Abstract
Context and Objective: Plumbagin (2-methyl, 5-hydroxy, 1, 4-naphthoquinone), an anticancer agent is encapsulated either as conventional or long circulating liposomal formulations to enhance its biological half-life and antitumor efficacy.
Methods: The liposomes were prepared by thin film hydration method and in vitro characterization was carried out to examine the particle size, zeta potential, drug encapsulation efficiency and in vitro release. The optimized formulations were tested for pharmacokinetic and pharmacodynamic efficacy against mice bearing B16F1 melanoma. Also in vivo toxicity studies were carried out.
Results and Discussion: The optimum particle size and entrapment efficiency was observed at drug to lipid molar ratio of 1:20. The in-vitro release of plumbagin from the liposomal formulations in phosphate-buffered saline (pH 7.4) showed biphasic release with an initial burst release followed by sustained release phase. Elimination half life (T1/2) of pegylated, conventional and free plumbagin was 1305.76 ± 278.16, 346.87 ± 33.82 and 35.89 ± 7.95 min respectively. Further, plumbagin exhibited better antitumor efficacy in vivo when administered as long circulating liposomes with no signs of normal tissue toxicity.
Conclusion: It can be concluded that the pegylated liposomes could provide a promising parenteral platform for plumbagin with enhanced plasma half-life and therapeutic efficacy.
Introduction
Plumbagin (PLB), a naturally occurring naphthoquinone widely distributed in the Plumbaginaceae family, has been reported to possess a wide spectrum of pharmacological properties (CitationPadhye et al., 2010). The anticancer activity of PLB has been established in human promyelocytic leukemia (CitationXu & Lu, 2010), prostate cancer (CitationAziz et al., 2008; CitationPowolny & Singh, 2008), ovarian (CitationThasni et al., 2008), melanoma (CitationWang et al., 2008), breast cancer (CitationAhmad et al., 2008), non-small cell lung carcinoma (CitationHsu et al., 2006) and cervical cancerous cell lines (CitationSrinivas et al., 2004). Also, PLB has shown radiosensitizing potential (CitationDevi et al., 1994; CitationPrasad et al., 1996; CitationNair et al., 2008). Besides, possessing anticancer activity, its roots have been used in Indian system of traditional medicine for the treatment of various ailments as antiparasitic (CitationLikhitwitayawuid et al., 1998), antimicrobial (Citationde Paiva et al., 2003), antiatherosclerotic properties (CitationMossa et al., 2004). PLB induces cell death by apoptosis via reactive oxygen species (ROS) induced oxidative stress (CitationWang et al., 2008; CitationXu & Lu, 2010), cause G2/M phase arrest through the induction of p21 (CitationHsu et al., 2006) and found to inhibit the activity of topoisomerase II (Topo II) through the stabilization of the Topo II–DNA cleavable complex (CitationKawiak et al., 2007; CitationPadhye et al., 2010). Extending its mechanism authors have reported the down regulation of transcription factor NF-kB (CitationAhmad et al., 2008) and its regulated anti-apoptotic (Bcl-2, Bcl-xL, IAP1, IAP2, cFLIP and survivin), proliferative (cyclin D1 and COX-2) and angiogenic (matrix metalloproteinase-9 and vascular endothelial growth factor) gene products upon PLB treatment (CitationSandur et al., 2006). Besides possessing anticancer potential, some reports suggest that PLB is associated with normal tissue toxicity and short biological half-life with rapid elimination in vivo (CitationChandrasekaran & Nagarajan, 1981). It has been reported that PLB has the tendency to cause hemorrhage on chronic administration due to competitive inhibition of vitamin K activity (CitationVijayakumar et al., 2006). Other toxic effects of PLB include diarrhea, skin rashes and hepatic toxicity (CitationSandur et al., 2006).
The utility of currently available conventional cancer chemotherapeutics is severely limited due to their lower therapeutic index, poor patient tolerance or due to their inability to reach the tumor site at therapeutic concentrations or rapid elimination in vivo leading to shorter biological half-life. Apart from the above, pharmaceutical issues like drug solubility problems make most of the effective cancer drugs to be administered with other solubilizing agents which themselves lead to severe side effects (CitationSharma & Sharma, 1997). In order to overcome these clinically related issues different approaches have been attempted to provide selective delivery only to those organs, tissues, or cells affected by the disease. Among the several promising drug-delivery systems, liposomal technology has resulted in a large number of formulations, encapsulating various hydrophilic and hydrophobic active molecules (CitationChrai et al., 2002; CitationImmordino et al., 2006) and high target efficiency and activity (CitationImmordino et al., 2006). Liposomes are considered to be very flexible in that their surfaces can be easily modified with a variety of functional moieties such as polyethylene glycol (PEG) molecules and targeting ligands (CitationKhalil et al., 2008). The clinical acceptance of liposomes as drug carriers has been reflected in a number of liposome-based formulations (CitationLasic, 1996; CitationPanchagnula, 1998; CitationDrummond et al., 1999; CitationMassing & Fuxius, 2000; CitationAllen & Cullis, 2004; CitationTorchilin, 2005). Literature review suggests that only few drug delivery approaches for PLB have been attempted such as niosomes, albumin and chitosan microspheres, PLGA based microparticles and thermosensitive liposomes in order to improve its anticancer potential and reduce the associated toxicity (CitationNaresh et al., 1996; CitationKini et al., 1997; CitationSingh & Udupa, 1997; CitationTiwari et al., 2002; CitationMandala Rayabandla et al., 2010; CitationPadhye et al., 2010). In the present study an attempt has been made to formulate PLB as long circulating liposomes to enhance its biological half-life and to evaluate its pharmacokinetic and pharmacodynamic efficacy in mice bearing B16F1 melanoma and efforts have also been made to evaluate the normal tissue toxicity.
Materials and methods
Chemicals
Plumbagin, HEPES buffer, soybean phosphatidylcholine (SPC), cholesterol (Chol) were obtained from Sigma Chemicals Co., St. Louis, MO, USA. 1, 2-distearoyl-sn-glycero-3-phosphoethanolamine [methoxy (polyethyleneglycol)-2000] (mPEG2000-DSPE) was a generous gift sample from Lipoid Company. Fetal bovine serum (FBS) purchased from Genetix Biotech Asia, India. Dimethylsulfoxide (DMSO), methanol and chloroform were obtained from Rankem laboratories, India. Methanol and all the other reagents used for HPLC analysis were of HPLC grade and procured from Merck, Mumbai, India.
