
Abstract
Effective clinical utilization of non-steroidal anti-inflammatory drugs such as diclofenac sodium (DS) is significantly limited by their ulcerogenic potential and poor bioavailability after oral administration, thus necessitating the need for a better carrier to minimize these obvious limitations. The objective of this study was to evaluate Eudragit® RS100/RL100 microspheres formulated by the solvent-evaporation technique for improved delivery of diclofenac. Three batches of (DF1, DF2 and DF3) microspheres were prepared using different ratios of Eudragit RS-100 and RL-100 polymers based on the solvent-evaporation method. The microspheres were characterized based on morphological properties, particle size analysis and encapsulation efficiency (EE%). In vitro release of DS was investigated in both 0.1 N HCl (pH 1.2) and phosphate-buffered saline (pH 7.4), while anti-inflammatory studies were evaluated in the rat model. Maximum EE% of 86.61 ± 0.11, 88.14 ± 0.16 and 85.50 ± 0.21 was obtained for DF1, DF2 and DF3, respectively. Discrete, smooth and brownish microspheres of size range 437 ± 0.01–479 ± 0.21 µm were obtained. Release of DS from the formulation depends on the polymer ratio. All the batches exhibited good anti-inflammatory activities. Microsphere formulations based on Eudragit® polymers would likely offer a reliable and alternative means of delivering DS orally.
Introduction
Diclofenac sodium (DS) is a non-steroidal anti-inflammatory drug that is used to relieve the symptoms of inflammation, stiffness, osteoarthritis and joint pain associated with rheumatoid arthritis (Attama et al., 2008). The therapeutic action of this drug with respect to it analgesic and anti-inflammatory activity is the ability of the drug to inhibit the secretion of arachidonic acid precursor known as prostaglandin (John & Linda, Citation2008). However, the main side effects of the drug especially in case of the long-term use include: abdominal bleeding, pain or cramps, blood clotting problems, peptic ulcers and vomiting. On the other hand, DS dissolves quickly in the intestinal alkaline condition so the effect of the long-term drug uptake is impossible because of the short biological half-life (Davies et al., Citation1997). Thus, total bioavailability of orally administered DS has few important challenges that cannot be clinically ignored. Several preparations of the DS were prepared to overcome the above short comings such as topical gel, cream and other topical applications (Zuniga et al., Citation2004).
Despite the improvement in the topical preparation of diclofenac, there is still much need for oral application as a result of obvious clinical needs such as post-operative pain, neuromuscular bone pain and its associated problem. Attempt to prevent the oral side effect led to the formulation of controlled release formulation of diclofenac. However, this formulation only reduced the gastrointestinal (GI) side effect especially for patient on short-term regimen. However, for patient on long-term regimen or in chronic case, the side effect is still much of health concern and need to be resolved. Based on this fact, alternative formulation becomes necessary for effective and more efficient delivery of this drug. More so, the oral route is widely accepted as the most common method of drug administration into the body due to accessibility, convenience, possibility of repeated self-administration by patients and the opportunity to achieve optimum absorption based on the large surface area available in the GI tract (Kidane & Bhatt, Citation2005).
Among various oral sustained drug delivery systems, polymeric microspheres are one of the options and have been studied in past few decades in order to achieve targeted delivery of drug molecules with associated benefits, including better oral bioavailability of drugs, reduction in side effects, decreased dosing frequency, and hence, improved patient compliance (Freiberg & Zhu, Citation2004; Basu & Adhiyaman, Citation2008; Desai et al., Citation2009).
Microspheres are considered to be one of the most promising drug delivery system targeted to prolong and control the delivery of certain drug molecule (Tanwar et al., Citation2007; Berchane et al., 2007). It has been previously mentioned that this delivery system has the capability to improve bioavailability and stability, and to target drug to a particular site in the body. Other advantages of this delivery system include limiting fluctuation within therapeutic range, reducing side effects, decreasing dosing frequency and improving patient compliance (Ofokansi & Adikwu, Citation2007; Ttrivedi et al., Citation2008). Several published works on polymeric materials used in the preparation of microsphere for controlled drug delivery were evaluated. It was found that Eudragit® polymers were more favored in the formulation of pH-sensitive drug molecules. The obvious advantages of these polymers include pH-independent release profiles; encapsulation of high amount of drug; release of the incorporated drug in controlled manner and high level of stability (Kishori et al., Citation2013). Generally, Eudragits® are copolymers of acrylic and methacrylic acid esters with quaternary ammonium groups. The ammonium groups are present as salts and make the polymers permeable (Haznedar & Dortunc, Citation2004).
