Abstract
The highly organized structure of the stratum corneum provides an effective barrier to the drug delivery into or across the skin. To overcome this barrier function, penetration enhancers are always used in the transdermal and dermal drug delivery systems. However, the conventional chemical enhancers are often limited by their inability to delivery large and hydrophilic molecules, and few to date have been routinely incorporated into the transdermal formulations due to their incompatibility and local irritation issues. Therefore, there has been a search for the compounds that exhibit broad enhancing activity for more drugs without producing much irritation. More recently, the use of biomaterials has emerged as a novel method to increase the skin permeability. In this paper, we present an overview of the investigations on the feasibility and application of biomaterials as penetration enhancers for transdermal or dermal drug delivery systems.
Introduction
Utilization of skin as a route for drug deliver has been an attracting alternative to conventional methods including injections and tablets (Prausnitz & Langer, Citation2008). Its advantages include the avoidance of the pain, elimination of hepatic first-pass metabolism, sustained drug release for a prolonged period, easy use and withdrawal in case of side effects (Tanner & Marks, Citation2008; Thomas & Finnin, Citation2004). However, the stratum corneum (SC) of the skin has a unique hierarchical structure filled with multiple lipid bilayers and the embedded corneocytes (Menon, Citation2002; Karande & Mitragotri, Citation2009), providing an outstanding transdermal barrier against the absorption of chemical and biological toxins (Naik et al., Citation2000; Hadgraft, Citation2004). In theory, there are three potential pathways in the skin: first, the transcelluar route which is through the cornecytes and the lipid matrix; second, the intercellular route which is only through the lipid domains between the cornecytes and third, the transappendageal route which is across the hair follicles, sebaceous glands or sweat glands (Sinico & Fadda, Citation2006; Guillard et al., Citation2009).
According to different therapeutic effect, drug administration via the skin could be classified into two categories: (i) transdermal delivery by which drugs diffuse through the various layers of the skin and finally enter the systemic circulation; (ii) dermal or topical delivery that carry drugs to the pathological sites within the skin (Brown, Citation2006). To facilitate transdermal or topical delivery of the drugs, skin penetration enhancers have been extensively studied (Williams & Barry, Citation2004). Up to date, more than 350 chemicals have been identified that can be used as penetration enhancers (Karande et al., Citation2005), including water, terpenes, sulfoxides, laurocapram, pyrrolidones, fatty acid, fatty alcohol glycol, surfactants, urea and so on. However, the success of these chemical enhancers has been limited, owing to their common skin irritancy and little enhancement effect on the large hydrophilic molecules (Bos & Meinardi, Citation2000). New technologies to overcome the skin barrier function are constantly being developed. Recently, biomaterials have emerged as novel penetration enhancers for enhanced transdermal delivery (Finnin & Morgan, Citation1999). Particularly, some of them exhibit excellent enhancing ability towards the transdermal absorption of macromolecules, which is often circumscribed by conventional chemical enhancers (Chen et al., Citation2006; Hsu & Mitragotri, Citation2011).
Although reviews on classic chemical enhancers have frequently been found in literatures (Walker & Smith, Citation1996; Sinha & Kaur, Citation2000; Williams & Barry, Citation2004; Pathan & Setty, Citation2009), an overview of the biomaterials that can be used as permeation enhancers has still been in lack. Therefore, in this article we reviewed the investigations on the biomaterials in the field of penetration enhancement, and grouped them into several categories () so that readers could find the details on a specific enhancer more easily. The paper would also be helpful to researchers in the selection of suitable enhancers for different drugs, especially for those hydrophilic macromolecules poorly absorbed in the skin.
Table 1. Biomaterials in enhanced transdermal drug delivery systems.
Biomaterials for enhanced skin penetration
Protein transduction domains-drug complex
Protein transduction domains (PTDs), also known as cell penetrating proteins, were initially designed to efficiently cross the biological membranes and facilitate the cell uptake of molecules to which they are attached. Among several different drug delivery routes, application of PTDs in topical and transdermal drug delivery systems has recently garnered tremendous attention in both cosmeutical and pharmaceutical research (Nasrollahi et al., Citation2012).
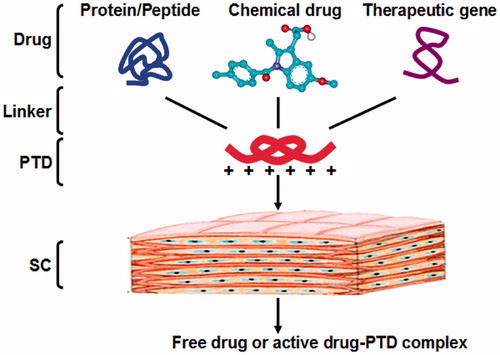
Structurally, most of PTDs are positively charged and have the typical structure of basic amino acids, i.e. arginine and lysine (Schwarze & Dowdy, Citation2000; Zaro & Shen, Citation2003). As shown in , PTDs could be covalently linked to the drugs and enhance the drug penetration into and across the skin. Under the physiologic pH within tissue, the PTD-drug complex could release the active drug, or remain unchanged to produce similar therapeutic effects as the free drug did.
Figure 1. Schematic representation of the skin delivery of PTD-drug complex. PTDs and drugs are conjugated via releasable or non-releasable chemical linker. In the body, the complex of PTD and cargo can exert its therapeutic effect after degradation into the free drug, or in the form of the conjugation with similar activity.
Arginine oligomers have been widely studied to enhance the cellular uptake of poorly membrane-permeable bioactive molecules (Gonçalves et al., Citation2005; Sakuma et al., Citation2010). As an example, heptamer oligomer of arginine (R7) was first suggested as a potential PTD in transdermal delivery of cyclosporine A (CsA) for treatment of inflammatory skin diseases (Rothbard et al., Citation2000). When administrated topically, CsA was poorly absorbed and exhibited little anti-inflammation effect. To improve its skin permeability, a R7-CsA conjugate was synthesized via a pH-sensitive linker, which was designed to release the active drug at physiologic pH within tissue. The penetration results showed that compared with unmodified CsA, R7-CsA could not only penetrate into cells in the deep skin layers of human and mouse skin, but also entered target dermal T lymphocytes. Under the physiological condition, the releasable R7-CsA conjugate was biologically active both in vitro and in vivo, leading to a functional inhibition of cutaneous inflammation. Although the water solubility of R7-CsA was much higher than the CsA, which might be an unfavorable factor to the drug delivery across the SC lipids, R7-CsA penetrated the skin as well as the peptide did. This special phenomenon could be related to its transport mechanism that is still being investigated.
The basic domain of human immunodeficiency virus type I (HIV-1) transactivator of transcription (TAT) has also been used for the introduction of exogenous conjugated macromolecules to various cells and even in animal tissues (Fawell et al., Citation1994; Vivès, Citation2003). Based on these observations, an alternative application of TAT in transdermal drug delivery was introduced to improve the skin penetration of a lipolytic peptide, glycine-lysine-histiding (GKH) (Lim et al., Citation2003). The study revealed that after conjugation with TAT, the amount of GKH that penetrated into and across the skin was both significantly increased. As expected, the lipolytic effect of GKH would not be changed by the chemical attachment with TAT, which was demonstrated by the in vitro lipolysis in cultured and isolated adipocytes. In addition, there was no cytotoxicity that could be observed in any dose concentration (10−4–10−7 mol/L) of the conjugate, TAT-GSK. The results indicated that TAT-GSK could be used as a new cosmetic ingredient in slimming products.