Animal model
C57BL/6J mice, procured from National Institute of Nutrition (Hyderabad, India) were maintained under controlled conditions of temperature (23 ± 2 °C), humidity (50 ± 5 %), and light (14 h and 10 h light and dark cycles respectively) and were allowed access to sterile food and water ad libitum. The animal care and handling were according to the guidelines of World Health Organization (Geneva, Switzerland) and the Indian National Science Academy (INSA) (New Delhi, India). For experimental purposes, eight to twelve week old mice from either sex, weighing 20 - 25 g from an inbred colony were selected. All the animal experiments were carried out with prior approval from the institutional animal ethics committee (Kasturba Medical College, Manipal, India).
Formulation development
Preparation of plumbagin (PLB) liposomes
PLB liposomes were prepared by thin film hydration method with minor modifications (CitationTalsma et al., 1989; CitationKaiser et al., 2003; CitationYang et al., 2007). The composition of lipids used for the formulation is represented in . Briefly, PLB, Cholesterol (Chol) and lipids (SPC) were accurately weighed, dissolved in chloroform and transferred into a suitable round bottom (RB) flask and submitted to evaporation under reduced pressure until a thin lipid film was obtained using BUCHI R-215 Rotavapor (Switzerland). The RB flasks were kept in vacuum desiccator for 6-8 h for final removal of traces of chloroform from the dried lipid film. Further, the lipid film was re-suspended in HEPES buffer (10mM, pH-6.5, 50mg lipid/ml) by rotating the flask at 200 rpm until the lipid film was completely hydrated using a rotary evaporator, at a temperature above the transition temperature (Tc) of the lipids (42 °C for SPC, 55 °C for mPEG2000-DSPE). The liposomal dispersion was then subjected to high pressure homogenization (HPH) (EmulsiFlex-C3, Avestin, Canada) equipped with thermostat to maintain product temperature during processing. The preformed liposomes were passed through HPH for 8 to10 cycles (flow rate of 40 ml/min) operated at pressure of about 12000 psi. The un-entrapped free PLB was separated from the liposomes by subjecting liposomal dispersion to ultracentrifugation at 4 °C (Sorvall WX Ultra Series Centrifuge, Thermo Scientific, USA) for 1 h at 160,000 × g. The supernatant was separated and the liposome pellet was re-suspended in HEPES buffer and stored at 4 °C in air tight glass (Type- I) containers for further experiments.
Table 1. Composition of liposomes and their respective particle size with zeta potential values.
Physicochemical characteristics of liposomes:
Negative-stain transmission electron microscopy (TEM)
The morphology of the prepared liposomes was studied by negative staining method. Briefly, the liposomal dispersion after appropriate dilution with normal saline was applied to 400 mesh formvar carbon-coated copper grids. Then the liposomes were stained with 1% w/v phosphotungistic acid solution (passed through 0.2 μm filters to eliminate impurities if any) for 2-3 min and rinsed with distilled water to washoff the excess stain and dried at room temperature for 10 min. The stained samples were observed using Morgagni 268 D transmission electron microscope (FEI, Netherlands) operated at 80 kV. The grid openings were randomly selected and viewed.
Particle size and zeta potential determination
The particle size and zeta potential of the prepared samples was determined after suitable dilutions using dynamic light scattering (DLS) technique with a Nano-ZS zeta sizer (Malvern Instruments, UK). The laser in this instrument was operated at 633 nm using a 173o angle between the incident and scattered laser beams. The instrument was validated prior to sample analysis using polystyrene standard beads. Data are reported as mean of two measurements along with poly dispersity index values.
Encapsulation efficiency determination
The amount of PLB incorporated into the liposomes was determined by HPLC (Shimadzu LC-2010, Kyoto, Japan) method. The liposomal pellet obtained after ultracentrifugation was dispersed in fresh HEPES buffer and the drug content in the supernatant and in the suspended pellet was determined. Briefly, the PLB was extracted by adding a mixture chloroform and methanol (2:1) and vortexing for 10 min. The samples were then centrifuged for 10 min at 5000 rpm and the resulting organic layer containing PLB was suitably diluted with mobile phase. The drug content analysis was performed using a reverse-phased Grace Vydac (Grace) C18 silica gel column (250 mm × 4.6 mm, 5 μm) operated at room temperature and the detector wavelength was set at 264 nm. Mixture of methanol: glacial acetic acid (0.1% v/v) (68:32) was used as the mobile phase at a flow rate of 1 ml/min. The percent entrapment efficiency (EE%) was calculated as EE% = (Dp/(Dp+Ds)) × 100, where Dp and Ds are drug concentration in pellet and in supernatant respectively.
In vitro drug release
The in vitro release of PLB from the liposomal formulations was performed by membrane dialysis bag (MW cutoff-12000) method with slight modifications (CitationXiong et al., 2005) in phosphate-buffered saline (PBS) at pH 7.4 containing 0.1% v/v Tween 80. Briefly, 1 ml aliquot of reconstituted PLB liposomes were transferred into dialysis tubing and placed in beaker (after sealing both ends) containing 50 ml of PBS maintained at 37 °C. At set time intervals, 1 ml sample was withdrawn, passed through 0.22 µM membrane filter and assayed for drug using UV-spectrophotometer at 422 nm. The removed samples were replaced with an equal quantity fresh PBS in order to maintain sink condition. The in vitro release studies were performed in triplicate for each prepared formulation.
Physical stability of liposomes
The optimized formulations F3b and C1 were evaluated for any particle size change by incubating in fetal bovine serum (CitationSahli et al., 1998; CitationSadzuka et al., 2003; CitationHan et al., 2006). The serum stability studies were performed by incubating the liposomes after dilution with 50% fetal bovine serum in PBS at 4 °C and 37 °C. The changes in the particle size were determined at predetermined time intervals using Nano-ZS zeta sizer (Malvern Instruments, UK).