In this work, a mixture of Eudragit® RL-100 and Eudragit® RS-100 was used to formulate microspheres by the solvent-evaporation technique for controlled delivery of DS, a model anti-inflammatory drug suitable for per oral administration. Liquid paraffin and acetone system were used for the preparation of microspheres. Magnesium stearate was used as a droplet stabilizer to prevent droplet coalescence in the oil medium and n-hexane was added as a non-solvent to the processing medium to solidify the microspheres. The effect of formulation factors such as polymer ratio of the microspheres was investigated. The prepared microspheres were evaluated for micromeritic properties, drug encapsulation, thermal properties, yield of production, particle size and size distribution, morphological study and in vitro drug release. The anti-inflammatory property of the formulations was also evaluated in the rat model.
Materials and methods
Materials
DS was a gift from AC Drugs Ltd. (Enugu, Nigeria). Eudragit® RS-100 and RL-100 were gifts from Evonik, GmbH (Darmstadt, Germany). Magnesium stearate was purchased from Acros Organics (MD, USA). Acetone, concentrated HCl, disodium hydrogen phosphate and sodium chloride (BDH, Poole, England), and all other reagents, and solvents were of analytical grade and were used without further purification. Distilled water was obtained from an all-glass still.
Preparation of microspheres
DS microspheres were prepared by the solvent-evaporation technique. Different ratios of Eudragit® RS and RL were dissolved in 15 ml acetone (solvent phase) with the help of magnetic stirrer. DS (50 mg) and magnesium stearate (100 mg), as a dispersing agent, were dispersed separately in 10 ml acetone and stirred as described earlier. These were added to the polymer solution, the total volume of acetone used was 25 ml. The resulting dispersion was emulsified drop-wise into 250 ml beaker, containing the mixture of 270 ml liquid paraffin light and 30 ml n-hexane-containing span 60, with gentle stirring. The emulsion was stirred with a mechanical stirrer with a double blade of 3 cm in diameter (Gallenport rotors, Germany) at 700 rpm for 1.5 h, until acetone evaporated completely. The microspheres formed were filtered using Whatman no.1 filter paper, washed three times with n-hexane and dried in a vacuum at room temperature for 24 h. Finally, the microspheres were collected and stored in a desiccator over fused calcium chloride for further use. Three different batches of the microspheres containing 1:1, 1:2 and 2:1 of Eudragit® RS:RL were formulated following the same procedure and labeled as DF1, DF2 and DF3, respectively. Unloaded sample (without drug) was similarly prepared with 1:1 ratio of Eudragit® and coded DF0.
Characterization of the microspheres
Differential scanning calorimetry
Thermal analysis and changes in heat capacity of the Eudragit® matrix and individual Eudragit® were determined using a differential scanning calorimeter (DSC) (NETZSCH DSC 204 F1, Germany). Approximately 4–5 mg of the individual Eudragit® and their formulations were weighed into an aluminum pan, sealed hermetically, and the thermal behavior determined in the range of 20–220 °C at a heating rate of 5 °C/min and then cooled back to 20 °C. The baselines were determined using an empty pan, and all the thermograms were baseline-corrected.
Micromeritic properties of microspheres
The flow properties of prepared microspheres were investigated by measuring the bulk density, tapped density, Carr’s index and packing factor. The bulk, tapped densities, flow rate were measured using standard methods (Marshal, 1987).
Determination of percentage yield
The formulated microspheres were recovered and weighed accurately. The yield of microspheres was determined by comparing the whole weight of formed microspheres with the combined weight of the copolymer and drug using the equation below:
Particle size analysis and morphology
The particle size and size distribution of the microspheres were evaluated using the sieve method. Standard sieves of various apertures (50–850 µm) were arranged starting from the smallest size to the biggest size upward. In each case, approximately 5.0 g quantity of the prepared microspheres was placed on the top sieve (biggest size) and shaken for 10 min, using a mechanical shaker (Geologists Syndicate Pvt. Ltd., India). The quantity of the microspheres retained on each sieve was then evaluated as the percentage of the initial amount of the microspheres used in the evaluation.