Further molecular modification of TAT has led to the discovery of a new penetrating peptide, YARA (YARAAARQARA), which has been demonstrated to transport in cells more effectively than TAT both in vitro and in vivo (Ho et al., Citation2001). The effect of YARA to penetrate the skin was studied in comparison with TAT by Lopes et al. (Citation2005). According to the fluorescence visualization of the penetrated skin, fluorescein isothiocyanate (FITC)-labeled YARA and TAT could both cross the SC and reach the viable layers of the porcine ear skin. Furthermore, the in vitro penetrating data revealed that the amount of YARA and TAT that penetrated into the skin was more than 30 times as much as that penetrated across the skin. Then, YARA and TAT was coupled with another peptide, phosphopepetide analog of the heat shock protein 20 (P20), and the topical and transdermal delivery of the conjugate was investigated. As a consequence, conjugated P20 showed a significantly higher penetrating activity than the nonconjugated P20. A small transdermal delivery of the conjugated peptide was also observed, which could not be achieved by the nonconjugated peptide at all. However, unlike cell transduction, no significant difference could be detected between the skin penetration of YARA and TAT.
Hsu & Mitragotri (Citation2011) identified a skin permeating and cell entering peptide (SPACE) with a sequence of AC-TGSTQHQ-CG. The study revealed that SPACE peptide penetrated well into the dermis of the porcine and human skin, as well as various cells including keratinocytes, fibroblasts and endothelial cells. Using SPACE as conjugated penetration enhancer, some siRNAs for treating atopic dermatitis have achieved significant delivery across SC and into the viable cell membranes, which provided a unique therapeutic opportunity in the treatment of dermatological diseases. In addition, it was noted that SPACE peptide was primarily effective in enhancing permeation of the conjugated but not the co-administrated cargoes. Cellular penetration was suggested for the potential transport mechanism of SPACE. Fourier transform infrared spectroscopy (FTIR) experiments with isolated human SC showed that the SPACE peptide likely changed the structure of the keratin of the corneocytes, instead of the SC lipids.
Non-conjugated peptides
In addition to conjugation with PTDs, drug delivery in the skin might also be improved by other penetrating peptides without the need of chemical attachment.
Magainin, originally isolated from the skin of the African clawed frog, Xenopus laevis, has 23 amino acid residues and a net charge up to +4 (Zasloff, Citation1987). Contrarily, the lipid membranes are negatively charged under physiological condition. Therefore, magainin could bind to the biological membranes by the way of electrostatic interaction and form pores in the lipid bilayers with the approximately estimated size of 1 nm (Matsuzaki et al., Citation1997). Such effect of magainin could also be used to make SC lipid bilayers leaky enough to enhance transdermal absorption (Kaushik, Citation2001). A recent study demonstrated that magainin could disrupt the SC lipid structure and significantly increase skin permeability (Kim et al., Citation2007). However, it is important to note that magainin’s enhancement requires co-administration with a surfactant chemical enhancer, such as N-lauroyl sarcosine, because of its weak penetrating activity to the SC. Besides the interaction with SC lipids, electrostatic forces between magainin peptides and drugs could also mediate drug transport across the skin (Kim et al., Citation2008). To examine this effect, the study chose two model drugs including fluorescein and granisetron, which had similar molecular weight and opposite charges. The pH and the concentration of added salt were investigated as influencing factor in the transdermal transport of charged molecules. The results displayed that increasing pH or shielding electrostatic interactions with 1–2 M NaCl solution both could decrease the transdermal penetration of the negative charged molecule, fluorescein. By contrast, such effect could lead to an increased transdermal permeability of the positive charged drug, granisetron. Therefore, it was obvious that the electrostatic interaction between the drug and magainin could also facilitate the transdermal delivery. Additionally, circular dichroism analysis, microscopy analysis and FTIR were respectively introduced to study the secondary structure of magainin and the lipid organization in the SC. Surprisingly, in the studied pH range, these characteristics did not alter significantly. Therefore, the most likely cause of its enhancement was that magainin partitioned into SC under the support of N-lauroyl sarcosine, and then created the diffused pores for charged molecules delivery across the skin.
TD-1 (ACSSSPSKHCG) is the only synthetic peptide specially designed as an enhancer for transdermal drug delivery. Chen et al. (Citation2006) found that when co-administrated with TD-1 on the rat skin, the systemic levels of insulin and human growth hormone could both be significantly improved. The mechanistic study demonstrated that TD-1 might create a transient opening on the skin barrier, which enabled the drug to pass through SC and reach the systemic circulation. In addition, a recent study also indicated that TD-1 could aid skin delivering of botulinum neurotoxin type A (BoNT-A) which was used to block the release of pain modulators in the periphery, and result in a lower level of PE evoked by saphenous nerve stimulation in the rat hind paw skin (Carmichael et al., Citation2010).
T2 peptide (LVGVFH) was another example for non-conjugated peptide enhancers, which was identified by phage display peptide library screening. T2 peptide could produce an excellent enhancing activity on the transdermal absorption of 5-fluorouracil without the aid of other skin penetration enhancers. The study also found that the enhancing effect of T2 was pH dependent, and closely associated with the c-terminal amino acid, histidine, in the T2. It was suggested that T2 peptide could interact with skin lipids and enhance the partitioning of molecules into the skin (Kumar et al., Citation2012).
Some PTDs were also used as non-covalent carriers to efficiently delivery therapeutic macromolecules through the skin. Hou et al. (Citation2007) found that some arginine-rich intracellular delivery (AID) peptides (TAT, R9, R9Z and SR9) could enhance the delivery of green fluorescent protein (GFP) in the animal skin without conjugation with the transported protein. The best permeation ability of the cargo protein and AID (SR9) mixture into cells was achieved with a ratio of 1:3. The cellular internalization and tissue penetration of other proteins (e.g. collagen and insulin) could also be mediated by AID peptides that were not covalently attached to the transported proteins. Further study suggested that the AID peptide did not produce cytotoxicity to animal cells, and the AID-mediated cellular entry and transdermal delivery involve macropinocytosis and action reorganization.
Metabolic modulators
The lipids in the skin SC, mainly composed of ceramides, cholesterol and fatty acids, are considered to play a key role in maintenance of the transdermal barrier function (Wertz & van den Beergh, Citation1998; de Jager et al., Citation2004). A decrease in the amount of any of these critical lipid species might alter the molar ratio required for normal barrier function, and break the skin barrier integrity. Therefore, it was suggested that the skin permeability can be mediated by lipid metabolic intervention (Patil et al., Citation1996; Elias et al., Citation2002).
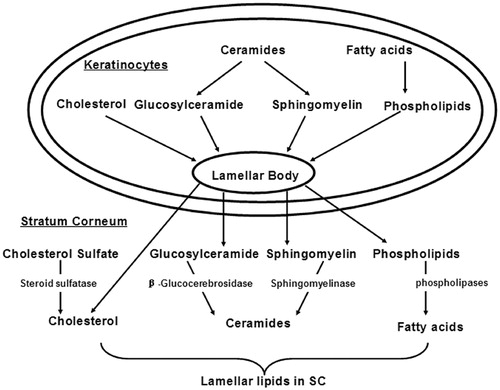
The segregation of lipids to the intercellular domains of the SC where they form multiple membrane bilayers underlies their putative role in the permeability barrier function of the skin (Menon et al., Citation1992). As shown in , the lamellar bodies generated during the keratinocyte differentiation are considered as the source of these lipids. In addition to phospholipids, glucosylcer-amides, sphingomyelin, and cholesterol, lamellar bodies also contain numerous enzymes, including lipid hydrolases, such as β-glucocerebrosidase, acidic sphingomyelinase, secretory phospholipase A2, and acidic/neutral lipases (Feingold Citation2009). Following the secretion of lamellar body, β-glucocerebrosidase converts glucosylceramides into ceramides, acidic sphingomyelinase converts sphingomyelin into ceramides, and phospholipases convert phospholipids into free fatty acids and glycerol (Feingold, Citation2007).
Figure 2. The formation of SC intercellular lipids that provide for the penetration barrier, adapted from Feingold (Citation2007, Citation2009).
