Abstract
Overexpression of drug efflux pump P-gp is one of the major reasons to cause multidrug resistance (MDR). To overcome P-gp mediated MDR, modulators, so called P-gp inhibitors, can be used to block efflux pump activity. Elacridar is one of the most potent P-gp inhibitors, which can cause irreversible and total P-gp blockage. Elacridar, among with other P-gp inhibitors, can be used in combination with anticancer drugs to enhance the effectiveness of chemotherapy against resistant tumor cells. On the other hand, P-gp is presented in normal tissues, thus non-selective blockage of P-gp can cause undesired side effects. Therefore, it is important to deliver P-gp inhibitor only to the tumor cells (along with anticancer drug) and limit its distribution in the body. In this study, we have developed PEG-PE-based long-circulating ca. 15 nm micelles co-loaded with elacridar and paclitaxel, and investigated their ability to overcome paclitaxel resistance in two cancer cell lines. Vitamin E, a common solubility enhancer for PEG-PE micelles, was found to have a negative effect on both particle size and encapsulation efficiencies. The human MDR1 gene-transfected and thus paclitaxel-resistant MDCKII-MDR1 P-gp overexpressing cells were used for cytotoxicity evaluation. Even though PEG-PE based micelles itself have a potential to enhance the cytotoxicity of paclitaxel, elacridar/paclitaxel-co-loaded micelles demonstrated the highest cytotoxicity compared to both free and micellar paclitaxel. The obtained results suggest that co-loading of paclitaxel and elacridar into micellar drug carriers results in promising preparations capable of overcoming paclitaxel resistance.
Introduction
The estimated number of new cancer cases in the US has reached up to 1.5 million (Jemal et al., Citation2008), but almost one third of those cases have drug resistance phenotype (Shabbits et al., Citation2001). Cancer cells exhibit drug resistance with various mechanisms, such as loss of a surface receptor, drug target mutations or differentiated metabolism. Most of these mechanisms can be overcome by using combination of anticancer drugs (Gottesman, Citation2002). On the other hand, most of the cases show resistance to a wide range of anticancer drugs that are chemically and structurally unrelated. This phenomenon described as multidrug resistance (MDR) (Gottesman et al., Citation1994; Szakács et al., Citation2006) and it is one of the major obstacles responsible for unsuccessful cancer treatment. The major mechanism of MDR in cultured cells, as well as in clinic, is the overexpression of P-gp, which is an efflux pump encoded by the MDR-1 gene (Chen et al., Citation1986; Ueda et al., Citation1987; Borst & Elferink, Citation2002). P-gp, a 170 kDa membrane phosphoglycoprotein, is one of the first identified members of ATP-binding cassette (ABC) family transporters (Juliano & Ling, Citation1976). This energy-dependent efflux pump can be found in many tissues and organs in the body, such as liver, kidney, small intestine, colon, placenta, uterus, and brain (Krishna & Mayer, Citation2000). While this wide localization indicates the physiological role of P-gp (Schinkel et al., Citation1997), the overexpression of this pump in the tumors like NSCLC, breast, kidney, and refractory ovarian cancer results in reduced cellular accumulation of anticancer drugs (Borst et al., Citation2000). P-gp can detect and bind many different, mainly hydrophobic, drugs as they enter the cell, and after the activation of its ATP-binding domains, the hydrolysis of one ATP molecule causes structural change in the P-gp shape. This change is responsible for drug efflux and decrease in intracellular drug amount (Sauna & Ambudkar, Citation2001). Many of anticancer drugs, like doxorubicin, vinblastine, vincristine, and paclitaxel are substrates of P-gp, and nearly 50% of cancer patients have P-gp overexpression (Clarke et al., Citation2005; Lage, Citation2006). Thus, maintaining the intracellular drug concentration is a crucial step for successful chemotherapy.
Paclitaxel (PTX) is an effective anticancer drug, which causes microtubule breakdown and acts as a mitotic inhibitor and apoptosis inducer of cancer cells (Torres & Horwitz, Citation1998). But PTX is also one of the substrates of P-gp (Allen et al., Citation2000) and is subjected to the efflux from P-gp overexpressing resistant cells, and drug resistance is often developed during the treatment. Even though there is a number of resistance mechanism for PTX, including altered metabolism of PTX, changes in microtubule (largely reviewed in (Orr et al., Citation2003)), or decreased sensitivity to death inducing stimuli, P-gp induced resistance still remains the most important mechanism of PTX resistance (Speicher et al., Citation1994; Walle & Walle, Citation1998).