Encapsulation efficiency of the microspheres
Approximately 50 mg of the prepared microspheres was weighed and crushed into fine powder by using a dispensing mortar and pestle. This powder was added to 5 ml of ethanol and was allowed to dissolve completely and 5 ml of phosphate buffer (pH 7.4) was added to this solution and mixed thoroughly. The resulting solution was analyzed for DS content by measuring absorbance in a UV-spectrophotometer (Jenway, Model 6405 Barloworld Scientific Ltd., England) at 238 nm. The amount of drug encapsulated in the microspheres was calculated by referring to a standard Beer’s plot and the encapsulation efficiency (EE) obtained using the following formula:
In vitro drug release study
DS release from the microspheres was evaluated in 0.1 N HCl (pH 1.2) and phosphate-buffered saline (PBS, pH 7.4) at 37 ± 1 °C using USP dissolution apparatus (Erweka, Germany). The polycarbonate dialysis membrane used as release barrier was pre-treated by soaking it in the dissolution medium for 24 h prior to the commencement of each release experiment. In each case, approximately 50 mg microspheres were placed in dialysis bag containing 5 ml release medium and tied to the paddle of dissolution apparatus. The rotation of paddle was adjusted to 50 rpm and 500 ml release medium was used. At predetermined time intervals, 2.5 ml volume of the release medium was withdrawn and replaced with equal amount of freshly prepared medium, to maintain a sink condition. The collected samples were filtered through 0.45 μm pore size Millipore filters (India) and analyzed spectrophotometrically (Jenway, Model 6405 Barloworld Scientific Ltd., England) at 238 nm. The amount of drug released at each time interval was determined with reference to the standard Beer’s plot for DS in phosphate buffer. This test was carried out in triplicate for all the batches.
Kinetic analysis of in vitro release profiles
The in vitro release data were analyzed using various kinetic models. The zero-order rate Equation (3) describes the systems where the drug release rate is independent of its concentration. The first order Equation (4) is the opposite of zero order, i.e. the release rate is concentration dependent. Higuchi is usually used to explain the release of drugs from insoluble matrix as a square root of time Equation (5). The Hixson–Crowell cube root law Equation (6) describes the release from systems where there is a change in surface area and diameter of particles or tablets.
where Q is the percent of drug released at time t, Q0 is the percent of drug remaining and K0, K1 and KH are the coefficients of the equations. Kp is constant incorporating structural and geometric characteristics of the release device, and n is the release exponent indicative of the mechanism of release.
Pharmacodynamic evaluation: in vivo anti-inflammatory efficacy
The anti-inflammatory activity of the DS-loaded microspheres was evaluated using the rat paw oedema test (Sinye et al., Citation2013). All animal studies were performed in accordance with the declaration of Helsinki and the European Community guidelines for the handling and use of laboratory animals. The protocols were approved by the Institutional Animal Care and Use Committee of the Faculty of Pharmaceutical Sciences, University of Nigeria. Adult Wistar rats of either sex (210–220 g) were divided into five groups of five animals per group. The animals were fasted and deprived of water for 12 h prior to the experiment. Microspheres equivalent to DS (20 mg/kg body weight) was administered orally to the rats. The reference group received 20 mg/kg of pure sample of DS, while the control group received normal saline 5 ml/kg. Thirty minutes post-treatment, 0.1 ml of 1% carrageenan in normal saline was injected into the sub-plantar surface of the left hind paw of the rats. The volumes of injected paws were measured before, and 1–8 h after injection of carrageenan by the plethysmographic method using plethysmometer (Ugo-basile, MD, USA). Inhibition of oedema was calculated using the following equation:
where Ic is the edema rate of the control group and It is the edema rate of the treated group.
Data and statistical analysis
The data were expressed as mean ± standard deviation (n = 3). The significance of the data was processed using a one-way analysis of variance. Results were considered significant at p < 0.05.
Results and discussion
DS-loaded microspheres based on a mixture of Eudragit® RS-100 and RL-100 polymer matrix were prepared by the emulsification solvent-evaporation technique. The effect of polymer ratio on the various parameters of the formulation evaluated and its ability to preserve the activity of the incorporated drug were investigated.