Pharmacokinetic studies
Pharmacokinetic studies were performed in C57BL/6J mice bearing B16F1 melanoma. For intravenous administration, PLB (1.2 mg/ml) was solubilized in sterile PBS containing 25% PEG-200. Liposomes after suitable dilution in PBS (1.2 mg/ml PLB) and free PLB were injected via tail vein at a dose of 6 mg/kg. Blood was withdrawn at appropriate time intervals (at each point, blood was collected from 4 mice) from the retro-orbital vein and plasma was separated by centrifugation (3 min, 13000 rpm) and stored in vials at -70 °C until HPLC analysis. Liquid-liquid extraction method using ethyl acetate as the extracting solvent was used to extract the PLB from the plasma samples as described in earlier method (CitationHsieh et al., 2006) with minor modifications. Briefly, 100 μl of plasma was vortexed for 15 min with 100 μl of 0.1N HCl and 2 ml of ethyl acetate using RIA vials, and centrifuged at 10000 rpm for 10 min at 4 °C. The upper organic layer obtained after centrifugation was transferred into another tube, dried with nitrogen gas, reconstituted in 150 μl of mobile phase prior to injection to the HPLC system. HPLC system (Shimadzu LC-2010, Kyoto, Japan) with reverse-phased Grace Vydac (Grace) C18 silica gel column (250 mm × 4.6 mm, 5 μm) was used at ambient temperature (25 °C) and the detector wavelength was set at 264 nm. The mobile phase consisted of methanol: glacial acetic acid (0.1% v/v) (68:32) delivered at a flow rate of 1.0 ml/min and 2, 3-dichloro-1, 4-naphthoquinone (10μl of 100μg/ml) was used as an internal standard. The limit of quantification (LOQ) was 0.1 μg/ml. In plasma, coefficient of variation (CV) of precision and accuracy was less than 15% and relative recovery was more than 62%. Pharmacokinetic parameters were calculated using the pharmacokinetic software PK Solutions 2.0 (Montrose, CO, USA).
Pharmacodynamic studies
Solid tumor model
Solid tumors were grown on the dorsal side of the mice by injection of 5 × 105 B16F1 melanoma cells intradermally. Once the tumors became palpable, the tumor diameter was measured using vernier caliper and the tumor volume was calculated (CitationDevi et al., 2000) using the formula V= π/6 (D1 × D2 × D3), where D1, D2 and D3 are the tumor diameters in three perpendicular planes. When the tumor volume became about 100 ± 10 mm3, liposomes diluted in PBS (pH-7.4) and free PLB (solubilized in PBS containing 25% PEG -200) at a dose of 2 mg/kg was intravenously administered as single dose via tail vein on 1, 3, 5, 7 and 10 days (n = 8 per treatment group). Control animals were injected with the vehicle (PBS containing 25% PEG-200). The tumor volume was measured every two days and the tumor response was assessed by volume doubling time (VDT) and growth delay (GD). Also, the animals were observed further for survival. The actuarial survival curves were plotted using Kaplan-Meier analysis. Some of the tumor bearing animals which were over stressed due to excess tumor burden was terminated from the study due to humane considerations and in the survival analysis these animals were considered as censored.
Toxicity assessment
The toxicity of free and long circulating liposomal formulations of PLB was evaluated after injecting the mice intravenously with 2 mg/kg for 5 consecutive days. Twenty four hours after the last dose, blood was collected by retro-orbital vein and evaluated for any hematological changes such as total RBC and WBC counts, hemoglobin, hematocrit values and platelet counts using automated blood cell counter Erma PCE-210 (Japan). These animals were then sacrificed and tissues of heart, liver, spleen and kidney were fixed in 10% formalin solution and embedded in paraffin. The paraffin embedded tissue sections were then processed and stained with hematoxylin–eosin (H&E) for microscopic examination.
Statistical analysis
The data obtained from the experiments was analyzed by GraphPAD InStat Software, USA. Statistical analysis was performed using Kaplan-Meier analysis and Logrank test method for survival, Student’s t-test, One-way ANOVA followed by Bonferroni’s post-hoc test for comparison between the groups. A p-value below 0.05 was considered to be significant.
Results
Formulation development study
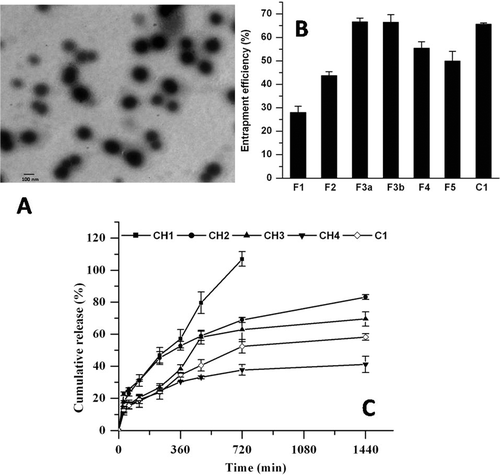
The present study was aimed at formulating PLB as long circulating liposomes to enhance its plasma half-life and anticancer potential using B16F1 melanoma as solid tumor model in vivo. The PLB liposomal formulations either conventional or long circulating were prepared by thin film hydration method using different molar ratios of drug to lipid. Negative staining TEM image ( shows that the prepared long circulating liposomes were discrete and round in structure.
Figure 1. (A) Transmission electron micrograph of pegylated liposomes (C1) of PLB. (B) Encapsulation efficiency (%) of PLB. F1-F5 represents conventional, whereas C1 represents long circulating liposomes. (C) Effect of cholesterol on in vitro release profile of conventional (CH1-CH4) and long circulating liposomes (C1) in PBS at 37 °C (n = 3).
Effect of phospholipid composition on entrapment efficiency and particle size
Initial studies were undertaken to examine the effect of phospholipid composition on the entrapment efficiency of liposomes. The liposomal formulations were prepared by varying molar ratios of PLB/lipid. clearly indicates an increase in the encapsulation efficiency of PLB with increase in molar ratio of PLB/lipid up to 1:20. Further increase in the PLB/lipid molar ratio from 1:20 to 1:40 resulted in a decreased entrapment efficiency of 49.99 ± 4.05% for F5 formulation. The highest entrapment efficiency for the conventional liposomes was found in F3a and F3b formulations with values of 66.62 ± 1.54 % and 66.47 ± 3.15 % respectively. The particle size also increased with increase in the PLB/lipid molar ratio as shown in .