To inhibit P-gp activity and reverse MDR, a number of agents with P-gp inhibitory activity have been identified. (Foxwell et al., Citation1989; Neumann et al., Citation1992; Stein et al., Citation1994; Krishna & Mayer, Citation2000). Among these inhibitors, Elacridar (ELC) is one of the most promising third generation model compounds, and has no affinity for other enzymes like CYP (Hyafil et al., Citation1993; Sparreboom et al., Citation1999). ELC can inhibit P-gp activity above 50 nM plasma concentrations and provide the total P-gp inhibition at around 0.5 µM (Witherspoon et al., Citation1996). Even with these advantages offered by ELC and other P-gp modulators, there is also a concern for inhibition of P-gp in normal tissues and organs in the body. To eliminate this non-specific effect, co-loading of P-gp inhibitors with anticancer drugs into the nano-sized drug delivery systems including nanoparticles, solid lipid nanoparticles and micelles have been investigated in recent years (Wang et al., Citation2005; Wong et al., Citation2006; Patil et al., Citation2009; Patel et al., Citation2011).
Based on these rationales, we have developed PTX- and ELC-co-loaded PEG-PE-based micelles. More recently, Patel et al. demonstrated the reversal of MDR by long-circulating liposomes, co-loaded with PTX and tariquidar (XR9576) (Patel et al., Citation2011). They found that long-circulating liposomes could encapsulate PTX and the third generation P-gp inhibitor tariquidar simultaneously and overcome the MDR in PTX-resistant SKOV3-TR cells.
In this study, we aimed to prove that it is possible to overcome MDR using inhibitor/drug-co-loaded PEG-PE-based micelles, which are known as suitable platforms for targeted drug delivery and formulation of low water soluble anticancer compounds (Torchilin et al., Citation2003; Torchilin, Citation2005; Sawant et al., Citation2008). By combining anticancer drug and a P-gp inhibitor, the effect of micellar carriers could be increased. Thus, we hypothesized that these long-circulating micelle-based delivery systems should provide clear benefits and increase the cytotoxicity by several simultaneously acting mechanisms such as: (i) Solubility of PTX would be increased by micellar encapsulation. This will prevent the use of excipients like Cremophor and alcohol, thus decrease the side-effects. Moreover, ELC, another water insoluble compound can be co-loaded into the hydrophobic micelle core along with PTX; (ii) Since PEG-PE-based long-circulating micelles have the size of about 20 nm, this drug carrier should be accumulated in the cancer tissue via the EPR effect. This accumulation will decrease the side effects of PTX, and also prevent the wide and non-specific distribution of P-gp inhibitor; (iii) ELC will be delivered into the cells simultaneously with PTX and prevent the efflux of drug from the resistant cells, so maximum combination effect can be achieved after uptake.
The prepared systems were characterized in terms of size, encapsulation efficiency, and drug release properties. Cytotoxicity studies were performed using MDCKII (MDR-1 and parental) cell lines. MDCKII-MDR1 cells are well investigated (Tang et al., Citation2002) and characterized as MDR-positive P-gp over-expressing cells (Evers et al., 1997) and have been thus chosen to investigate the potential MDR reversal effect of the formulations.
Materials and methods
Materials
1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy(polyethylene glycol)-2000] (PEG–PE) was purchased from Avanti Polar Lipids (Alabaster, AL, USA) and used without further purification. Paclitaxel (PTX) was from AppliChem (Darmstadt, Germany). Vitamin E and sodium salicylate (SS) were from Sigma (St. Louis, MO, USA). Elacridar base (ELC) was purchased from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Water-soluble tetrazolium salt WST-1 premixed cell proliferation assay kit was from Clontech (Mountain View, CA, USA). MDCKII cells (MDR-1 and Parental) were generously gifted by Prof. Piet Borst and Prof. Alfred Schinkel from The Netherlands Cancer Institute (NKV). All other reagents were of analytical grade.
Preparation of micelles
Micelles were prepared by the film-forming method. In this method, lipids and other micelle forming materials in organic solvents were dried by using a rotary evaporator. After removing of the solvents, a thin film was obtained on the surface of the flask. To enhance the loading of PTX and ELC into the micelles, vitamin E was used in some formulations (), and the molar ratio between PEG-PE and vitamin E was kept at 90:10. PTX-loaded micelles were prepared by adding PTX solution in methanol (1 mg/mL) to the PEG-PE or PEG-PE: vitamin E mixture in chloroform. Initial PTX amount added into micelles was 5% or 10% of the polymer weight. To prepare PTX and ELC-co-loaded micelles, ELC solution (0.5 mg/mL) in methanol: acetonitrile mixture (1:1 v/v) was added to above PTX/micelle components to give the final theoretical concentration of 25 µM. Organic solvents were removed under vacuum by using a rotary evaporator, following freeze-drying for the residual solvent removal. Drug-loaded micelles were formed after hydrating the film in phosphate buffered saline (PBS), pH 7.4, to give the final concentration of micelle materials of 1 mM. Non-incorporated PTX or PTX and ELC were separated by filtering the micelle formulations through 0.2 µm filter, and characterization studies were carried out with filtered micelles. Compositions of the studied micelle formulations are given in .