Thermal properties of the polymer and the loaded formulations
The melting peaks for Eudragit® RS are 60.2 °C and 81.8 °C, whereas the corresponding enthalpy was −6.052 and −4.834 mW/mg, respectively. Eudragit® RL showed similar melting endothermic peaks of 61.1 °C and 85.0 °C with corresponding enthalpy of −7.205 and −7.431 mW/mg, respectively. The polymers seem to be amorphous in nature. However, 1:1 ratio mixture of the polymer (Eudragit® RS/RL = unloaded or no drug) that was used as the matrix base showed two different melting endothermic peaks () of 61.1 °C and 85.0 °C with corresponding two different enthalpies of −7.205–7.431 mW/mg, respectively (). This is an indication that the matrix base lower the crystallinity of the polymer due to disorientation of the structure of the polymer thereby create numerous space for possible drug entrapment. However, the DSC trace of pure DS powder () shows a peak at 90.0 °C with an enthalpy of −32.79 mW/mg. The formulated microspheres based on varying ratios of polymer matrix showed different DSC characteristics. Microsphere formulated with 1:1 (DF-1) ratio of RS/RL loaded with 50 mg of DS gave two different melting endothermic peaks () of 62.9 °C and 78.5 °C with a corresponding enthalpy of −4.319 and −2.978 mW/mg, respectively, whereas microsphere formulated base on 1:2 (DF-2) showed a single melting peak () of 62.2 °C with an enthalpy of −3.328 mW/mg. Batch formulated with 2:1 of RS/RL (DF-3) gave two varying melting endothermic peaks () of 62.7 °C and 204.6 °C with corresponding enthalpy of −2.342 and −2.879 mW/mg, respectively (). The implication of the DSC characteristics implies that lower enthalpy as seen in this formulation suggests less crystallinity and the possibility for retention of an entrapped drug over time, which is one of the goals of this research study. For the purpose of comparison () showed the combined thermographs of the DSC studied.
Figure 1. DSC thermogram of unloaded matrix (DF-0) contain (RS/RL 1:1 without drug).
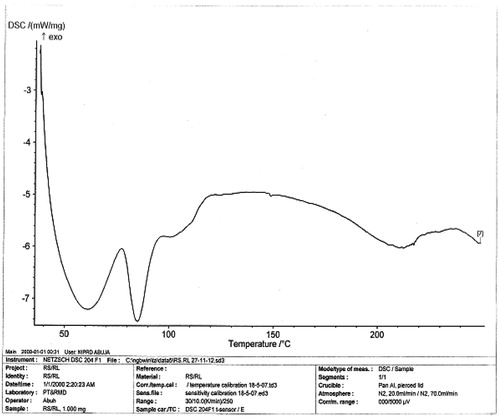
Figure 2. DSC thermogram of pure DS.
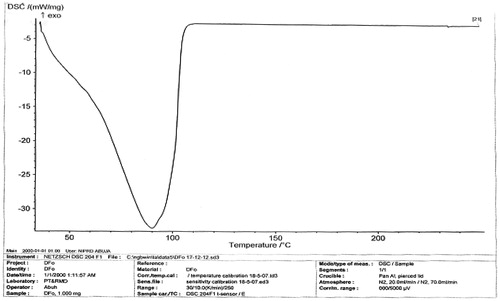
Figure 3. DSC thermogram of microsphere (DF-1) contains RS/RL (1:1 plus 50 mg of DS).
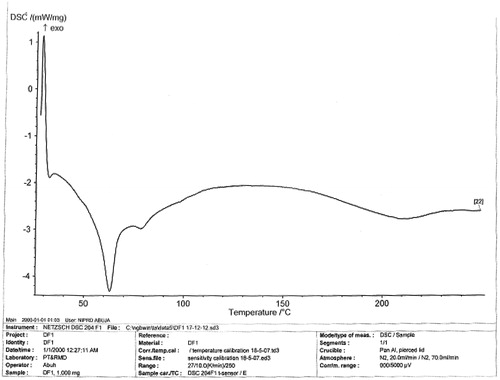
Figure 4. DSC thermogram of microsphere (DF-2) contains RS/RL (1:2 plus 50 mg of DS).
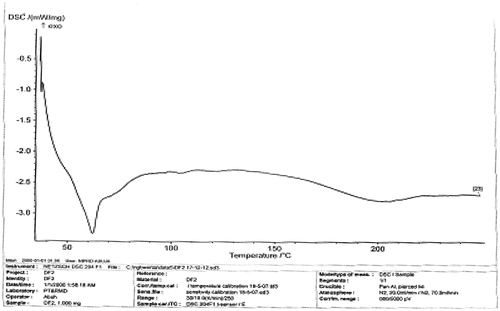
Figure 5. DSC thermogram of microsphere (DF-3) contains RS/RL (2:1 plus 50 mg of DS).