Effect of cholesterol composition on particle size and entrapment efficiency
After the optimization of the initial batches, the cholesterol content in the formulation F3 was altered in order to observe its effect on particle size and entrapment efficiency. From it can be inferred that there was not much effect on the particle size when the SPC/Chol content increased from 9:1 to 9:2 molar ratio, while at 9:5 (CH4) molar ratio, the particle size increased. The entrapment efficiency increased with increase in the cholesterol content up to 9:2 molar ratio of SPC/Chol. Further increase in the cholesterol content did not improve the incorporation efficiency of PLB in liposomes ().
Table 2. Effect of cholesterol on physicochemical characteristics of liposomes.
In vitro release
The effect of cholesterol on the release rate profile of PLB for the prepared formulations (CH1-CH4) was also examined. From it can be observed that the release rate decreased with increase in the cholesterol content. The formulation with SPC/Chol molar ratio of 9:1 showed faster cumulative drug release of 106.4 ± 4.46 % in 12 h whereas the formulation with 9:2 and 9:3 showed 83.2 ± 1.55% and 69.55 ± 4.44% drug release in 24 h respectively. However, the formulation with increased cholesterol content i.e. with 9:5 molar ratio of SPC/Chol showed decreased cumulative drug release of 41.16 ± 5.10 % in 24 h. The optimized long circulating liposomal formulation (C1) had a cumulative drug release of 58.27 ± 3.42 % in 24 h.
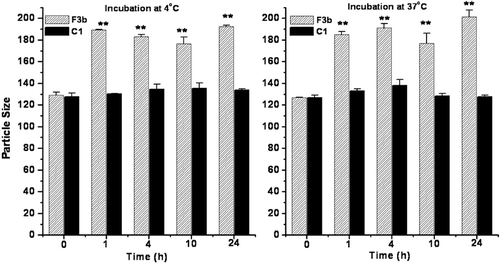
Serum stability
The physical stability of pegylated liposomes in fetal bovine serum was evaluated by measuring the change in particle size. The particle size of conventional liposomes increased after incubation in serum suggesting aggregation and/or fusion of liposomes in serum as shown in . It was observed that there was not much change in the particle size of the pegylated liposomes up to 24 h.
Figure 2. Physical stability of conventional (F3b) and long circulating liposomes (C1) in fetal bovine serum as a function of particle size up to 24 h incubation. Significant values: **p < 0.01 in comparison to particle size at initial time point.
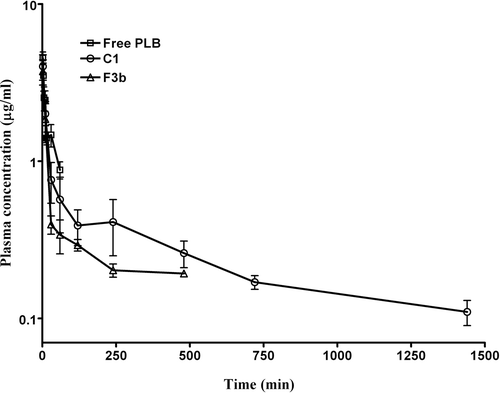
Pharmacokinetic studies
represents the plasma drug concentration-time profile of free PLB, conventional and long circulating liposomes. The pharmacokinetics of free PLB was characterized with AUC0-∞ of 152.89 ± 10.05 µg.min/ml, elimination half-life (T1/2) of 35.89 ± 7.95 min and a plasma clearance of 0.03 ± 0.002 L/min (). The plasma levels of PLB decreased sharply with time when administered by intravenous route. The plasma levels of PLB were detectable up to 24 h for long circulating liposomes whereas for conventional liposomes, the plasma levels were found up to 8 h only following intravenous administration. Compared to free PLB, the long circulating liposomes exhibited an increased plasma half-life of 1305.76 ± 278.16 min (36.38 fold increase in comparison to free PLB) and AUC0-∞ value 479.73 ± 80.11 µg.min/ml (3.13 fold increase in comparison to free PLB). Long circulating liposomes (C1) showed the highest values of MRT, AUC0-∞, t1/2 whereas CL value was significantly less compared to free PLB and conventional liposomes ().
Table 3. Pharmacokinetic parameters of PLB, free and encapsulated as conventional and pegylated liposomes, after intravenous bolus administration at a dose of 6 mg/kg to tumor bearing mice (n = 4).
Figure 3. Plasma concentration of PLB obtained from mice after intravenous administration of free PLB, conventional (F3b) and pegylated liposomes (C1) at a dose of 6 mg/kg. The data represents mean ± SD of four animals.
Pharmacodynamic studies
The optimized liposomal formulations were further evaluated for its antitumor efficacy against B16F1 melanoma cells grown in vivo in C57BL/6J mice. The animals were administered with 2 mg/kg of free PLB or conventional and long circulating liposomes once the tumor size reached about 100 ± 10 mm3. The tumor volume in the vehicle treated control group grew rapidly and reached about 5000 mm3 in about 12 days, while in the treated groups the tumor growth was significantly delayed (. All the treated groups have shown significant (p<0.001) reduction in the tumor volume when compared to the vehicle treated group. In comparison to free PLB, the tumor inhibition for the groups treated with pegylated and conventional liposomes were higher (p<0.01). This could be further justified by increase in the tumor volume doubling time (VDT) of 4.15 ± 0.67 and 3.77 ± 0.61 days for long circulating and conventional liposomes in comparison to free PLB treated group having VDT value of 2.35 ± 0.17 days. Also the growth delay values for the pegylated liposomes treated group were significantly higher () in comparison to free PLB group.
Table 4. Tumor growth parameters for free PLB and liposomal formulations (n = 8).
Figure 4. Effect of free PLB and formulated liposomes as conventional or pegylated (A) on tumor growth inhibition and (B) survival of C57BL/6J mice inoculated with B16F1 melanoma cells. Significant levels - ** p < 0.001 compared with vehicle treated group; a p < 0.01, b p < 0.001 compared to free plumbagin treated group. When tumor volume became about 100 ± 10 mm3, free PLB, conventional liposomes (F3b) or pegylated liposomes was intravenously injected (containing 2 mg/kg PLB each time on days 1, 3, 5, 7 and 10 (total dose of 10 mg/kg)). Control animals were injected with the vehicle (PBS containing 25% PEG-200). Each data represents the mean ± SD (n = 8).