Several studies have shown that applications of fatty acid synthesis inhibitor (e.g. cerulenin and 5-(tetradecyloxy)-2-furancarboxylic acid), cholesterol synthesis inhibitor (e.g. atorvastatin and fluvastain) or ceramide synthesis inhibitor (e.g. β-chloroalanine) on the organic solvent treated in vivo skin could delay the normalization of the lipid barrier function, and enhance the transdermal delivery of different drugs, such as caffeine and lidocaine hydrochloride (Tsai et al., Citation1996), levodopa (Babita & Tiwary, Citation2002, Citation2004a, Citationb) and 5-fluorouracil (Gupta et al., Citation2004). ATR-FTIR experiments revealed that rat skin after treatment with lipid synthesis inhibitors demonstrated a significant decrease in the peak height and area for C-H stretching absorbance of the SC lipids, suggesting that the lipids in SC was removed by application of lipid synthesis inhibitors. By contrast, an insignificant shift in the frequency of these stretching absorption peaks indicated that no fluidization of the SC lipids was produced by lipid synthesis inhibitors (Babita et al., Citation2005). In addition, synthesis of skin lipid was a calcium induced process, which could be disturbed by calcium cannel blocker, but promoted by co-application of excessive extracellular Ca2+ (Babita & Tiwary, Citation2004b).
Trypsin is a proteolytic enzyme that has extensively been used for in vitro epidermal separation and keratinocyte* isolation. However, its enhancement effects on transdermal drug delivery are rarely reported. Li et al. (Citation2008) first found that the trypsin could be used as a biochemical enhancer that could promote the transdermal absorption of insulin in vitro and increase the hypoglycemic effect in vivo. In a further study, FITC-labeled dextrans and FITC-insulin were respectively selected as macromolecule and polypeptide penetrants (Li et al., Citation2009). Permeation experiments were carried out in a three-dimensional cultured human epidermis model. Fluorescence microscopy examination was used to explore the effect of trypsin on the transport pathway for peptides. The study revealed that trypsin increased the penetration via the intercellular pathway and the hair follicular delivery. Attenuated total reflection-Fourier transform infrared spectroscopy (ATR-FTIR) experiments indicated that trypsin altered the SC protein structure, thereby affecting skin barrier properties and resulting in the enhancing effect.
Chitosan and its derivatives
Chitosan is a non-toxic, biocompatible polymer that can enhance the drug absorption across mucosa epithelia. However, it is not effective as permeation enhancer at neutral and alkaline pH values due to its poor aqueous solubility when pH > 6.5 (Thanou et al., Citation2001). To improve its solubility and effectiveness, N-trimethyl chitosan (TMC) has been developed and investigated, which exhibits considerably good permeation enhancement effect in basic environments when using Caco-2 cells as a model (Kotzé et al., Citation1997; Thanou et al., Citation2000). In addition, the molecular weight, deacetylation degree and the charge of TMC represented by the degree of quaternization, are considered as important factors determining its enhancing activity (Jonker et al., Citation2002; Di Colo et al., Citation2004; Giuseppina et al., Citation2005).
Based on the cationic nature of chitosan and the similarity between epithelial cells and SC, namely the fixed negative charges in the tight junction between their respective cells, it is therefore reasonable to speculate that chitosan and its derivatives could also be used to enhance the penetration through the skin. Permeation enhancement effect of chitosan has been reported by Sapra et al. (Citation2008) in the transdermal delivery of carvedilol.
To seek more potent penetration enhancers, N-trimethyl chitosans (TMC) with quaternization degree of 40% and 60%, i.e. TMC40 and TMC60, were investigated for their permeation enhancing ability through the skin (He et al., Citation2008). Both the in vitro and in vivo experiments revealed that the transdermal enhancing ability of TMCs increased with quaternization degree when using testosterone as transdermal penetrant. Compared with 2% Azone, 5% TMC60 exhibited a greater enhancement (p < 0.05) while 5% TMC40 had a similar effect with Azone. ATR-FTIR study suggested that TMCs increased the transdermal permeability by alternating the secondary structure of keratin in SC. Further mechanistic study suggested that chitosan and its derivatives could also increase the water content in SC, decrease HaCaT cells membrane potentials and enhance HaCaT cells membrane fluidity significantly (He et al., Citation2009).
To avoid the drawbacks of high molecular weight and low solubility in water or organic vehicles, low molecular weight chitosans (LMWCs) was studied as transdermal enhancers for the penetration of baicalin (Zhou et al., Citation2010). As the two units in the copolymer structure of chitosans, the individual enhancement effect of β-D-GlcNAc and β-D-GlcNH2 was also investigated. It was interesting to find that β-D-GlcNH2 could increase the enhancement factor to 7.9, while β-D-GlcNAc had no significant effect. When CS80-1000 was used as a transdermal enhancer, the highest enhancement effect was observed, providing an enhancement factor of 15.9. Hence, it was speculated that β-D-GlcNH2 or its oligomers were the effective fraction for the transdermal enhancement of chitosans, and the deacetylation degree of chitosans might be an important factor that affected the LMWC enhancement effect. As expected, when the deacetylation degree of chitosans decreased from 14% to 80%, the enhancement activity decreased significantly, and even no effect was observed at the deacetylation degree of 41%. The effect of pH on the LMWC enhancement effect, which might influence the dissociation equilibrium of the amino group in β-D-GlcNH2, was then considered in the study. The best pH value for β-D-GlcNH2 and CS80-1000 were observed respectively at 7.0 and 7.5. In addition, the molecular weight and concentration of chitosan could also affect the LMWC enhancement effect. When the molecular weight increased from 1000 Da to 5000Da, the enhancement factor of chitosan decreased from 15.9 to 1.0. With the increase of the concentration of β-D-GlcNH2 or CS80-1000, the enhancement effect improved first, and then decreased when the maximum was achieved. Based on these findings, it was concluded that chitosan molecular weight, degree of deacetylation, pH of donor baicalin solutions, and enhancer concentration all affected LMWC enhancement effects.
In the study of Lv et al. (Citation2011), a biomimetic chitosan derivative, N-Arginine chitosans (N-Arg-CSs), were prepared and studied as penetration enhancer for transdermal delivery of adefovir. N-Arg-CSs were synthesized by chemical coupling of chitosans and L-arginine, mimicking the arginine-rich PTD in order to get the cellular uptake functions of typical PTDs. The influence of molecular weight, enhancer concentration and degrees of arginine substitution (DS) was investigated. The most effective enhancer was found as 2% (w/v) concentration of 10 kDa N-Arg-CS with 6% DS. Similar with the study of Zhou et al. (Citation2010), the best pH for the enhancement effect of N-Arg-CS was observed at about 7.0, and its transdermal effect of N-Arg-CS was parabolic depended on the enhancer concentration. For N-Arg-CS with different molecular weight, the accumulative penetrated amount of adefovir with N-Arg-CS (MW, 10 kDa) was higher than that of N-Arg-CS with molecular weight of 5 and 10 kDa. The study also indicated that chemical conjugation of arginine and chitosan had a significantly stronger effect on the skin permeability than the physical mixing. A complex of N-Arg-CS and adefovir might be formed by the interaction between positive and negative charge, which played a significant role for the transdermal enhancement effect of N-Arg-CS.
Cyclodextrins
Cyclodextrins (CDs) are cyclic oligosaccharides, consisting of (α-1,4)-linked α-D-glucopyranose units, with a hydrophilic outer surface and a somewhat lipophilic central cavity (Loftsson & Masson, Citation2001). The three parent CDs (α-CD, β-CD and γ-CD), as well as various kinds of chemically modified CD derivatives have been widely used as penetration enhancers for transdermal delivery (Matsuda & Arima, Citation1999). The roles of CDs in the enhanced transdermal drug delivery can be summarized as follows.