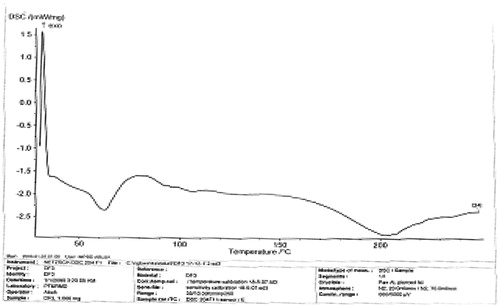
Figure 6. DSC thermograms (overlayed) of the various formulations.
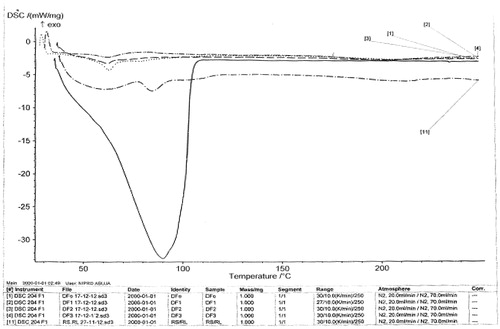
Table 1. Thermal properties of DS microspheres.
Physical flow properties of microspheres
The physical properties that determine the flow characteristics of microspheres are expressed in terms of Carr’s index and angle of repose (). The Carr’s indices for all formulations were less than 20, which indicate an excellent flow compared with the original drug crystals. However, DF-0 which contained no drug gave CI greater than 20. This may be attributed to the agglomeration of the polymer due to interparticulate interaction of the polymer. Also, the microspheres were found to exhibit higher packing properties than the unloaded batch DF0. The improvement in flow properties suggests that the microspheres can be easily handled during processing and formulation.
Table 2. Physical characteristics and micromeritics profiles of the microspheres.
The bulk and tapped densities as well as the compressibility index and angle of repose of the microspheres formulated with different ratios of the Eudragit® RS/RL-loaded DS are presented in . The polymer type and ratio mixtures used for the preparation of the microspheres as well as the sieve screening processes affected the bulk and tapped densities and compressibility index of the formulations, which ultimately affected the angle of repose. The bulk and tapped densities of the formulated microsphere in the batch DF-2 RS/RL (1:2) were slightly lower than DF-1, DF-3 and the unloaded batch DF-0. This may be as result of the polymer combination and the ratio used. The permeability of the polymer may also have contributed, since the particle size depends more on the nature of the polymer (Staniforth, Citation2003). All these affected the structural composition and the interaction during the formulation processing.
There were also slight variation in the compressibility indices and the angle of repose of the loaded microsphere in comparison with unloaded batch (DF-0). The difference in compressibility index and angle of repose among the loaded microsphere DF-1, DF-2 and DF-3 was insignificant (p > 0.05). This indicates that the method of this formulation did not affect the microspheres. The slight variation observed in the physical parameters of the microspheres, i.e. the bulk and tapped densities, and the compressibility indices and angle of repose may be attributed to the variation in the ratio and type of the polymer. Of most important is the structural difference in the Eudragit® RS and RL, and partly the process of drying and the sieving method.
In all cases, the yield of the microspheres from all the formulations as presented in was generally high indicating that the formulation procedures and parameters employed in formulating the microspheres are very effective and efficient. The percentage loss was less than 3–5%, this might arise during the filtration process, transferring or during drying.
Particles size analysis and morphology
The particle size ranges of the microspheres were narrow and showed an insignificant different among the loaded microspheres, whereas there was a significant (p < 0.05) differences between the loaded microspheres in comparison with the unloaded batch (DF-0). Particle size played a significant role in the release of drug as well as in drug stability. The particle sizes of the microspheres as measured by digital photomicroscope were in the range of 437 ± 0.01 to 479 ± 0.21 µm for loaded microspheres and 398 ± 0.61 µm for the unloaded microspheres. The microspheres formulated with Eudragit® RS/RL in the ratio of 2:1 had the highest particle size compared with other formulations.
Photomicroscope studies are presented in , DF-0–DF-3 shows that particles made of Eudragit RL100 and RS100 were spherical and not aggregated. The surface of the drug-loaded microspheres manifested the presence of drug particles, clearly visible from outside at high magnification (DF-0–DF-3). However, according to the type and concentration of the polymer, there were no significant differences in morphology of the formulated microspheres (DF-0–DF-3). The surface morphology and the internal structures of DS-loaded microspheres showed a typical porous structure.