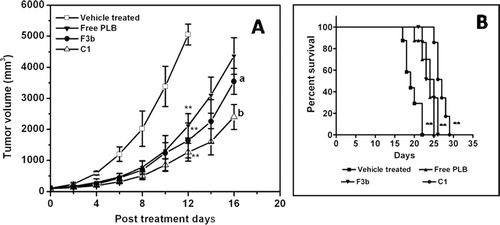
Survival of tumor bearing mice in response to the different treatments was also determined. The survival plots using Kaplan-Meier analysis for different treatment groups is shown in . The median survival values for PLB alone, conventional and pegylated PLB liposomes were found to be 24, 25 and 27 days respectively in comparison to 19 days for vehicle treated control.
Toxicity assessment
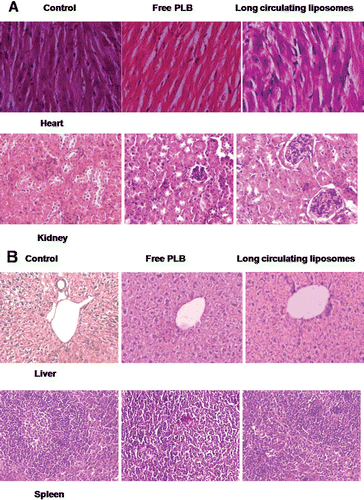
In order to study the toxicity produced by intravenous administration of PLB in the free or liposomal encapsulated forms, if any, the hematological parameters of the animals were determined one day after completion of the treatment and these values were then compared with that of the values obtained from the vehicle treated control mice. The results of the hemograms of the animals are shown in . It is observed that, there were no significant changes in the blood related parameters upon administration of either free PLB or formulated liposomes at repeated doses of 2 mg/kg b. wt. for 5 consecutive days indicating its non-toxic nature. Also, the histopathological examination of all treated groups showed no signs of toxic pathologic changes in liver, heart, kidney and spleen in comparison to vehicle treated group ( and ).
Table 5. Hematological changes after intravenous administration of free PLB and pegylated liposomes (C1) for 5 consecutive days.
Figure 5. Toxicity assessment of free PLB and long circulating liposomes (C1) in C57BL/6J mice. The histopathologic examination of tissue sections of (A) heart and kidney, (B) liver and spleen was made after mice were sacrificed and staining with H&E stain. All the treatment groups showed no signs of toxic pathological change; magnification 400X; 100X for liver.
Discussion
Liposomes are considered as one of the useful drug delivery systems owing to their structural advantages of possessing large aqueous interior favoring the incorporation of water soluble drugs and at the same time have the ability to retain hydrophobic moieties as well. Further, its biocompatibility property and non toxic nature offers an effective means of drug delivery to the desired site of action (CitationSharma & Sharma, 1997; CitationMiller et al., 1998). However, conventional liposomes, due to their rapid clearance from blood stream and ineffective targeting issues stimulated the researchers to explore more effective pegylated liposomes.
In the present study the liposomes were prepared by adopting thin film hydration method and subsequent size reduction by high pressure homogenization. Initial batches were taken with varying molar ratios of drug to lipid and the effect of lipid ratio on particle size and entrapment efficiency was examined. Conventional liposomes were prepared using soya lecithin and cholesterol and mPEG2000-DSPE was incorporated to formulate long circulating liposomes. During the formulation development it was noticed that as the drug to lipid molar ratio increased there was a corresponding increase in the entrapment efficiency up to 1:20 whereas, further increase in the lipid content did not result in an increment of entrapment of PLB into liposomes. The present results corroborate the earlier findings wherein the formulation containing paclitaxel and lipids in the molar ratio of 1:30 had good incorporation efficiency, while further increase in paclitaxel/lipid molar ratio or in the cholesterol content decreased the entrapment as well as the physical stability of the prepared formulations (CitationCrosasso et al., 2000). Similar observations were made in earlier reports where the incorporation efficiency of 9-nitrocamptothecin did not improve upon increase in the drug/lipid molar ratio (CitationChen et al., 2006). Hence for further optimization process the drug to lipid molar ratio was chosen as 1:20. The next step in formulation development was to study the effect of cholesterol on entrapment and particle size. Cholesterol is known to improve the fluidity of the bilayer (CitationKirby et al., 1980) and reduces the permeability of entrapped molecules by providing mechanical stiffness to the membrane and provides the stability to the bilayer membrane in presence of biological fluids (CitationVemuri & Rhodes, 1995). It can be seen that the percent entrapment increases up to a specific concentration of cholesterol and thereafter it became constant. Similar results were observed by other investigators wherein, they demonstrated the rigidity of the prepared liposomes reached plateau level at higher concentration of cholesterol and thereafter, further increase in cholesterol content did not had much effect on incorporation efficiency (CitationGulati et al., 1998).
The effect of cholesterol on the in vitro PLB release from the liposomes was also studied. The drug release from the prepared liposomes decreased with increase in the cholesterol content. This decrease in the release may be due to the increased rigidity of the lipidic bilayer at higher levels of cholesterol. The release of PLB showed initial burst release phase, releasing approximately 20% of PLB during the first 2 h followed by sustained drug release over the next 24 h. The initial burst effect could be due to the release of liposomal surface bound free drug and sustained drug release could be due to the stabilization of lipidic bilayers by cholesterol and thus an sustained release effect could be expected using the prepared conventional and pegylated liposomes.
The serum stability of the optimized liposomal formulations was also tested up to 24 h. It is reported that the protein adsorption to the surface of the liposomes is an important indication for the stability of the liposomes in serum (CitationHan et al., 2006). It was observed that there was not much change in the particle size of the pegylated liposomes up to 24 h which could be due to steric stabilization of the prepared liposomes by the presence of PEG chains which might have prevented their aggregation/fusion whereas there was a significant increase in the particle size of conventional liposomes due to aggregation and fusion of liposomes in the presence of bovine serum.