Table 1. Micelle formulations and characterization parameters.
Characterization of micelles
Micelle size
The micelle size (hydrodynamic diameter of the micelles) was measured by the DLS using Malvern Zeta Sizer Nano Series ZS (Malvern Instruments, Worchestershire, UK) instrument. Formulations were diluted with ultrapure MilliQ water to achieve same count rates around 500–750 kcps and attenuator value of 6–7. The particle sizes of all formulations were measured in triplicate.
CMC determination
CMC values of micelle formulations of PEG-PE and PEG-PE: vitamin E were estimated by the Pyrene method (Goddard et al., Citation1985; Zhao et al., Citation1990; Dabholkar et al., Citation2006). To determine the CMC value, known amounts of pyrene crystals were placed into the glass tubes and different concentrations of micelle formulations varied from 10 nM to 10 mM were added. Following 24 h of incubation at room temperature with shaking, the solutions were filtered through 0.2 µm PTFE filters to remove free pyrene. The fluorescence of the samples was measured at the excitation wavelength of 339 nm and emission wavelength of 390 nm using spectrofluorophotometer (Shimadzu RF-5301PC, Japan). A sharp increase in the fluorescence corresponds the CMC value of the given micelle formulation. The results obtained from pyrene method was confirmed by size-exclusion chromatography by using Shodex® Protein KW-804 (Shoko Co., LTD, Japan) column, phosphate buffer (100 mM phosphate, 150 mM Na2SO4) pH 7.0 as mobile phase and flow rate of 1 mL/min.
Loading efficiency
Loading efficiency of PTX or PTX and ELC into micelles was determined after filtration of the loaded micelle dispersions through 0.2 µm PTFE filters to remove non-incorporated drugs. Clear micelle dispersions were then diluted with 70:30 (v/v) acetonitrile: acetic acid buffer (pH 3.1), which dissolved the micelle structures as well as the incorporated drugs. Then, samples were injected into the HPLC system to evaluate the encapsulation efficiencies. Agilent 1200 HPLC system equipped with quaternary pump, diode array (DAD), and fluorescence detectors was used to simultaneously quantify PTX and ELC. A C18 reverse phase column, 4.6 × 150 mm, 3 µm (Fortis Technologies, Cheshire, UK) was eluted with 60:40 (v/v) acetonitrile: acetic acid buffer (pH 3.1) mobile phase containing 10 mM final concentration of ion-coupling agent 1-octane sulfonic acid (1-OSA) at 0.5 mL/min flow rate. The injection volume was 50 µL, and the analysis was carried out at 30°C for 9 min. DAD 229 nm (PTX) and Ex/Em: 265/485 (ELC) was used. The investigated validation parameters were specificity, linearity, precision (repeatability, intermediate precision and reproducibility), accuracy and stability.
In vitro release studies
PTX release from micelle formulations was monitored by the membrane dialysis at 37°C against 1 M sodium salicylate (SS) solution under sink conditions. SS was used to enhance the PTX solubility in water according to the hydrotropic effect (Lee et al., Citation2003). 1 M concentration was chosen after micelle stability studies at different SS concentrations in 0.5–4 M range, and was found as the optimum that gives desired solubility of PTX (17.6 µg/mL in 1 M SS solution) while keeps the micelle structures intact (Sarisozen et al., Citation2012). 0.5 mg PTX-containing micelle dispersion was placed in Spectra/Por regenerated cellulose dialysis bag (MWCO 2000 Da, Spectrum Labs, Rancho Dominquez, CA, USA) and dialyzed for 24 h while stirring at 50 rpm on magnetic stirrer. At determined time points, 1 mL of sample has been withdrawn and replaced with fresh medium. PTX amount in samples were analyzed by validated HPLC method given above.
Cytotoxicity and MDR reversal of formulations
The cytotoxicity of the micelles was tested using MDCKII-MDR1 and MDCKII-parental cell lines. The medium for cells cultivating was prepared by adding FBS, penicillin-streptomycin, and L-glutamine to the DMEM to the final concentration of 10% v/v, 50 U/mL–50 µg/mL and 2 mM, respectively. Cells were incubated in an incubator (5% CO2, 37°C) until approximately 80% confluency. Then, the cells were trypsinized, and 100 µL of cell suspension containing 5 × 103 cells was seeded into each well of the 96-well, flat-bottomed plates, and cells were kept overnight in the incubator to allow them to attach to the wells. The following day, the medium was removed, and 100 µL of micelle formulations in the culture medium was added into wells (n = 6). Before applying the formulations, the PTX and ELC amounts in the micelles were analyzed to make the correct dilutions regarding the PTX and ELC concentrations. After 12 h, 10 µL of WST-1 cell proliferation reagent was added into wells and tests were carried on according to manufacturer’s protocol.
