins. Free Radic Biol Med 2007;43:1255–1262.
- Tam HL, Shiu SW, Wong Y, Chow WS, Betteridge DJ, Tan KC. Effects of atorvastatin on serum soluble receptors for advanced glycation end-products in type 2 diabetes. Atherosclerosis 2010;209:173–177.
- Jinnouchi Y, Yamagishi S, Takeuchi M, Ishida S, Jinnouchi Y, Jinnouchi J, Imaizumi T. Atorvastatin decreases serum levels of advanced glycation end products (AGEs) in patients with type 2 diabetes. Clin Exp Med 2006;6:191–193.
- Mahajan N, Bahl A, Dhawan V. C-reactive protein (CRP) up-regulates expression of receptor for advanced glycation end products (RAGE) and its inflammatory ligand EN-RAGE in THP-1 cells: inhibitory effects of atorvastatin. Int J Cardiol 2010;142:273–278.
- Yoon SJ, Yoon YW, Lee BK, Kwon HM, Hwang KC, Kim M, et al. Potential role of HMG CoA reductase inhibitor on oxidative stress induced by advanced glycation endproducts in vascular smooth muscle cells of diabetic vasculopathy. Exp Mol Med 2009;41:802–811.
- Feng B, Xu L, Wang H, Yan X, Xue J, Liu F, Hu JF. Atorvastatin exerts its anti-atherosclerotic effects by targeting the receptor for advanced glycation end products. Biochim Biophys Acta 2011;1812:1130–1137.
- Saltzman EA, Guay AT, Jacobson J. Improvement in erectile function in men with organic erectile dysfunction by correction of elevated cholesterol levels: a clinical observation. J Urol 2004;172:255–258.
- Rizvi K, Hampson JP, Harvey JN. Do lipid-lowering drugs cause erectile dysfunction?A systematic review. Fam Pract 2002;19:95–98.
- Trivedi D, Kirby M, Wellsted DM, Ali S, Hackett G, O’Connor B, van Os S. Can simvastatin improve erectile function and health-related quality of life in men aged ≥ 40 years with erectile dysfunction? Results of the Erectile Dysfunction and Statins Trial [ISRCTN66772971]. BJU Int 2012 [Epub ahead of print].
- Castro MM, Rizzi E, Rascado RR, Nagassaki S, Bendhack LM, Tanus-Santos JE. Atorvastatin enhances sildenafil-induced vasodilation through nitric oxide-mediated mechanisms. Eur J Pharmacol 2004;498:189–194.
- Dadkhah F, Safarinejad MR, Asgari MA, Hosseini SY, Lashay A, Amini E. Atorvastatin improves the response to sildenafil in hypercholesterolemic men with erectile dysfunction not initially responsive to sildenafil. Int J Impot Res 2010;22:51–60.
- Herrmann HC, Levine LA, Macaluso J Jr, Walsh M, Bradbury D, Schwartz S, et al. Can atorvastatin improve the response to sildenafil in men with erectile dysfunction not initially responsive to sildenafil?Hypothesis and pilot trial results. J Sex Med 2006;3:303–308.
- Bank AJ, Kelly AS, Kaiser DR, Crawford WW, Waxman B, Schow DA, Billups KL. The effects of quinapril and atorvastatin on the responsiveness to sildenafil in men with erectile dysfunction. Vasc Med 2006; 11:251–257.
- Fibbi B, Morelli A, Marini M, Zhang XH, Mancina R, Vignozzi L, et al. Atorvastatin but not elocalcitol increases sildenafil responsiveness in spontaneously hypertensive rats by regulating the RhoA/ROCK pathway. J Androl 2008;29:70–84.
- Forbes JM, Thorpe SR, Thallas-Bonke V, Pete J, Thomas MC, Deemer ER, et al. Modulation of soluble receptor for advanced glycation end products by angiotensin-converting enzyme-1 inhibition in diabetic nephropathy. J Am Soc Nephrol 2005;16:2363–2372.
- Schlimmer N, Kratz M, Böhm M, Baumhäkel M. Telmisartan, ramipril and their combination improve endothelial function in different tissues in a murine model of cholesterol-induced atherosclerosis. Br J Pharmacol 2011; 163:804–814.
- Böhm M, Baumhäkel M, Teo K, Sleight P, Probstfield J, Gao P, et al.; ONTARGET/TRANSCEND Erectile Dysfunction Substudy Investigators. Erectile dysfunction predicts cardiovascular events in high-risk patients receiving telmisartan, ramipril, or both: the ONgoing Telmisartan Alone and in combination with Ramipril Global Endpoint Trial/Telmisartan Randomized AssessmeNt Study in ACE iNtolerant subjects with cardiovascular Disease (ONTARGET/TRANSCEND) Trials. Circulation 2010;121:1439–1446.
- Simm A, Wagner J, Gursinsky T, Nass N, Friedrich I, Schinzel R, et al. Advanced glycation endproducts: a biomarker for age as an outcome predictor after cardiac surgery?Exp Gerontol 2007;42:668–675.
- Simonsen U. Interactions between drugs for erectile dysfunction and drugs for cardiovascular disease. Int J Impot Res 2002;14:178–188.
- Kloner RA, Henderson L. Sexual function in patients with chronic angina pectoris. Am J Cardiol2013 [Epub ahead of print].
- Timmis AD, Chaitman BR, Crager M. Effects of ranolazine on exercise tolerance and HbA1c in patients with chronic angina and diabetes. Eur Heart J 2006;27:42–48.