First, CDs can form hydrophilic inclusion complexes with lipophilic compounds in aqueous solution to improve drug solubility without changing their intrinsic ability to permeate the lipophilic barrier (Zi et al., Citation2008). During the complex formation, no covalent bonds are formed or broken, and in the aqueous solution the complexes are readily dissociated to release the free drug (Loftsson et al., Citation1998). Up to date, most of CD and its derivatives have demonstrated their potential as solubilizers to promote percutaneous absorption of many different drugs (Loftsson et al., Citation1995; Ventura et al., Citation2006; Zi et al., Citation2008). In addition, it was indicated that the solubility enhancing effect was closely depended on the concentration of CDs applied in the donor vehicle. When the drug was saturated in the suspension, the flux was increased as the CD concentration was increased. Maximum flux through the skin was obtained when just enough CD was used to keep all the drugs in solution (Loftsson & Sigurdardóttir, Citation1997). After all the drugs were dissolved, the drug flux decreased with the concentrations of CDs in the solution, which might be the reason for the inactivity of hydroxypropyl-β-cyclodextrin (HP-β-CD) towards the in vitro transdermal permeation of corticosterone (Shaker et al., Citation2003).
Second, CDs are able to extract significant amounts of some lipophilic components from the skin. Legendre et al. (Citation1995) investigated the extracting effect of β-CD, randomly methylated β-CD (RAMEB) and on the different components of skin lipids. Results showed that for the isolated SC, RAMEB extracted all the major lipid classes from the isolated SC, while HP-β-CD mainly extracted cholesterol, triglycerides and a small amount of cholesterol esters without affecting the content of ceramides and free fatty acid in the isolated SC. For the full thickness rat skin, the ability of HP-β-CD in removing cholesterol was 2-fold higher than RAMEB. Due to the effect of lipid extraction, all these CDs exhibited the ability to destabilize the liposomes that served as the skin lipid model. Further study showed that the DSC profiles and FTIR spectrum remained unchanged after skin treatment with different CDs, indicating that CDs mainly interacted with SC surface to extract skin component without producing any lipid fluidizing effect (Legendre et al., Citation1995).
Third, CDs have the potential to modify the drug release from polymeric systems (Arima et al., Citation1996). For example, Orienti et al. (Citation1991) investigated the effect of β-CD and HP-β-CD on the ketoprofen release from two kinds of dermal bases, i.e. carbopol gel and petrolatum/lanolin (9:1) ointment. The complexing CDs initially decreased the drug release from the two bases, while for longer periods the loss of the diffusible drug lowered the release rate more significantly in the case of pure ketoprofen than the drug-CD complex. As a consequence, the drug release in the presence of complexing CDs exhibited a more controllable and constant profile. Additionally, Umemura et al. (Citation1990) demonstrated that different release profiles could be achieved by using different CD derivatives. The release rate of nitroglycerin from ointment was accelerated by complexing with 2,6-diethyl-β-CD (DE-β-CD), but retarded by complexing with β-CD. Therefore combination of DE-β-CD and β-CD might be applicable for sustained release preparation for percutaneous administration.
Forth, the skin irritancy of chemicals can be alleviated by CDs in the formulation. For example, the skin irritation of prochlorperazine (PCP) was found to be reduced by complexation with β- or γ-CD, particularly by γ-CD, where the irritability of 80 mM PCP with 80 mM γ-CD was almost the same as that of 16 mM PCP alone. On the contrary, α-CD could enhance the irritative reaction of PCP, following the erythema with the eschar formation after application of 80 mM PCP with 80 mM α-CD (Uekama et al., 1982). Another study also pointed out that in the presence of 2,6-dimethyl-β-CD (DM-β-CD) and HP-β-CD, the drug celecoxib could preferentially interacted with the macrocycle, reducing its lesive action on the skin tissue (Ventura, Citation2006).
In addition, CDs can also be used as stabilizers in the formulation to reduce the drug decomposition (Davaran et al., Citation2006) in the formulation, protect against skin metabolism (Lopez et al., Citation2000) and depress the loss of volatile drug (Umemura et al., Citation1990).
Dendrimers
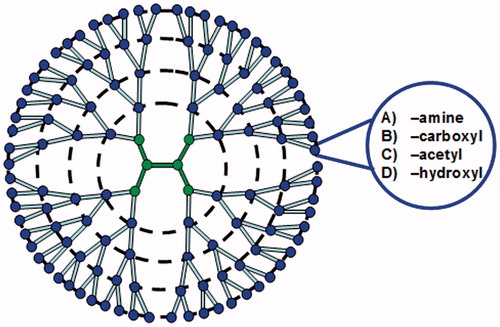
Dendrimers are hyperbranched, ordered, monodisperse polymers with a large number of surface functional groups. They can be used to encapsulate drug inside the core by a host-guest interaction, or by conjugation with the surface functional groups (Duncan & Izzo, Citation2005; Cheng et al., Citation2007). So far, dendrimers with different functionalities have widely been used in biomedical, pharmaceutical and biopharmaceutical fields. Among them, the most investigated one for transdermal drug delivery is the polyamidoamine (PAMAM) dendrimers. In terms of molecular size, PAMAM dendrimers () can be classified as generation (G) 0–10, and their surfaces can be modified to be amine-, carboxyl-, acetyl- or hydroxyl-terminated for different surface charge (Nanjwade et al., Citation2009).
Figure 3. Schematic representation of generation (G) 4 dendrimers. In terms of molecular size, PAMAM dendrimers can be classified as G 0-10, and their surfaces can be modified to be amine-, carboxyl-, acetyl- or hydroxyl- terminated for different surface charge.
In recent era, PAMAM dendrimers have found application as penetration enhancers in transdermal delivery of various bioactives (Mignani et al., Citation2013). An earlier investigation reported that PAMAM dendrimers could slightly enhance the absorption of tamsulosin from the polyhydroxyalkanoate (PHA)-based transdermal patches (Wang et al., Citation2003a). Further mechanistic study revealed that tamsulosin crystals had highly ordered orientation in the dendrimer-PHA matrix, which could limit the diffusion direction of tamsulosin and facilitate the drug permeation through the skin (Wang et al., Citation2003b).
Cheng et al. (Citation2007) suggested that PAMAM dendrimer (G5-NH2) could covalently conjugate with different NSAIDs (ketoprofen or diflunisal), significantly improve the drug permeation through the skin and achieve a prolonged pharmcodynamic profile. An in vivo pharmacokinetic assay showed that the blood concentration of indometacin in rats was increased in the presence of PAMAM dendrimers (G4-NH2 and G4-OH) (Chauhan et al., Citation2003). In addition, transdermal delivery of other drugs, such as 8-methoxypsoralen (8-MOP), could also be enhanced by the PAMAM dendrimers (G3-NH2 and G4-NH2) both in vitro and in vivo. Confocal images revealed that 8-MOP could transport into the deeper layers of the skin than the single drug solution (Borowska et al., Citation2010, Citation2012).
The molecular size, surface charge and concentration could all affect the penetration enhancement of PAMAM dendrimers (Venuganti & Perumal, Citation2008, Citation2009). In the transdermal delivery of the hydrophilic drug 5-fluorouracil, the permeability coefficient was inversely related to the molecular weight of the dendrimers, and lower generation dendrimers (G2-NH2) enhanced the drug penetration to a greater extent than the higher ones (G4-NH2 or G6-NH2). The drug flux nonlinearly increased with the dendrimer concentration. For PAMAM dendrimers with different surface charge, the enhancement effect was in the following decreasing order: G4- NH2 > G4-OH > G3.5-COOH. Transepidermal water loss, skin resistance measurement and ATR-FTIR studies revealed that the cationic dendrimers could more easily interact with the SC lipids and influence drug permeation through the skin (Venuganti & Perumal, Citation2009). In another study, Filipowicz et al. (Citation2011) also reported that the small-sized G2-NH2 dendrimer might be a more potent penetration enhancer than the larger ones for the transdermal absorption of the hydrophilic drug (riboflavin) from the emulsions or hydrogels. Additionally, use of iontophoresis could further increase the skin penetration of cationic and neutral dendrimers into viable epidermis, which provided an effective method to delivery macromolecules into and across the skin (Venuganti et al., Citation2011).