Results and discussion
Particle size of the micelles
The mean particle size and polydispersity values of the micelle formulations are given in . Vitamin E was added to micelle formulations to enhance the encapsulation efficiency of the hydrophobic drugs by enlarging the micelle core (Torchilin, Citation2005). The formation of a larger micelle core results in larger particle sizes of micelles. More interestingly, while the results showed similar z-average values for size, the formulations with vitamin E demonstrated two different size peaks. This result shows multimodal and wide size distribution and causes a higher PDI value. Since two drugs were encapsulated into the same micelle formulation, two different size peaks could indicate two separate micelle populations, one encapsulating PTX and the other one encapsulating ELC. Even though our previous study indicates that it is not the issue when PTX and another P-gp inhibitor Cyclosporine A (CycA) were co-loaded in the PEG-PE based mixed micelles (Sarisozen et al., Citation2012), Bae et al. (Citation2007) and Jule et al. (Citation2004) have reported that co-loading of drugs into micelle structures can result in formation of the mixed micellar dispersion with two different micelle populations. On the other hand, formulations without vitamin E gave good particle size results and the PDI values were below 0.2. Also there were no significant differences (p > 0.05) between 5% and 10% PTX loading (Formulations M3 and M4). According to the obtained results, M3 and M4 were considered as the optimum formulation candidates.
CMC determination
CMC value of a micelle-forming compound is one of the indicators of its in vitro and in vivo stability. Micelles are known to dissociate upon dilution and below their CMC concentration, micelle-forming materials are present in the aqueous solutions as unimers. As a consequence, they can not encapsulate or solubilize any substance (Torchilin, Citation2001). Micelle formulations intended to be used parenterally, will be facing extreme dilution in the blood. In these conditions, it is very important to stay at the concentrations above CMC to avoid early release of the micelle-incorporated drugs, as well as to allow them to reach the site of action. The CMC values of PEG-PE alone and PEG-PE/vitamin E micelles were found to be 1.2 × 10−3 mM and 2.7 × 10−4 mM, respectively. The obtained CMC values are in accordance with the previously published data related to these systems (Trubetskoy & Torchilin, Citation1995; Sawant et al., Citation2008). These CMC values are around 100-fold lower than the conventional surfactants. The results can indicate that upon the dilution in blood, our PEG-PE micelles will stay intact and active substances will be encapsulated in the micelle structures. Studies with PEG-PE micelle systems revealed that, low CMC values of these systems provide them to preserve their integrity after extreme dilutions and thus, led them to be accumulated in the tumorous tissues (Lukyanov et al., Citation2002; Weissig et al., Citation1998).
Loading of active substances
PTX and ELC were successfully co-loaded into the PEG-PE-based micelles. Formulation codes and loading efficiencies are given in . In this study, the final micelle-forming material concentration was 1 mM, and results showed that the amount of drug loaded into the micelles was sufficient for characterization and cytotoxicity studies.
Table 2. Encapsulation efficiencies of the formulations.
We have already demonstrated that PEG-PE-based micelles can simultaneously incorporate PTX and first generation P-gp inhibitor CycA (Sarisozen et al., Citation2012). In this study, ELC, a third generation P-gp inhibitor with different chemical structure than CycA, was co-loaded with PTX into PEG-PE micelles. Unlike in some other published studies, the encapsulation enhancer effect of vitamin E was not observed with ELC. It has been reported that vitamin E can enhance the loading efficiencies of drugs into micelles due to forming larger micelle core (Gao et al., Citation2003; Sawant et al., Citation2008), but in our case vitamin E had no significant solubility enhancer effect for active substances. Similar to particle size results, formulations without vitamin E gave better encapsulation efficiencies. More interestingly, when comparing formulations of M3 and M4, an increase was found in the amount of loaded ELC with increasing amount of PTX, even though the initial ELC amounts were the same (25 µM). However, this phenomenon is not valid for vitamin E containing formulations M1 and M2. Without making a general assumption, these results suggest that PTX caused increase in ELC encapsulation in micelle formulations which did not contain vitamin E. It was noted that ELC can cause total P-gp inhibition at around 0.5 µM concentrations (Witherspoon et al., Citation1996), thus the initial loading concentration should be adjusted for achieving the required dose of ELC after dilutions for cytotoxicity studies. Pre-formulation studies indicated that 25 µM of ELC starting concentration was appropriate and sufficient to have ELC-co-loaded micelles with optimum concentrations for cytotoxicity experiments. Particle size and loading experiment results showed that M4 was the optimum formulation. Therefore it was chosen for further studies.