Figure 7. (DF0-DF3) Photomicrographs of representative batches of microspheres containing different ratios of RS:RL 100 and 50 mg of DS: DF-1, DF-2 and DF-3 contain 1:1, 1:2 and 2:1 of polymer ratio, respectively.

Encapsulation efficiency
The drug EE increased with increasing concentration of the Eudragit® RL as seen in batch DF-2 such that the maximum percentage drug encapsulation was 88.14 ± 0.16% (DF-2), whereas the minimum percentage drug encapsulation of 85.50 ± 0.21% was found in batch DF3 as shown in . It shows that the span-60 which serves as the solubilizer, magnesium stearate, in addition to the Eudragit® polymer-enhanced drug solubilization especially when the Eudragit® RL was increased. The obvious explanation for this effect was the permeability nature of Eudragit® RL which is easily hydrated thereby incorporating more drug than Eudragit® RS polymer. In all cases, the polymer matrix accommodated high amount of drug, an indication that the choice of the polymer was well selected for this formulation. The low crystallinity of the matrix as shown in the DSC results is also an added advantage to these polymers.
In vitro release study
The in vitro release study was carried out by using two different media, i.e. PBS (pH 7.4) and 0.1 N HCl (pH 1.2) to mimic the GI conditions. In each case, the release study was evaluated for 18 h. The release of DS in the acidic condition was found to be low in all cases, Eudragit® RS/RL in ratio (1:2) DF-2 had the highest amount of drug released followed by (1:1) DF-1 and least in (2:1) DF-3. The maximum amount of DS released was found to be 24% and the least was 19% at the end of 18 h in 0.1 N HCl (pH 1.2), shown in ; whereas in the PBS (pH 7.4), the reverse was the case with respect to the maximum amount of drug released. Maximum release was obtained in Eudragit® RS/RL mixed in a ratio of (1:2) compared with (1:1) and (2:1) as depicted in . However, contrary to our earlier preliminary study using either of the polymers alone, it was observed that mixtures of Eudragit® RS100 and Eudragit® RL100 showed greater effect on the release rate of the drug. Increasing the concentration of Eudragit® RL100 caused an increase in release of the drug which was significant (p < 0.05). Similarly, ratio 1:1 of Eudragit® RS/RL followed the same trend, but less amount of drug was released compared with Eudragit® 1:2 (DF-2). However, the release was significantly (p < 0.05) different from when the Eudragit® RS/RL was combined in 2:1 as seen in batch DF-1. This variation may be as a result of the amount of quaternary ammonium compound in Eudragit® RL-100 is higher (10%) compared with Eudragit® RS-100 (5%), which makes it more permeable to the dissolution medium.
Figure 8. In vitro release of DS from Eudragit RS/Eudragit RL microspheres in pH 1.2 buffer solution: DF-1 = (◊) RS/RL (1:1) + 50 mg of DS, DF-2 = (×)RS/RL (1:2) + 50 mg of DS and DF-3 = (▴) RS/RL (2:1) + 50 mg of DS. Each data point represents the mean ± SEM from n = 3.
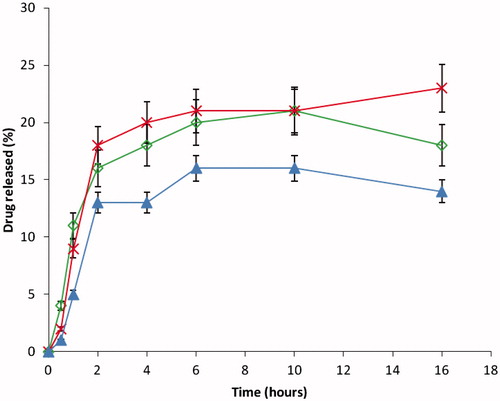
Figure 9. In vitro release of DS from Eudragit RS/Eudragit RL microspheres in pH 7.4 buffer solution: DF-1 = (◊) RS/RL (1:1) + 50 mg of DS, DF-2 = (×) RS/RL (1:2) + 50 mg of DS and DF-3 = (▴) RS/RL (2:1) + 50 mg of DS. Each data point represents the mean ± SEM from n = 3.