In addition to PAMAM, dendritic PEGylated polyglycerol amine has also been used for transdermal delivery of bioactive molecules. The nanoparticles based on it have demonstrated significant ability to penetrate the skin through the follicular route (Küchler et al., Citation2009).
Three different possible mechanisms of dendrimers were very recently reviewed and discussed by Y. Zhao et al. First, dendrimers could act as a drug release modifier and boost the drug dissolution from the vehicle. Second, via particle engineering the properties of dendrimers can preferably penetrate the skin via the follicular route. Third, certain low generation dendrimers may impair the SC barrier function, particularly in the presence of a potent vehicle (Sun et al., Citation2012).
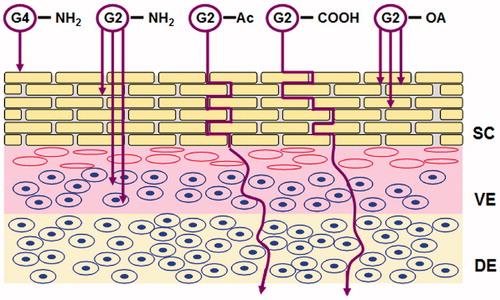
In a recent mechanistic study (), it was suggested that smaller PAMAM dendrimers (G2-NH2) could be absorbed into the SC as well as the underlying viable epidermis, more efficiently than the larger one (G4-NH2) which was only retained at the outmost layer of SC. Although the permeation of G2-NH2 in the receiver solution was also better than G4, the amount was too low to make an accurate quantification. By contrast, PAMAM dendrimers modified either by acetylationb (G2-Ac) or by carboxylation (G2-COOH) exhibited increased skin permeation and likely diffused through an extracelluar pathway. Confocal images showed that G2-NH2 could be internalized into the individual cell in both epidermal and dermal layers, while neither acetyl nor carboxyl modified PAMAM interacted with the cell. The study also found that conjugation of oleic acid to G2-NH2 could further increase the drug absorption and retention in the skin (Yang et al., Citation2012).
Figure 4. Schematic illustration of skin penetration of PAMAM dendrimers with different molecular size and surface attachments, SC: stratum corneum; VE: viable epidermis; DE: dermis, adapted from Yang et al. (Citation2012).
Oligodimethylsiloxanes
Oligodimethylsiloxanes (ODMSs) which themselves hardly permeated in the skin have also been designed as transdermal penetration enhancers to reduce the systemic side effect, owing to their large molecular volume and high lipophilicity (Akimoto & Nagase, Citation2003). This idea was originated from some cationic polymers, including benzalkonium chloride and hexadecylpyridinium bromide-type polymers, which exhibited significant enhancing effect towards the percutaneous absorption of 5-fluorouracil without producing sever skin irritation and discomfort (Aoyagi et al., Citation1990, Citation1991). Based on this hypothesis, oligodimethylsiloxanes (ODMSs) was selected as a bulky and inert backbone and then was covalently linked with polar and ionic substituent group at one chain end, such as pyridinio (Aoyagi et al., Citation1992b, d), quaternary amino (Aoyagi et al., Citation1992a), carboxyl (Nagase et al., Citation1992). In order to avoid the interaction between enhancers and dissociated drugs, oligodimethylsiloxanes with a non-ionic end group were also synthesized with phosphoramide (Aoyagi et al., Citation1992c) and 1-alkyl-2-pyrrolidon-3-yl (Aoyagi et al., Citation1996). However, it was suggested that these ODMSs were effective only for hydrophobic drugs, but not active enough for hydrophilic drugs. To explore polymeric enhancers for hydrophilic drugs, a hydrophilic PEG segment was introduced and obtained PEG-PDMS block copolymer containing an ammonium moiety at one chain en (Akimoto et al., Citation1997). The results showed that the introduction of PEG segment was more effective to enhance the hydrophilic antipyrine than the hydrophobic indomethacin. The enhancing activity of the copolymer was influenced by the chain length of PEG and PDMS segments, and a suitable balance of chain lengths would exist to appear the highest enhancing effect. By determination of kinetic parameters in the drug permeation, it was found that the enhancing activity was due to an increase of the partition coefficient of the drug into the SC.
Additionally, a different type of oligodimethylsiloxanes (ODMSs) with a sugar moiety as polar end group was also investigated, including cellobiosyl terminated ODMS (Cell-ODMS), glucopiranosyl terminated ODMS (GluO-ODMS) and its thioether analog (GlcS-ODMS) (Akimot et al., Citation2000). The results showed that the permeation of antipyrine was increased by the addition of GlcS-ODMS and GlcO-ODMS, but not by Cell-ODMS. A further study demonstrated that GluO-ODMS was also more effective for hydrophilic drugs than the lipophilic ones, and its activity was decreased with the molecular weight when the degrees of polymerization of ODMS segment were more than 3 (Akimoto et al., Citation2001).
To identify enhancers for both hydrophilic and lipophilic drugs, a series of alkyldisiloxanes containing sugar moiety with various alkyl chain lengths were synthesized (Akimoto & Nagase, Citation2003). These novel compounds generally showed good enhancing activity on the transdermal absorption for both hydrophilic and hydrophobic drugs. Their promoting effect was influenced by the alkyl chain length. It was proposed that a suitable balance of polarity would be necessary to obtain a good enhancing ability. Ocyl and decyl derivatives exhibited the best effect. Additionally, the results revealed that these compounds could increase both of the partition and diffusion parameters, and also exhibit fat extraction effect.
From the above, the synthesized copolymers with N-methylpyridinio, quaternary amino, phosphoramide, carboxyl, 1-alkyl-2-pyrrolidon-3-y were proved to be more effective for the lipophilic drugs than the hydrophilic ones, while corresponding PEG and β-D-glucopyranosyl derivatives which possessed more hydrophilic groups had reversed effect. The reason might be that the latter would condense in the SC, alter its solvency to the fat soluble materials and advance the drug partition in the SC. Experiments above suggested that ODMSs was definitely indispensible for enhancing effect regardless of which group was connected to it.
Further consideration
Use of biomaterials as potential penetration enhancers is a new and developing method for enhanced transdermal drug delivery, and there are still many problems that need further research.
Although these biomaterials have frequently been studied for their in vitro activity, relative little is known about their in vivo effect in animals or humans. Due to the complexity of the skin, it is difficult to establish a reliable correlation between the in vitro and in vivo skin permeability. Therefore, more work is necessary to find out the feasibility of these biomaterials for in vivo skin application. In addition, the efficacy of novel penetration enhancers is generally investigated in the solution vehicle. There are few studies revealing that whether these enhancers currently under investigation can be formulated into an acceptable pharmaceutical product, such as patches or semisolid formulations.
Sever skin damage is often caused by the application of penetration enhancers, and biomaterials are no exception. The events occurring in primary skin irritation represent a complex series of chemical and physiologic changes (Bason et al., Citation1991). In general, it is accepted that two different pathogenetic pathways may be involved in the process of skin irritation, firstly via damage to the lipid barrier of the SC and secondly via the direct irritation effect on cells of the skin (Berardesca & Distante, Citation1994; Welss et al., Citation2004). The skin irritation of a penetration enhancer would be likely more reversible if it only interacts with the intercellular lipids of the SC without affecting keratinocytes or others cells in the epidermis and dermis (Zesch, Citation1983). However, the above mentioned biomaterials, such as PTDs and amine-terminated PAMAM dendrimers could generally penetrate into the cells in the epidermis and dermis of the skin, or interact with the keratin of the keratinocytes, which might lead to a major concern about their potential cell toxicity. Therefore, a cellular and molecular insight into the skin irritation would still be required for further study.