In vitro release studies
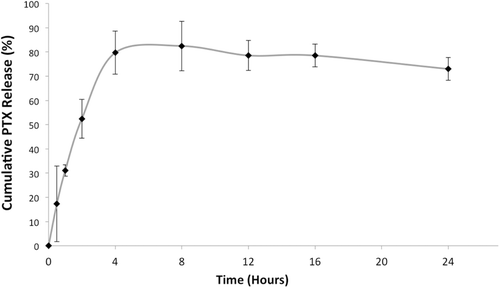
PTX release from micelles was monitored by the membrane-dialysis method. SS solution was used as a release medium for micelle formulations because of its hydrotropic property (Cho et al., Citation2004; Musacchio et al., Citation2009; Li et al., Citation2010). 1 M concentration was chosen after PEG-PE-based micelle stability studies and found to be the optimum (Sarisozen et al., Citation2012). ELC release was not monitored during the release studies, since the amount of the released ELC from the micelles was lower than the LOQ of the analytical method. The release profile for PTX is presented in the . As can be seen from the figure, the highest PTX release was slightly above 80% after 5–6 h.
Figure 1. PTX release from PTX- and ELC-co-loaded micelles under sink conditions (n = 3, mean ± SD).
Cytotoxicity of micelles
Cytotoxicity of micelles (Formulation M4) was investigated using MDCKII (MDR1 and parental) cell lines. MDCKII-MDR1 cells (although, not human) were used in this study because they provide an excellent model for the P-gp-mediated resistance (Irvine et al., Citation1999; Tang et al., Citation2002). Their parental cell line, MDCKII-parental cells, can serve as a positive control in cytotoxicity studies, since they do not express P-gp on their membrane hence they are sensitive to PTX even at low concentrations.
Micelles were diluted to give 0.5 µM ELC concentrations and PTX concentrations were normalized among different formulations. Cytotoxicity data are presented in and as the % inhibition corresponding to control group (significant differences between the groups are marked with the * sign and p values are given). The results for MDCKII-parental cells () showed that even the empty micelles were slightly cytotoxic. However, since this cell line has no P-gp efflux activity, there was no difference in the cytotoxicity between free PTX solution and micellar PTX, and between the PTX micelles and PTX-ELC-co-loaded micelles. Thus, PTX can easily enter the sensitive cells and could not be pumped back due to lack of P-gp activity.
Figure 2. Cytotoxicity of different formulations after 12 h towards MDCKII-parental cells. Error bars indicate mean ± SD, n = 6.
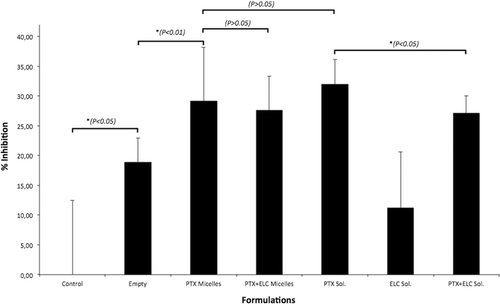
Figure 3. Cytotoxicity of different formulations after 12 h towards MDCKII-MDR1 cells. Error bars indicate mean ± SD, n = 6.
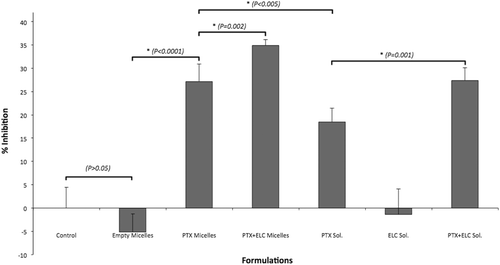
presents the cytotoxicity results with MDCKII-MDR1 resistant cells after 12 h. Free PTX solution showed some cytotoxicity in the resistant cells, even though this value was lower than that of sensitive cells. At the same time, micellar PTX showed significantly higher activity, indicating that micellar encapsulation can enhance the anticancer activity of PTX against MDR cell lines. There are several studies with polymeric micelles demonstrating the P-gp inhibition by micelle materials or by micellar delivery of drugs into the cells. While some groups have used Pluronic P85 (Batrakova et al., Citation2001; Kabanov et al., Citation2002) or Pluronic P123/F127 (Zhang et al., Citation2010), others used PEG-PE (Dabholkar et al., Citation2006) or mPEG-PLA and Pluronic L61 (Li et al., Citation2010) micelles to circumvent P-gp-mediated MDR.
Co-delivery of anticancer drugs together with P-gp inhibitors has recently become more popular to overcome MDR. In some cases, anticancer drugs were encapsulated into the nanocarriers and P-gp inhibitors were administered as free solutions (Krishna et al., Citation2000). However, when administered in a free form, P-gp inhibitors are distributed to all tissues and organs, which can cause the non-specific inhibition of P-gp. To prevent this, P-gp inhibitors should be targeted to their site of action, i.e. to cancer cells. There are quite a few studies, which indicate that anticancer drug- and P-gp inhibitor-co-loaded nanocarriers can overcome the MDR. Wong et al. (Citation2006) simultaneously encapsulated ELC with Doxorubicin into the polymer-lipid hybrid nanoparticles, and they found that dual agent-loaded nanoparticles were able to reverse MDR. Same results were achieved with PLGA nanoparticles encapsulating combinations of PTX-tariquidar (Patil et al., Citation2009) and vincristine-verapamil (Song et al., Citation2009).