From the pharmacokinetic study it can be observed that the plasma levels of free PLB decreased sharply with time when administered by intravenous route. The results were consistent with the findings of Chandresekaran and Nagarajan (1981) where PLB levels were undetectable in blood up to 24 h in rats whereas the drug was detected in urine within 4 h after administration with a major portion of PLB excreted via urination by 24 h. In contrast the plasma levels of PLB were detectable up to 24 h for long circulating liposomes whereas for conventional liposomes, the plasma levels were found up to 8 h only following intravenous administration. This could be due to the uptake of the conventional liposomes by reticuloendothelial system (RES), thereby its clearance from the systemic circulation or, it might be due to the lower concentration of PLB below the lower limit of HPLC detection. Compared to free PLB, the long circulating liposomes exhibited 36.38 fold increase in plasma half-life in comparison to free PLB and 3.13 fold increase in AUC in comparison to free PLB. This significant increase in the plasma half-life (p<0.001) of PLB in long circulating liposomes could be attributed to the stealth effect leading to slower removal of PLB loaded liposomes from the plasma compartment. The results of pegylated liposomes group showed that the long circulating carrier system increased the exposure of PLB in the plasma, and prolonged the half-life of PLB in comparison with those of conventional or free PLB. It is reported that the pegylation process leads to the formation of hydrophilic coat on the liposomal surface and there by repel opsonin penetration and adsorption (CitationAllen & Hansen, 1991; CitationPapahadjopoulos et al., 1991; CitationSharma & Sharma, 1997; CitationDrummond et al., 1999). This reduced marking by the opsonins leads to slower uptake of the pegylated liposomes by the cells of RES in comparison to the conventional liposomes and hence extended systemic circulation half-life.
The antitumor efficacy of the optimized liposomal formulations was evaluated against B16F1 melanoma cells grown in vivo in C57BL/6J mice. All the treated groups have shown significant (p<0.001) reduction in the tumor volume when compared to the vehicle treated group. In comparison to free PLB, the tumor inhibition for the group treated with pegylated liposomes was higher and found to be significant (p<0.001). Survival studies revealed an increased life span of 12.5% for the pegylated liposomal treated group when compared to free PLB treated group. This increase in the tumor inhibitory effect observed with long circulating liposomes could be by virtue of prolonged circulation time of pegylated liposomes leading to increased local concentration of PLB at the tumor site via EPR effect. Also, it is documented that the extravasation of liposomes and its penetration into the inflammation or tumor site is directly correlated to their size. The liposomes of smaller size (around 100 nm in diameter) have improved chances of tumor site localization where the blood vessels are irregular in shape, inherently leaky as a result of large number of fenestrae and wide inter-endothelial junctions with poor lymphatic drainage (CitationJain, 1987; CitationIyer et al., 2006). Earlier study has shown enhanced antitumor effect of PLB by formulating it as thermosensitive liposomes (CitationTiwari et al., 2002) wherein a combination of liposomal administration and localized hyperthermia (at the tumor site) exhibited better antitumor efficacy when compared with the free PLB treated group with or without hyperthermia against mice bearing B16F1 melanoma. Also many other studies demonstrated improved antitumor efficacy with reduced toxicity of PLB using various drug delivery systems. PLB encapsulated as chitosan based microspheres have exhibited significant antitumor efficacy when compared to free PLB treated groups in mice bearing B16F1 melanoma (CitationMandala Rayabandla et al., 2010). However, the optimized long circulating liposomes at a dose of 10 mg/kg (total cumulative dose) in the present study have exhibited better antitumor efficacy when compared to the chitosan based microspheres administered by intramuscular route (18 mg/kg, total cumulative dose) of PLB when tested against B16F1 melanoma bearing mice.
CitationSingh et al (1997) have demonstrated reduced toxicity and enhanced therapeutic efficacy of PLB in mice bearing Sarcoma-180 tumor after poly (lactic-co-glycolic) acid (PLGA) based gel implantation subcutaneously. In another study, the niosomal and albumin microspheres of PLB have exhibited marginal antitumor activity when compared to free PLB treated groups when tested against B16F1 melanoma grown in vivo (CitationKini et al., 1997).
Also, the free PLB and optimized long circulating formulation were tested for normal tissue toxicity by performing hematological and histopathological studies. The results demonstrated no signs of normal tissue toxicity in both the free PLB and pegylated liposomal treated group indicating its non-toxic nature. The non-toxic nature of plumbagin at the doses tested corroborates the earlier reports, wherein PLB did not exhibit any significant normal tissue toxicity when tested at a dose of 2 mg/kg per day against melanoma A375.S2 cells implanted in nude mice even at a total dose of 56 mg/kg b.wt. (CitationWang et al., 2008). Similar observations were made in another study where the authors have observed no signs of normal tissue toxicity when tested in vivo using human promyelocytic leukemia NB4 tumor xenograft in NOD/SCID mice at a total dose of 42 mg/kg b.wt. (CitationXu & Lu, 2010). These studies clearly substantiate the safe levels of PLB supporting the need for further clinical exploration as a large body of preclinical studies convincingly demonstrated the anticancer potential of plumbagin.
Conclusion
The long circulating PLB formulation designed in the present study exhibited optimal entrapment efficiency and in vitro drug release. Further, these liposomes have shown enhanced plasma half-life and exhibited better antitumor efficacy compared to free and conventional PLB liposome formulations. Also the free as well as pegylated liposomes of PLB have shown no signs of normal tissue toxicity. Therefore, it may be useful as a promising parenteral formulation for the effective delivery of PLB.
Acknowledgement
The financial support from Indian Council of Medical Research (ICMR), New Delhi, India (IRIS No: 2005-00150), to carry out this study is gratefully acknowledged. The authors are thankful to Dr. K. Satyamoorthy, Director, Manipal Life Sciences Centre, Manipal University, Manipal for providing the facilities and encouragement during this study. The authors are also thankful to Dr. Kanthi Latha Pai, Professor and Head, Department of Pathology, KMC International Centre, Manipal University, Manipal for her kind help in carrying out the histopathological toxicity studies.
Declaration of interest
The authors have no conflict of interest.
References
- Ahmad, A., Banerjee, S., Wang, Z., Kong, D., Sarkar, F.H. (2008). Plumbagin-induced apoptosis of human breast cancer cells is mediated by inactivation of NF- B and Bcl-2. J Cell Biochem. 105:1461–1471.