The precise mechanism responsible for the skin enhancement of these biomaterials is controversial and may vary among different compounds. Unlike the normal cell membrane, the outmost layer of the skin is composed of dead cells rich in keratin, and the endocytosis of these biomaterials which plays an essential role in transmucosal enhancement is not expected for SC (Lopes et al., Citation2005; Hsu & Mitragotri, Citation2011). Therefore, the lipids and keratins in SC would still be the main acting sites for biomaterial penetration enhancers. The electronic interaction with the tight junction in SC lipid might be responsible for the penetration enhancement of biochemical compounds, such as arginine oligomers, chitosans (Ohtake et al., Citation2003; Taveira et al., Citation2009). As a consequence, the charged properties of these biomaterials are of great significance to the transdermal enhancing activity. Uchida et al. (Citation2011) have demonstrated that increased transdermal siRNA delivery can be achieved by using a tight junction modulator, AT1002. On the other hand, some peptide enhancers such as SPACE, chitosan and trypsin, have also been reported to alter the secondary structure of the keratin in SC to increase the permeability of the skin (He et al., Citation2009; Li et al., Citation2009; Hsu & Mitragotri, Citation2011). Furthermore, the complex interactions between the drug, enhancer and the component in SC are involved in the enhancing process, which could not be explained by mechanistic studies only focusing on the alteration of the SC (Kim et al., Citation2008).
Conclusion
The paper has given an overview of the use of biomaterials as novel skin penetration enhancers. This delivery technology has broad potential for enhancing the penetration of a wide range of therapeutic agents into and across the skin barrier. As a promising approach, biochemical penetration enhancers will continue to be a popular research topic in transdermal and topical drug delivery system. At the same time, additional investigations are still needed to extend the insights into their potential skin irritancy, in vivo efficacy, as well as their enhancing mechanisms.
Declaration of interest
This work was supported by the National Natural Science Foundation of China (No: 30973654 and No: 81173007).
The authors report no declarations of interest.
References
- Akimoto T, Aoyagi T, Minoshima J, Nagase Y. (1997). Polymeric percutaneous drug penetration enhancer: synthesis and enhancing property of PEG/PDMS block copolymer with a cationic end group. J Control Release 49:229–41
- Akimoto T, Kawahara K, Nagase Y, Aoyagi T. (2000). Preparation of oligodimethylsiloxanes with sugar moiety at a terminal group as a transdermal penetration enhancer. Macromol Chem Phys 201:2729–34
- Akimoto T, Kawahara K, Nagase Y, Aoyagi T. (2001). Polymeric transdermal drug penetration enhancer: the enhancing effect of oligodimethylsiloxane containing a glucopyranosyl end group. J Control Release 77:49–57
- Akimoto T, Nagase Y. (2003). Novel transdermal drug penetration enhancer: synthesis and enhancing effect of alkyldisiloxane compounds containing glucopyranosyl group. J Control Release 88:243–52
- Aoyagi T, Akimoto T, Nagase Y. (1992a). Novel silicones for transdermal therapeutic system, IV. Modified route to prepare oligodimethylsiloxane containing cationic end-group and its property as transdermal penetration enhancer. Makromo Chem 193:2821–8
- Aoyagi T, Nakamura T, Yabuchi Y, Nagase Y. (1992b). Novel silicones for transdermal therapeutic system III: Preparation of pyridinio or ammonio-terminated polydimethylsiloxanes and the evaluation as transdermal penetration enhancer. Polymer 24:545–53
- Aoyagi T, Tadenuma R, Nagase Y. (1996). Novel silicones for transdermal therapeutic system, 6. Preparation of oligodimethylsiloxane containing 2-pyrrolidone moiety as a terminal group and its enhancing effect on transdermal drug penetration. Macromol Chem Phys 197:677–86
- Aoyagi T, Takamura K, Nagase Y. (1992c). Novel silicones for transdermal therapeutic system. II. Preparation of oligodimethylsiloxanes containing diethyl phosphoramidate group at the chain end and the the evaluation as transdermal penetration enhancer. Polymer J 24:375–81
- Aoyagi T, Takamura Y, Nakamura T, et al. (1992d). Novel silicones for transdermal therapeutic systems: 1. Synthesis of 1-methyl-4-pyridinio-terminated polydimethylsiloxane and evaluation as a transdermal penetration enhancer. Polymer 33:2203–7
- Aoyagi T, Terashima O, Nagase Y, Matsui K. (1991). Preparation of a polymer containing hexadecylpyridinium bromide groups and its utilization as a transdermal drug penetration enhancer. Polymer 32:2106–11
- Aoyagi T, Terashima O, Suzuki N, et al. (1990). Polymerization of benzalkonium chloride-type monomer and application to percutaneous drug absorption enhancer. J Control Release 13:63–71
- Arima H, Miyaji T, Irie T, et al. (1996). Possible enhancing mechanism of the cutaneous permeation of 4-biphenylylacetic acid by beta-cyclodextrin derivatives in hydrophilic ointment. Chem Pharm Bull 44:582–6
- Babita K, Rana V, Tiwary AK. (2005). Lipid synthesis inhibitors: effect on epidermal lipid conformational changes and percutaneous permeation of levodopa. AAPS PharmSciTech 6:E473–81
- Babita K, Tiwary AK. (2004a). Skin lipid synthesis inhibition: a possible means for enhancing percutaneous delivery of levodopa. Curr Drug Deliv 1:397–403
- Babita K, Tiwary AK. (2004b). Transcutaneous delivery of levodopa: enhancement by fatty acid synthesis inhibition. Mol Pharm 2:57–63
- Babita SG, Tiwary AK. (2002). Role of sphingosine synthesis inhibition in transcutaneous delivery of levodopa. Int J Pharm 238:43–50
- Bason MM, Gordon V, Maibach HI. (1991). Skin Irritation. Int J Dermatol 30:623–6
- Berardesca E, Distante F. (1994). The modulation of skin irritation. Contact Dermatitis 31:281–7
- Borowska K, Laskowska B, Magoń A, et al. (2010). PAMAM dendrimers as solubilizers and hosts for 8-methoxypsoralene enabling transdermal diffusion of the guest. Int J Pharm 398:185–9
- Borowska K, Wołowiec S, Rubaj A, et al. (2012). Effect of polyamidoamine dendrimer G3 and G4 on skin permeation of 8-methoxypsoralene – in vivo study. Int J Pharm 426:280–3
- Bos JD, Meinardi MM. (2000). The 500 Dalton rule for the skin penetration of chemical compounds and drugs. Exp Dermatol 9:165–9
- Brown MB. (2006). Dermal and transdermal drug delivery systems: current and future prospects. Drug Deliv 13:175–87
- Carmichael NM, Dostrovsky JO, Charlton MP. (2010). Peptide-mediated transdermal delivery of botulinum neurotoxin type A reduces neurogenic inflammation in the skin. Pain 149:316–24
- Chauhan AS, Sridevi S, Chalasani KB, et al. (2003). Dendrimer-mediated transdermal delivery: enhanced bioavailability of indomethacin. J Control Release 90:335–43
- Chen Y, Shen Y, Guo X, et al. (2006). Transdermal protein delivery by a coadministered peptide identified via phage display. Nat Biotechnol 24:455–60
- Cheng Y, Man N, Xu T, et al. (2007). Transdermal delivery of nonsteroidal anti-inflammatory drugs mediated by polyamidoamine (PAMAM) dendrimers. J Pharm Sci 96:595–602