Long-circulating liposomes co-loaded with PTX and tariquidar have also been studied (Patel et al., Citation2011). Liposomes with uniform size distribution and high loading efficacy provided a good nanopreparation against PTX-resistant cells and successfully reversed MDR in SKOV3-TR cell line. With these results in mind, we wanted to demonstrate that drug-loaded PEG-PE-based micelles, which represent a convenient delivery system for poorly soluble drugs, can also be a good delivery platform for MDR reversal. There are several available data on drug co-loading into the micellar structures (Diezi & Kwon, Citation2012) and on the successful action of PEG-PE-based micelles co-loaded with an anticancer drug and first generation P-gp inhibitor (Sarisozen et al., Citation2012). As can be seen from , co-loaded PEG-PE-based micelles provided the highest cytotoxicity against P-gp overexpressing MDR cells, thus indicating that micelle co-loading of PTX and ELC together can reverse MDR and make the cells sensitive to PTX again, even at short exposure times.
Conclusions
In summary, ELC and PTX simultaneously encapsulated into the PEG-PE micelles with desired particle size, loading values and release properties and these systems resulted in highest cytotoxicity in MDCKII-MDR1 resistant cell line. Even though PEG-PE based micelles have a potential to enhance the cytotoxicity of PTX in resistant cells, co-loading of P-gp inhibitor is a more promising option to get successful response to PTX and overcome the MDR.
Acknowledgement
MDCKII cells (Parental and MDR-1) were generously gifted by Prof. Piet Borst and Prof. Alfred Schinkel from The Netherlands Cancer Institute (NKV).
Declaration of interest
This study was supported by grant 109S106 from The Scientific and Technological Research Council of Turkey (TUBITAK).
References
- Allen JD, Brinkhuis RF, van Deemter L, Wijnholds J, Schinkel AH. Extensive contribution of the multidrug transporters P-glycoprotein and Mrp1 to basal drug resistance. Cancer Res 2000;60:5761–5766.
- Bae Y, Diezi TA, Zhao A, Kwon GS. Mixed polymeric micelles for combination cancer chemotherapy through the concurrent delivery of multiple chemotherapeutic agents. J Control Release 2007;122:324–330.
- Batrakova EV, Miller DW, Li S, Alakhov VY, Kabanov AV, Elmquist WF. Pluronic P85 enhances the delivery of digoxin to the brain: in vitro and in vivo studies. J Pharmacol Exp Ther 2001;296:551–557.
- Borst P, Evers R, Kool M, Wijnholds J. A family of drug transporters: the multidrug resistance-associated proteins. J Natl Cancer Inst 2000;92:1295–1302.
- Borst P, Elferink RO. Mammalian ABC transporters in health and disease. Annu Rev Biochem 2002;71:537–592.
- Chen CJ, Chin JE, Ueda K, Clark DP, Pastan I, Gottesman MM, Roninson IB. Internal duplication and homology with bacterial transport proteins in the mdr1 (P-glycoprotein) gene from multidrug-resistant human cells. Cell 1986;47:381–389.
- Cho YW, Lee J, Lee SC, Huh KM, Park K. Hydrotropic agents for study of in vitro paclitaxel release from polymeric micelles. J Control Release 2004;97:249–257.
- Clarke R, Leonessa F, Trock B. Multidrug resistance/P-glycoprotein and breast cancer: review and meta-analysis. Semin Oncol 2005;32:S9–15.
- Dabholkar RD, Sawant RM, Mongayt DA, Devarajan PV, Torchilin VP. Polyethylene glycol-phosphatidylethanolamine conjugate (PEG-PE)-based mixed micelles: some properties, loading with paclitaxel, and modulation of P-glycoprotein-mediated efflux. Int J Pharm 2006;315:148–157.
- Diezi TA, Kwon G. Amphotericin B/sterol co-loaded PEG-phospholipid micelles: effects of sterols on aggregation state and hemolytic activity of amphotericin B. Pharm Res 2012;29:1737–1744.
- Foxwell BM, Mackie A, Ling V, Ryffel B. Identification of the multidrug resistance-related P-glycoprotein as a cyclosporine binding protein. Mol Pharmacol 1989;36:543–546.
- Gao Z, Lukyanov AN, Chakilam AR, Torchilin VP. PEG-PE/phosphatidylcholine mixed immunomicelles specifically deliver encapsulated taxol to tumor cells of different origin and promote their efficient killing. J Drug Target 2003;11:87–92.
- Goddard ED, Turro NJ, Kuo PL, Ananthapadmanabhan KP. Fluorescence probes for critical micelle concentration determination. Langmuir 1985;1:352–355.