- Allen, T.M, Cullis, P.R. (2004). Drug delivery systems: entering the mainstream. Science. 303:1818–1822.
- Allen, T.M., Hansen, C. (1991). Pharmacokinetics of stealth versus conventional liposomes: effect of dose. Biochim Biophys Acta. 1068:133–141.
- Aziz, M.H., Dreckschmidt, N.E., Verma, A.K. (2008). Plumbagin, a medicinal plant-derived naphthoquinone, is a novel inhibitor of the growth and invasion of hormone-refractory prostate cancer. Cancer Res. 68:9024–9032.
- Chandrasekaran, B., Nagarajan, B. (1981). Metabolism of echitamine and plumbagin in rats. J Biosci. 3:395–400.
- Chen, J., Ping, Q.N., Guo, J.X., Chu, X.Z., Song, M.M. (2006). Effect of phospholipid composition on characterization of liposomes containing 9-nitrocamptothecin. Drug Dev Ind Pharm. 32:719–726.
- Chrai, S.S., Murari, R., Ahmad, I. (2002). Liposomes: A Review. Pharm Tech. 26:28–34.
- Crosasso, P., Ceruti, M., Brusa, P., Arpicco, S., Dosio, F., Cattel, L. (2000). Preparation, characterization and properties of sterically stabilized paclitaxel-containing liposomes. J Control Release. 63:19–30.
- de Paiva, S.R., Figueiredo, M.R., Aragao, T.V., Kaplan, M.A. (2003). Antimicrobial activity in vitro of plumbagin isolated from Plumbago species. Mem Inst Oswaldo Cruz. 98:959–961.
- Devi, P.U., Kamath, R., Rao, B.S. (2000). Radiosensitization of a mouse melanoma by withaferin A: in vivo studies. Indian J Exp Biol. 38:432–437.
- Devi, P.U., Solomon, F.E., Sharada, A.C. (1994). In vivo tumor inhibitory and radiosensitizing effects of an Indian medicinal plant, Plumbago rosea on experimental mouse tumors. Indian J Exp Biol. 32:523–528.
- Drummond, D.C., Meyer, O., Hong, K., Kirpotin, D.B., Papahadjopoulos, D. (1999). Optimizing liposomes for delivery of chemotherapeutic agents to solid tumors. Pharmacol Rev. 51:691–743.
- Gulati, M., Grover, M., Singh, M., Singh, S. (1998). Study of azathioprine encapsulation into liposomes. J Microencapsul. 15:485–494.
- Han, H.D., Lee. A., Song, C.K., Hwang, T., Seong, H., Lee, C.O., Shin, B.C. (2006). In vivo distribution and antitumor activity of heparin-stabilized doxorubicin-loaded liposomes. Int J Pharm. 313:181–188.
- Hsieh, Y.J., Lin, L.C., Tsai, T.H. (2006). Measurement and pharmacokinetic study of plumbagin in a conscious freely moving rat using liquid chromatography/tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 844:1–5.
- Hsu, Y.L., Cho, C.Y., Kuo, P.L., Huang, Y.T., Lin, C.C. (2006). Plumbagin (5-hydroxy-2-methyl-1, 4-naphthoquinone) induces apoptosis and cell cycle arrest in A549 cells through p53 accumulation via c-Jun NH2-terminal kinase-mediated phosphorylation at serine 15 in vitro and in vivo. J Pharmacol Exp Ther. 318:484–494.
- Immordino, M.L., Dosio, F., Cattel, L. (2006). Stealth liposomes: review of the basic science, rationale, and clinical applications, existing and potential. Int J Nanomedicine. 1:297–315.
- Iyer, A.K., Khaled, G., Fang, J., Maeda, H. (2006). Exploiting the enhanced permeability and retention effect for tumor targeting. Drug Discov Today. 11:812–818.
- Jain, R.K. (1987). Transport of molecules across tumor vasculature. Cancer Metastasis Rev. 6:559–593.
- Kaiser, N., Kimpfler, A., Massing, U., Burger, A.M., Fiebig, H.H., Brandl, M., Schubert, R. (2003). 5-Fluorouracil in vesicular phospholipid gels for anticancer treatment: entrapment and release properties. Int J Pharm. 256:123–131.
- Kawiak, A., Piosik, J., Stasilojc, G., Gwizdek-Wisniewska, A., Marczak, L., Stobiecki, M., Bigda, J., Lojkowska, E. (2007). Induction of apoptosis by plumbagin through reactive oxygen species-mediated inhibition of topoisomerase II. Toxicol Appl Pharmacol. 223:267–276.
- Khalil, I.A., Kogure, K., Futaki, S., Harashima, H. (2008). Octaarginine-modified liposomes: enhanced cellular uptake and controlled intracellular trafficking. Int J Pharm. 354:39–48.
- Kini, D.P., Pandey, S., Shenoy, B.D., Singh, U.V., Udupa, N., Umadevi, P., Kamath, R. (1997). Antitumor and antifertility activities of plumbagin controlled release formulations. Indian J Exp Biol. 35:374–379.
- Kirby, C., Clarke, J., Gregoriadis, G. (1980). Effect of the cholesterol content of small unilamellar liposomes on their stability in vivo and in vitro. Biochem J. 186:591–598.
- Lasic, D.D. (1996). Doxorubicin in sterically stabilized liposomes. Nature. 380:561–562.
- Likhitwitayawuid, K., Kaewamatawong, R., Ruangrungsi, N., Krungkrai, J. (1998). Antimalarial naphthoquinones from Nepenthes thorelii. Planta Med. 64:237–241.
- Mandala Rayabandla, S.K., Aithal, K., Anandam, A., Shavi, G., Nayanabhirama, U., Arumugam, K., Musmade, P., Bhat, K., Bola Sadashiva, S.R. (2010). Preparation, in vitro characterization, pharmacokinetic, and pharmacodynamic evaluation of chitosan-based plumbagin microspheres in mice bearing B16F1 melanoma. Drug Deliv. 17:103–113.
- Massing, U., Fuxius, S. (2000). Liposomal formulations of anticancer drugs: selectivity and effectiveness. Drug Resist Updat. 3:171–177.