- Davaran S, Hanaee J, Rashidi MR, et al. (2006). Influence of poly (ethylene glycol)-γ-cyclodextrin complexes on stabilization and transdermal permeation of ascorbic acid. J Biomed Mater Res A 78:590–4
- de Jager MW, Gooris GS, Dolbnya IP, et al. (2004). Modelling the stratum corneum lipid organisation with synthetic lipid mixtures: the importance of synthetic ceramide composition. BBA-Biomembranes 1684:132–40
- Di Colo G, Burgalassi S, Zambito Y. (2004). Effects of different N-trimethyl chitosans on in vitro/in vivo ofloxacin transcorneal permeation. J Pharm Sci 93:2851–62
- Duncan R, Izzo L. (2005). Dendrimer biocompatibility and toxicity. Adv Drug Deliv Rev 57:2215–37
- Elias PM, Tsai J, Menon GK, et al. (2002). The potential of metabolic interventions to enhance transdermal drug delivery. J Investig Dermatol Symp Proc 7:79–85
- Fawell S, Seery J, Daikh Y, et al. (1994). TAT-mediated delivery of heterologous proteins into cells. Proc Natl Acad Sci USA 91:664–8
- Feingold KR. (2007). Thematic review series: skin lipids. The role of epidermal lipids in cutaneous permeability barrier homeostasis. J Lipid Res 48:2531–46
- Feingold KR. (2009). The outer frontier: the importance of lipid metabolism in the skin. J Lipid Res 50:s417–22
- Filipowicz A, Wołowiec S. (2011). Solubility and in vitro transdermal diffusion of riboflavin assisted by PAMAM dendrimers. Int J Pharm 408:152–6
- Finnin BC, Morgan TM. (1999). Transdermal penetration enhancers: applications, limitations, and potential. J Pharm Sci 88:955–8
- Giuseppina S, Silvia R, Maria CB, et al. (2005). Buccal penetration enhancement properties of N-trimethyl chitosan: influence of quaternization degree on absorption of a high molecular weight molecule. Int J Pharm 297:146–55
- Gonçalves E, Kitas E, Seelig J. (2005). Binding of oligoarginine to membrane lipids and heparan sulfate: structural and thermodynamic characterization of a cell-penetrating peptide. Biochemstry 44:2692–702
- Guillard EC, Tfayli A, Laugel C, Baillet-Guffroy A. (2009). Molecular interactions of penetration enhancers within ceramides organization: A FTIR approach. Eur J Pharm Sci 36:192–9
- Gupta M, Mahajan A, Babita K, et al. (2004). Inhibition of skin sphingosine synthesis: enhanced percutaneous permeation of 5-fluorouracil. Pharmzie 59:212–6
- Hadgraft J. (2004). Skin deep. Eur J Pharm Biopharm 58:291–9
- He W, Guo X, Xiao L, Feng M. (2009). Study on the mechanisms of chitosan and its derivatives used as transdermal penetration enhancers. Int J Pharm 382:234–43
- He W, Guo X, Zhang M. (2008). Transdermal permeation enhancement of N-trimethyl chitosan for testosterone. Int J Pharm 356:82–7
- Ho A, Schwarze SR, Mermelstein SJ, et al. (2001). Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo. Cancer Res 61:474–7
- Hou YW, Chan MH, Hsu HR, et al. (2007). Transdermal delivery of proteins mediated by non-covalently associated arginine-rich intracellular delivery peptides. Exp Dermatol 16:999–1006
- Hsu T, Mitragotri S. (2011). Delivery of siRNA and other macromolecules into skin and cells using a peptide enhancer. Proc Natl Acad Sci USA 108:15816–21
- Jonker C, Hamman JH, Kotzé AF. (2002). Intestinal paracellular permeation enhancement with quaternized chitosan: in situ and in vitro evaluation. Int J Pharm 238:205–13
- Karande P, Jain A, Ergun K, et al. (2005). Design principles of chemical penetration enhancers for transdermal drug delivery. Proc Natl Acad Sci USA 102:4688–93
- Karande P, Mitragotri S. (2009). Enhancement of transdermal drug delivery via synergistic action of chemicals. Biochim Biophysica Acta 1788:2362–73
- Kaushik S, Krishnan A, Prausnitz MR, Ludovice PJ. (2001). Magainin-mediated disruption of stratum corneum lipid vesicles. Pharm Res 18:894–6
- Kim YC, Late S, Banga AK, et al. (2008). Biochemical enhancement of transdermal delivery with magainin peptide: modification of electrostatic interactions by changing pH. Int J Pharm 362:20–8
- Kim YC, Ludovice PJ, Prausnitz MR. (2007). Transdermal delivery enhanced by magainin pore-forming peptide. J Control Release 122:375–83
- Kotzé AF, Luessen HL, Kotzé HL, de Leeuw BJ, et al. (1997). N-trimethyl chitosan chloride as a potential absorption enhancer across mucosal surface: in vitro evaluation in intestinal epithelial cells (Caco-2). Pharm Res 14:1197–202
- Küchler S, Radowski MR, Blaschke T, et al. (2009). Nanoparticles for skin penetration enhancement – a comparison of a dendritic core-multishell-nanotransporter and solid lipid nanoparticles. Eur J Pharm Biopharm 71:243–50
- Kumar S, Sahdev P, Perumal O, Tummala H. (2012). Identification of a novel skin penetration enhancement peptide by phage display peptide library screening. Mol Pharm 9:1320–30
- Legendre JY, Rault I, Petit A, et al. (1995). Effects of β-cyclodextrins on skin: implications for the transdermal delivery of piribedil and a novel cognition enhancing-drug, S-9977. Eur J Pharm Sci 3:311–22
- Li Y, Quan Y, Zang L, et al. (2009). Trypsin as a novel potential absorption enhancer for improving the transdermal delivery of macromolecules. J Pharm Pharmacol 61:1005–12
- Li Y, Quan Y, Zang L, et al. (2008). Transdermal delivery of insulin using trypsin as a biochemical enhancer. Biol Pharm Bull 31:1574–9
- Lim JM, Chang MY, Park SG, et al. (2003). Penetration enhancement in mouse skin and lipolysis in adipocytes by TAT-GKH, a new cosmetic ingredient. J Cosmet Sci 54:483–91
- Loftsson T, Masson M. (2001). Cyclodextrins in topical drug formulations: theory and practice. Int J Pharm 225:15–30
- Loftsson T, Olafsson JH. (1998). Cyclodextrins: new drug delivery systems in dermatology. Int J Dermatol 37:241–6
- Loftsson T, Sigurđardóttir AM, ólafsson JH. (1995). Improved acitretin delivery through hairless mouse skin by cyclodextrin complexation. Int J Pharm 115:255–8
- Loftsson T, Sigurđardóttir AM. (1997). Cyclodextrins as skin penetration enhancers. In: Szejtli J, Szente L, eds. Proceedings of the eighth international symposium on cyclodextrins. Dordrecht, Netherlands: Kluwer Academic Publishers, 403–6
- Lopes L B, Brophy CM, Furnish E, et al. (2005). Comparative study of the skin penetration of protein transduction domains and a conjugated peptide. Pharm Res 22:750–7
- Lopez RFV, Collett JH, Bentley MV. (2000). Influence of cyclodextrin complexation on the in vitro permeation and skin metabolism of dexamethasone. Int J Pharm 200:127–32
- Lv H, Zhang Z, Wang X, et al. (2011). A biomimetic chitosan derivates: preparation, characterization and transdermal enhancement studies of N-arginine chitosan. Molecules 16:6778–90
- Matsuda H, Arima H. (1999). Cyclodextrins in transdermal and rectal delivery. Adv Drug Deliver Rev 36:81–99
- Matsuzaki K, Nakamura A, Murase O, et al. (1997). Modulation of magainin 2-lipid bilayer interactions by peptide charge. Biochemistry 36:2104–11
- Menon GK, Ghadially R, Williams ML, Elias PM. (1992). Lamellar bodies as delivery systems of hydrolytic enzymes: implications for normal and abnormal desquamation. Br J Dermatol 126:337–45
- Menon GK. (2002). New insights into skin structure: scratching the surface. Adv Drug Deliver Rev 54:S3–17
- Mignani S, Kazzouli SE, Bousmina M, Majoral JP. (2013). Expand classical drug administration ways by emerging routes using dendrimers drug delivery systems: a concise overview. Adv Drug Deliv Rev http://dx.doi.org/10.1016/j.addr.2013.01.001.