- Gottesman MM, Ambudkar SV, Ni B, Aran JM, Sugimoto Y, Cardarelli CO, Pastan I. Exploiting multidrug resistance to treat cancer. Cold Spring Harb Symp Quant Biol 1994;59:677–683.
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med 2002;53:615–627.
- Hyafil F, Vergely C, Du Vignaud P, Grand-Perret T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridonecarboxamide derivative. Cancer Res 1993;53:4595–4602.
- Irvine JD, Takahashi L, Lockhart K, Cheong J, Tolan JW, Selick HE, Grove JR. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J Pharm Sci 1999;88:28–33.
- Jemal A, Siegel R, Ward E, Hao Y, Xu J, Murray T, Thun MJ. Cancer statistics, 2008. CA Cancer J Clin 2008;58:71–96.
- Jule E, Yamamoto Y, Thouvenin M, Nagasaki Y, Kataoka K. Thermal characterization of poly(ethylene glycol)-poly(D,L-lactide) block copolymer micelles based on pyrene excimer formation. J Control Release 2004;97:407–419.
- Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976;455:152–162.
- Kabanov AV, Batrakova EV, Alakhov VY. Pluronic block copolymers for overcoming drug resistance in cancer. Adv Drug Deliv Rev 2002;54:759–779.
- Krishna R, Mayer LD. Multidrug resistance (MDR) in cancer. Mechanisms, reversal using modulators of MDR and the role of MDR modulators in influencing the pharmacokinetics of anticancer drugs. Eur J Pharm Sci 2000;11:265–283.
- Krishna R, St-Louis M, Mayer LD. Increased intracellular drug accumulation and complete chemosensitization achieved in multidrug-resistant solid tumors by co-administering valspodar (PSC 833) with sterically stabilized liposomal doxorubicin. Int J Cancer 2000;85:131–141.
- Lage H. MDR1/P-glycoprotein (ABCB1) as target for RNA interference-mediated reversal of multidrug resistance. Curr Drug Targets 2006;7:813–821.
- Lee J, Lee SC, Acharya G, Chang CJ, Park K. Hydrotropic solubilization of paclitaxel: analysis of chemical structures for hydrotropic property. Pharm Res 2003;20:1022–1030.
- Li X, Li P, Zhang Y, Zhou Y, Chen X, Huang Y, Liu Y. Novel mixed polymeric micelles for enhancing delivery of anticancer drug and overcoming multidrug resistance in tumor cell lines simultaneously. Pharm Res 2010;27:1498–1511.
- Lukyanov AN, Gao Z, Mazzola L, Torchilin VP. Polyethylene glycol-diacyllipid micelles demonstrate increased acculumation in subcutaneous tumors in mice. Pharm Res 2002;19:1424–1429.
- Musacchio T, Laquintana V, Latrofa A, Trapani G, Torchilin VP. PEG-PE micelles loaded with paclitaxel and surface-modified by a PBR-ligand: synergistic anticancer effect. Mol Pharm 2009;6:468–479.
- Neumann M, Wilisch A, Diddens H, Probst H, Gekeler V. MDR hamster cells exhibiting multiple altered gene expression: effects of dexniguldipine-HCl (B859-35), cyclosporin A and buthionine sulfoximine. Anticancer Res 1992;12:2297–2302.
- Orr GA, Verdier-Pinard P, McDaid H, Horwitz SB. Mechanisms of Taxol resistance related to microtubules. Oncogene 2003;22:7280–7295.
- Patel NR, Rathi A, Mongayt D, Torchilin VP. Reversal of multidrug resistance by co-delivery of tariquidar (XR9576) and paclitaxel using long-circulating liposomes. Int J Pharm 2011;416:296–299.
- Patil Y, Sadhukha T, Ma L, Panyam J. Nanoparticle-mediated simultaneous and targeted delivery of paclitaxel and tariquidar overcomes tumor drug resistance. J Control Release 2009;136:21–29.
- Sarisozen C, Vural I, Levchenko T, Hincal AA, Torchilin VP. PEG-PE-based micelles co-loaded with paclitaxel and cyclosporine A or loaded with paclitaxel and targeted by anticancer antibody overcome drug resistance in cancer cells. Drug Deliv 2012;19:169–176.
- Sauna ZE, Ambudkar SV. Characterization of the catalytic cycle of ATP hydrolysis by human P-glycoprotein. The two ATP hydrolysis events in a single catalytic cycle are kinetically similar but affect different functional outcomes. J Biol Chem 2001;276:11653–11661.
- Sawant RR, Sawant RM, Torchilin VP. Mixed PEG-PE/vitamin E tumor-targeted immunomicelles as carriers for poorly soluble anti-cancer drugs: improved drug solubilization and enhanced in vitro cytotoxicity. Eur J Pharm Biopharm 2008;70:51–57.