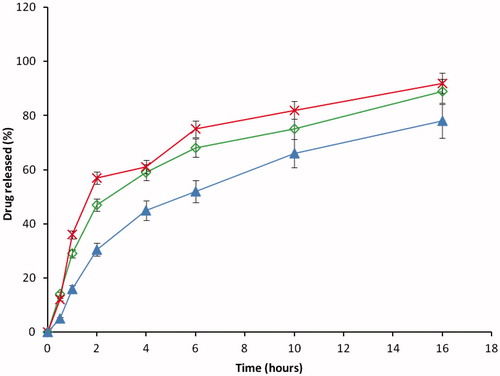
On the other hand, the increase in Eudragit® RS concentration in the DF-3 led to the formation of highly compacted microspheres, which resulted in an increased diffusional path length and consequent retardation in the release of the encapsulated drug. It was observed that the increase in the diffusional length prevented the ease of drug migration as a result of polymer-drug complex formation, which takes longer time to hydrate before the drug can be expelled or migrate from the core matrix. The results here are in agreement with earlier research (Sunit et al., Citation2005; Bipul et al., Citation2011), as they also based their finding on the variation on the ammonia component in the Eudragit® RS and RL. The general dividing rule here is that the DS had a better release in PBS (pH 7.2) than in 0.1 N HCl (pH 1.2) and that the release depends on the combination ratio of the polymer matrix.
The results of the kinetic analysis of the in vitro release of the DS from the prepared microspheres are shown in . The results revealed that, in all cases, the various batches of the formulation predominantly followed the Higuchi model of release. However, DF2 followed both the Higuchi and Hixson–Crowell models of release. This indicates that the DS encapsulated in the polymer matrix were disorientated when in contact with liquid medium and allowed the DS to diffuse out of the hydrated Eudragit® matrix. More so, all the formulation DE1–DF3 showed affinity toward Higuchi kinetic release, showing that diffusion-controlled release mechanism predominated. Although, the Eudragit® matrix was not soluble in water, but ability to hydrate in the medium paved way for the drug to diffuse due to structural relaxation that led to the formation of pores through which the drug eventually diffused in a controlled manner. The variation in the kinetic results is traceable to the various ratios of polymer used in the formulation.
Table 3. Kinetics of release of diclofenac from microspheres in phosphate buffer (7.4).
Anti-inflammatory properties
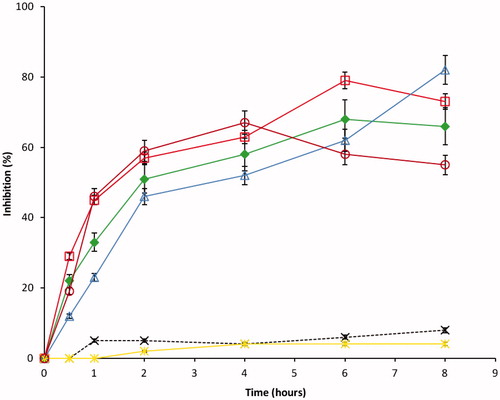
In all cases, the anti-inflammatory results presented, in , show that DS-loaded microspheres exhibited good anti-inflammatory activity that was significantly higher than that of the control group (p < 0.05). The oedema maximum inhibition ranged from 35.3% to 53.3% for DS-loaded microspheres (DF-1–DF-3) while the reference drug showed inhibition of 25–47%. The DS-loaded microspheres showed higher inhibition of oedema than the reference 25–47% for positive control (MKT-S) and 2–7% for negative control (NS). This may be due to increased absorption of drug in vivo in the presence of the Eudragit® matrix used as the carrier. The anti-inflammatory profiles were observed to be dependent on the composition of the microspheres. Microspheres prepared from combinations of Eudragit® RS and Eudragit® RL in a ratio of (1:2) showed a greater percentage of anti-inflammatory effect which was significant (p < 0.05) in comparison with those prepared from a ratio of 1:1 and 2:1 of Eudragit® RS/RL. The in vivo result supports our findings in the in vitro study; this effect could be due to high permeability action of the Eudragit® RL polymer that allowed the ease of diffusion of the drug into the biological system and subsequently enhances drug absorption. The poor permeability of the Eudragit® RS polymer also manifested in the in vivo study, it shows that the slow action of batch DF-3 on the inflammatory site may be due to the amount of the drug that was able to release at the initial stage. However, there was a sharp change at 6–8 h, where the DF-3 maintained a steady increase in the activity compared with the DF-2 and DF-1.
Figure 10. Anti-inflammatory profiles of the formulations: DF-1 = (▪) RS/RL (1:1) + 50 mg of DS, DF-2 = (□) RS/RL (1:2) + 50 mg of DS, DF-3 = (Δ) RS/RL (2:1) + 50 mg of DS, DF-0 = (×) RS/RL (1:1) no drug, NS = (*) normal saline and MKT-S = (0) market sample. Each data point represents the mean ± SEM from n = 3.