- Nagase Y, Aoyagi T, Akimoto T, Fuchikami T. (1992). Novel silicones for transdermal therapeutic system, V. A novel method of preparation of carboxy-terminated polydimethylsiloxane and its enhancing effect on transdermal drug penetration. Makromol Chem 13:441–6
- Naik A, Kalia YN, Guy RH. (2000). Transdermal drug delivery: overcoming the skin’s barrier function. Pharm Sci Technol Today 3:318–26
- Nanjwade BK, Bechra HM, Derkar GK, et al. (2009). Dendrimers: emerging polymers for drug-delivery systems. Eur J Pharm Sci 38:185–96
- Nasrollahi SA, Taghibiglou C, Azizi E, Farboud ES. (2012). Cell-penetrating peptides as a novel transdermal drug delivery system. Chem Biol Drug Des 80:639–46
- Ohtake K, Maeno T, Ueda H, et al. (2003). Poly-L-arginine predominantly increases the paracelullar permeability of hydrophilic macromolecules across rabbit nasal epithelium in vitro. Pharm Res 20:153–60
- Orienti I, Zecchi V, Bertasi V, Fini A. (1991). Release of ketoprofen from dermal bases in presence of endrimersns: effect of the affinity constant determined in semisolid vehicles. Arch Pharm 324:943–7
- Pathan IB, Setty CM. (2009). Chemical penetration enhancers for transdermal drug delivery systems. Trop J Pharm Res 8:173–9
- Patil S, Singh P, Szolar-Platzer C, Maibach H. (1996). Epidermal enzymes as penetration enhancers in transdermal drug delivery? J Pharm Sci 85:249–52
- Prausnitz MR, Langer R. (2008). Transdermal drug delivery. Nat Biotechnol 26:1261–8
- Rothbard JB, Garlington S, Lin Q, et al. (2000). Conjugation of arginine oligomers to endrimersn A facilitates topical delivery and inhibition of inflammation. Nat Med 6:1253–7
- Sakuma S, Suita M, Masaoka Y, et al. (2010). Oligoarginine-linked polymers as a new class of penetration enhancers. J Control Release 148:187–96
- Sapra B, Jain S, Tiwary AK. (2008). Transdermal delivery of carvedilol containing glycyrrhizin and chitosan as permeation enhancers: biochemical, biophysical, microscopic and pharmacodynamic evaluation. Drug Deliv 15:443–54
- Schwarze SR, Dowdy SF. (2000). In vivo protein transduction: intracellular delivery of biologically active proteins, compounds and DNA. Trends Pharmacol Sci 21:45–8
- Shaker DS, Ghanem AH, Li SK, et al. (2003). Mechanistic studies of the effect of hydroxypropyl-β-cyclodextrin on in vitro transdermal permeation of corticosterone through hairless mouse skin. Int J Pharm 253:1–11
- Sinha VR, Kaur MP. (2000). Permeation enhancers for transdermal drug delivery. Drug Dev Ind Pharm 26:1131–40
- Sinico C, Fadda AM. (2006). Vesicular carriers for dermal drug delivery. Exp Opin Drug Deliv 6:813–25
- Sun M, Fan A, Wang Z, Zhao Y. (2012). Dendrimer-mediated drug delivery to the skin. Soft Matter 8:4301–5
- Tanner T, Marks R. (2008). Delivering drugs by the transdermal route: review and comment. Skin Res Technol 14:249–60
- Taveira SF, Nomizo A, Lopez RFV. (2009). Effect of the iontophoresis of a chitosan gel on doxorubicin skin penetration and cytotoxicity. J Control Release 134:35–40
- Thanou M, Verhoef JC, Junginger HE. (2001). Chitosan and its derivatives as intestinal absorption enhancers. Adv Drug Deliv Rev 50:S91–101
- Thanou MM, Kotzé AF, Scharringhausen T, et al. (2000). Effect of degree of quaternization of N-trimethyl chitosan chloride for enhanced transport of hydrophilic compounds across intestinal Caco-2 cell monolayers. J Control Release 64:15–25
- Thomas BJ, Finnin BC. (2004). The transdermal revolution. Drug Discov Today 9:697–703
- Tsai JC, Guy RH, Thornfeldt CR, et al. (1996). Metabolic approaches to enhance transdermal drug delivery. 1. Effect of lipid synthesis inhibitors. J Pharm Sci 85:643–8
- Uchida T, Kanazawa T, Takashima Y, Okada H. (2011). Development of an efficient transdermal delivery system of small interfering RNA using functional peptides, Tat and AT-1002. Chem Pharm Bull 59:196–201
- Umemura M, Ueda H, Tomono K, Nagai T. (1990). Effect of diethyl-beta-cyclodextrin on the release of nitroglycerin from formulations. Drug Des Deliv 6:297–310
- Ventura CA, Tommasini S, Falcone A, et al. (2006). Influence of modified endrimersns on solubility and percutaneous absorption of celecoxib through human skin. Int J Pharm 314:37–45
- Venuganti VV, Sahdev P, Hildreth M, et al. (2011). Structure--skin permeability relationship of dendrimers. Pharm Res 28:2246–60
- Venuganti VVK, Perumal OP. (2008). Effect of poly(amidoamine) (PAMAM) endrimers on skin permeation of 5-fluorouracil. Int J Pharm 361:230–8
- Venuganti VVK, Perumal OP. (2009). Poly(amidoamine) dendrimers as skin penetration enhancers: influence of charge, generation, and concentration. J Pharm Sci 98:2345–56
- Vivès E. (2003). Cellular uptake of the Tat peptide: an endocytosis mechanism following ionic interactions. J Mol Recognit 16:265–71
- Walker RB, Smith EW. (1996). The role of percutaneous penetration enhancers. Adv Drug Deliver Rev 18:295–301
- Wang Z, Itoh Y, Hosaka Y, et al. (2003a). Novel transdermal drug delivery system with polyhydroxyalkanoate and starburst polyamidoamine endrimers. J Biosci Bioeng 95:541–3
- Wang Z, Itoh Y, Hosaka Y, et al. (2003b). Mechanism of enhancement effect of endrimers on transdermal drug permeation through polyhydroxyalkanoate matrix. J Biosci Bioeng 96:537–40
- Welss T, Basketter DA, Schröder KR. (2004). In vitro skin irritation: facts and future. State of the art review of mechanisms and models. Toxicol In Vitro 18:231–43
- Wertz PW, van den Beergh B. (1998). The physical, chemical and functional properties of lipids in the skin and other biological barriers. Chem Phys Lipids 91:85–96
- Williams AC, Barry BW. (2004). Penetration enhancers. Adv Drug Deliver Rev 56:603–18
- Yang Y, Sunoqrot S, Stowell C, et al. (2012). Effect of size, surface charge, and hydrophobicity of poly(amidoamine) dendrimers on their skin penetration. Biomacromolecules 13:2154–62
- Zaro J, Shen WC. (2003). Quantitative comparison of membrane transduction and endocytosis of oligopeptides. Biochem Biophys Res Commun 307:241–7
- Zasloff M. (1987). Magainins, a class of antimicrobial peptides from Xenopus skin-isolation, characterization of two active forms, and partial c-DNA sequence of a precursor. Proc Natl Acad Sci USA 84:5449–53
- Zesch A. (1983). Skin irritation by topical drugs. Derm Beruf Umwelt 31:74–8
- Zhou X, Liu D, Liu H, et al. (2010). Effect of low molecular weight chitosans on drug permeation through mouse skin: 1. Transdermal delivery of baicalin. J Pharm Sci 99:2991–8
- Zi P, Yang X, Kuang H, et al. (2008). Effect of HPβCD on solubility and transdermal delivery of capsaicin through rat skin. Int J Pharm 358:151–8