- Miller, C.R., Bondurant, B., McLean, S.D., McGovern, K.A., O’Brien, D.F. (1998). Liposome- Cell Interactions in Vitro: Effect of Liposome Surface Charge on the Binding and Endocytosis of Conventional and Sterically Stabilized Liposomes. Biochemistry. 37:12875–12883.
- Mossa, J.S., El-Feraly, F.S., Muhammad, I. (2004). Antimycobacterial constituents from Juniperus procera, Ferula communis and Plumbago zeylanica and their in vitro synergistic activity with isonicotinic acid hydrazide. Phytother Res. 18:934–937.
- Nair, S., Nair, R.R., Srinivas, P., Srinivas, G., Pillai, M.R. (2008). Radiosensitizing effects of plumbagin in cervical cancer cells is through modulation of apoptotic pathway. Mol Carcinog. 47:22–33.
- Naresh, R., Udupa, N., Devi, P.U., Pillai, G.K. (1996). Niosomal plumbagin with reduced toxicity and improved anticancer activity in BALB/C mice. J Pharm Pharmacol. 48:1128–1132.
- Padhye, S., Dandawate, P., Yusufi, M., Ahmad, A., Sarkar, F.H. (2010). Perspectives on medicinal properties of plumbagin and its analogs. Med Res Rev. (DOI: 10.1002/med.20235, In press).
- Panchagnula, R. (1998). Pharmaceutical aspects of paclitaxel. Int J Pharm. 172:1–15.
- Papahadjopoulos, D., Allen, T.M., Gabizon, A., Mayhew, E., Matthay, K., Huang, S.K., Lee, K.D., Woodle, M.C., Lasic, D.D., Redemann, C. (1991). Sterically stabilized liposomes: improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc Natl Acad Sci U S A. 88:11460–11464.
- Powolny, A.A., Singh, S.V. (2008). Plumbagin-induced apoptosis in human prostate cancer cells is associated with modulation of cellular redox status and generation of reactive oxygen species. Pharm Res. 25:2171–2180.
- Prasad, V.S., Devi, P.U., Rao, B.S., Kamath, R. (1996). Radiosensitizing effect of plumbagin on mouse melanoma cells grown in vitro. Indian J Exp Biol. 34:857–858.
- Sadzuka, Y., Nakade, A., Tsuruda, T., Sonobe, T. (2003). Study on the characterization of mixed polyethyleneglycol modified liposomes containing doxorubicin. J Control Release. 91:271–280.
- Sahli, A., Cansell, M., Tapon-Bretaudiere, J., Letourneur, D., Jozefonvicz, J., Fischer, A.M. (1998). The stability of heparin-coated liposomes in plasma and their effect on its coagulation. Colloids Surf B Biointerfaces. 10:205–215.
- Sandur, S.K., Ichikawa, H., Sethi, G., Ahn, K.S., Aggarwal, B.B. (2006). Plumbagin (5-hydroxy-2-methyl-1, 4-naphthoquinone) suppresses NF- B activation and NF- B-regulated gene products through modulation of p65 and I B kinase activation, leading to potentiation of apoptosis induced by cytokine and chemotherapeutic agents. J Biol Chem. 281:17023–17033.
- Sharma, A., Sharma, U.S. (1997). Liposomes in drug delivery: progress and limitations. Int J Pharm. 154:123–140.
- Singh, U.V., Udupa, N. (1997). Reduced toxicity and enhanced antitumor efficacy of betacyclodextrin plumbagin inclusion complex in mice bearing Ehrlich ascites carcinoma. Indian J Physiol Pharmacol. 41:171–175.
- Srinivas, P., Gopinath, G., Banerji, A., Dinakar, A., Srinivas, G. (2004). Plumbagin induces reactive oxygen species, which mediate apoptosis in human cervical cancer cells. Mol Carcinog. 40:201–211.
- Talsma, H., Ozer, A.Y., Van Bloois, L., Crommelin, D.J.A. (1989). The size reduction of liposomes with a high pressure homogenizer (microfluidizer TM). Characterization of prepared dispersions and comparison with conventional methods. Drug Dev Ind Pharm. 15:197–207.
- Thasni, K.A., Rakesh, S., Rojini, G., Ratheeshkumar, T., Srinivas, G., Priya, S. (2008). Estrogen-dependent cell signaling and apoptosis in BRCA1-blocked BG1 ovarian cancer cells in response to plumbagin and other chemotherapeutic agents. Ann Oncol. 19:696–705.
- Tiwari, S.B., Pai, R.M., Udupa, N. (2002). Temperature sensitive liposomes of plumbagin: characterization and in vivo evaluation in mice bearing melanoma B16F1. J Drug Target. 10:585–591.
- Torchilin, V.P. (2005). Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 4:145–160.
- Vemuri, S., Rhodes, C.T. (1995). Preparation and characterization of liposomes as therapeutic delivery systems: a review. Pharm Acta Helv. 70:95–111.
- Vijayakumar, R., Senthilvelan, M., Ravindran, R., Devi, R.S. (2006). Plumbago zeylanica action on blood coagulation profile with and without blood volume reduction. Vascul Pharmacol. 45:86–90.
- Wang, C.C.C., Chiang, Y.M., Sung, S.C., Hsu, Y.L., Chang, J.K., Kuo, P.L. (2008). Plumbagin induces cell cycle arrest and apoptosis through reactive oxygen species/c-Jun N-terminal kinase pathways in human melanoma A375. S2 cells. Cancer Lett. 259:82–98.
- Xiong, X.B., Huang, Y., Lu, W., Zhang, X., Zhang, H., Nagai, T., Zhang, Q. (2005). Enhanced intracellular delivery and improved antitumor efficacy of doxorubicin by sterically stabilized liposomes modified with a synthetic RGD mimetic. J Control Release. 107:262–275.
- Xu, K.H., Lu, D.P. (2010). Plumbagin induces ROS-mediated apoptosis in human promyelocytic leukemia cells in vivo. Leuk Res. 34:658–665.
- Yang, T., Choi, M.K., Cui, F.D., Kim, J.S., Chung, S.J., Shim, C.K., Kim, D.D. (2007). Preparation and evaluation of paclitaxel-loaded PEGylated immunoliposome. J Control Release. 120:169–177.