Conclusions
Polymeric carriers could be exploited to improve the delivery of therapeutic molecules. In this study, DS-loaded microspheres based on biodegradable Eudragits were successfully prepared by the solvent-evaporation technique, which is simple, cheap and reproducible. All the formulations showed good physicochemical properties. The release of drug from microspheres based on Eudragit® RS and RL (2:1) was very slow compared with Eudragit® RS-100 and RL-100 (1:2). The anti-inflammatory properties of the drug were retained in all the formulations. It follows that this delivery system could offer a better and more promising approach for the treatment of inflammation than the conventional formulation.
Declaration of interest
The authors state no conflicts of interest and have received no funding from any agency neither from an individual for the research or in the preparation of this manuscript.
References
- Attama AA, Reichl S, Müller-Goymann CC. (2008). Diclofenac sodium delivery to the eye: in vitro evaluation of novel solid lipid nanoparticle formulation using human cornea construct. Int J Pharm 355:307–13
- Basu SK, Adhiyaman R. (2008). Preparation and characterization of nitrndipine-loaded Eudragit RL 100 microspheres by an emulsion-solvent evaporation method. Trop J Pharm Res 7:1033–41
- Berchane NS, Carson KH, Rice-Ficht AC, et al. (2007). Effect of mean diameter andpolydispersity of PLG microspheres on drug release: experiment and theory. Int J Pharm 337:118–26
- Bipul N, Lila K, Pradeep K. (2011). Preparation and in vitro dissolution profile of zidovudine loaded microspheres made of Eudragit RS 100, RL 100 and their combinations. Acta Poloniae Pharmaceutica n Drug Res 68:415--422
- Davies NM, Sharkey KA, Asfaha S, et al. (1997). Aspirin causes rapid up-regulation of cyclooxygenase-2 expression in the stomach of rats. Aliment Pharmacol Ther 11:1101–8
- Desai JV, Patil JS, Kulkarni RV, et al. (2009). Alginate-based microparticulate oral drug delivery system for rifampicin. Res J Pharm Tech 2:301–3
- Freiberg S, Zhu XX. (2004). Polymer microspheres for controlled drug release. Int J Pharm 2:1–18
- Haznedar S, Dortunç B. (2004). Preparation and in vitro evaluation of Eudragit microspheres containing acetazolamide. Int J Pharm 269:131–40
- John LW, Linda V. (2008). NSAID-induced gastrointestinal damage and the design of GI-sparing NSAIDs. Curr Opin Invest Drugs 9:1151–6
- Kidane A, Bhatt PP. (2005). Recent advances in small molecule drug delivery. Curr Opin Chem Biol 9:347–51
- Kishori LD, Nilima AT, Paraag SG. (2013). Formulation and development of tinidazole microspheres for colon targeted drug delivery system. J Pharm Res 5:158–60
- Marshal K. (1987). Compression and consolidation of powdered solids. In: Lachman L, Lieberman HA, Kanig JL, eds. Theory and practice of industrial pharmacy, 3rd ed. (Indian ed.), Bombay: Varghese Publishing House, 66–7
- Ofokansi KC, Adikwu MU. (2007). Formulation and evaluation of microspheres based on gelatin-mucin admixtures for the rectal delivery of cefuroxime sodium. Trop J Pharm Res 6:825–32
- Sunit KS, Abdul AM, Barik BB, et al. (2005). Formulation and in vitro evaluation of Eudragit microspheres of stavudine. Trop J Pharm Res 4:369–75
- Sinye AB, Salome AC, Anthony AA, et al. (2013). In vitro and in vivo characterisation of piroxicam-loaded dika wax lipospheres. Trop J Pharm Res 12:33–8
- Staniforth, J. (2003). Particle-size analysis. In: Aulton ME, ed. The science of dosage form design, 2nd ed. Toronto: Churchill Livingstone, 152–65
- Tanwar YS, Naruka PS, Ojha GR. (2007). Development and evaluation of floating microspheres of verpamil hydrochloride. Braz J Pharm Sci 43:529–34
- Ttrivedi P, Verma AML, Garud N. (2008). Preparation and characterization of aceclofenac microspheres. Asian J Pharm 2008:110–5
- Zuniga JR, Phillips CL, Shugars D, et al. (2004). Analgesic safety and efficacy of diclofenac sodium softgels on postoperative third molar extraction pain. J Oral Maxillofac Surg 62:806–15