- Schinkel AH, Mayer U, Wagenaar E, Mol CA, van Deemter L, Smit JJ, van der Valk MA, Voordouw AC, Spits H, van Tellingen O, Zijlmans JM, Fibbe WE, Borst P. Normal viability and altered pharmacokinetics in mice lacking mdr1-type (drug-transporting) P-glycoproteins. Proc Natl Acad Sci USA 1997;94:4028–4033.
- Shabbits JA, Krishna R, Mayer LD. Molecular and pharmacological strategies to overcome multidrug resistance. Expert Rev Anticancer Ther 2001;1:585–594.
- Song XR, Cai Z, Zheng Y, He G, Cui FY, Gong DQ, Hou SX, Xiong SJ, Lei XJ, Wei YQ. Reversion of multidrug resistance by co-encapsulation of vincristine and verapamil in PLGA nanoparticles. Eur J Pharm Sci 2009;37:300–305.
- Sparreboom A, Planting AS, Jewell RC, van der Burg ME, van der Gaast A, de Bruijn P, Loos WJ, Nooter K, Chandler LH, Paul EM, Wissel PS, Verweij J. Clinical pharmacokinetics of doxorubicin in combination with GF120918, a potent inhibitor of MDR1 P-glycoprotein. Anticancer Drugs 1999;10:719–728.
- Speicher LA, Barone LR, Chapman AE, Hudes GR, Laing N, Smith CD, Tew KD. P-glycoprotein binding and modulation of the multidrug-resistant phenotype by estramustine. J Natl Cancer Inst 1994;86:688–694.
- Stein WD, Cardarelli C, Pastan I, Gottesman MM. Kinetic evidence suggesting that the multidrug transporter differentially handles influx and efflux of its substrates. Mol Pharmacol 1994;45:763–772.
- Szakács G, Paterson JK, Ludwig JA, Booth-Genthe C, Gottesman MM. Targeting multidrug resistance in cancer. Nat Rev Drug Discov 2006;5:219–234.
- Tang F, Horie K, Borchardt RT. Are MDCK cells transfected with the human MDR1 gene a good model of the human intestinal mucosa? Pharm Res 2002;19:765–772.
- Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release 2001;73:137–172.
- Torchilin VP, Lukyanov AN, Gao Z, Papahadjopoulos-Sternberg B. Immunomicelles: targeted pharmaceutical carriers for poorly soluble drugs. Proc Natl Acad Sci USA 2003;100:6039–6044.
- Torchilin VP. Lipid-core micelles for targeted drug delivery. Curr Drug Deliv 2005;2:319–327.
- Torres K, Horwitz SB. Mechanisms of Taxol-induced cell death are concentration dependent. Cancer Res 1998;58:3620–3626.
- Trubetskoy VS, Torchilin VP. Use of polyoxyethylene-lipid conjugates as long-circulating carriers for delivery of therapeutic and diagnostic agents. Adv Drug Deliv Rev 1995;16:311–320.
- Ueda K, Cardarelli C, Gottesman MM, Pastan I. Expression of a full-length cDNA for the human “MDR1” gene confers resistance to colchicine, doxorubicin, and vinblastine. Proc Natl Acad Sci USA 1987;84:3004–3008.
- Walle UK, Walle T. Taxol transport by human intestinal epithelial Caco-2 cells. Drug Metab Dispos 1998;26:343–346.
- Wang J, Goh B, Lu W, Zhang Q, Chang A, Liu XY, Tan TM, Lee H. In vitro cytotoxicity of Stealth liposomes co-encapsulating doxorubicin and verapamil on doxorubicin-resistant tumor cells. Biol Pharm Bull 2005;28:822–828.
- Weissig V, Whiteman KR, Torchilin VP. Accumulation of protein-loaded long-circulating micelles and liposomes in subcutaneous Lewis lung carcinoma in mice. Pharm Res 1998;15:1552–1556.
- Witherspoon SM, Emerson DL, Kerr BM, Lloyd TL, Dalton WS, Wissel PS. Flow cytometric assay of modulation of P-glycoprotein function in whole blood by the multidrug resistance inhibitor GG918. Clin Cancer Res 1996;2:7–12.
- Wong HL, Bendayan R, Rauth AM, Wu XY. Simultaneous delivery of doxorubicin and GG918 (Elacridar) by new polymer-lipid hybrid nanoparticles (PLN) for enhanced treatment of multidrug-resistant breast cancer. J Control Release 2006;116:275–284.
- Zhang W, Shi Y, Chen Y, Yu S, Hao J, Luo J, Sha X, Fang X. Enhanced antitumor efficacy by paclitaxel-loaded pluronic P123/F127 mixed micelles against non-small cell lung cancer based on passive tumor targeting and modulation of drug resistance. Eur J Pharm Biopharm 2010;75:341–353.
- Zhao CL, Winnik MA, Riess G, Croucher MD. Fluorescence probe techniques used to study micelle formation in water-soluble block copolymers. Langmuir 1990;6:514–516.