
Abstract
Context: Zidovudine (AZT) is employed against AIDS and hepatitis; its use is limited by active efflux transporters (AETs) that induce multidrug resistance for intracellular therapies and hamper AZT to reach the brain. Ursodeoxycholic acid (UDCA) conjugation with AZT (prodrug UDCA–AZT) allows to elude the AET systems.
Objective: To investigate the effect of the Pluronic F68 coating on the loading, release and stability of poly(D,L lactide-co-glicolide) nanoparticles (NPs) embedded with UDCA–AZT.
Materials and methods: The mean diameter of the NP prepared by nanoprecipitation or emulsion/solvent evaporation methods was determined using both photon correlation spectroscopy and sedimentation field–flow fractionation; particle morphology was detected by scanning electron microscope. The stability of the free and encapsulated UDCA–AZT was evaluated in rat liver homogenates by high-performance liquid chromatography analysis.
Results and discussion: The mean diameter of the NPs was found to be ∼600 nm with a relatively high polydispersity. The NPs obtained by emulsion/solvent evaporation were not able to control the prodrug release, differently from NPs obtained by nanoprecipitation. The presence of the Pluronic coating did not substantially modify the kinetics of the drug release, or the extent of the burst effect that were instead only influenced by the preparation parameters. UDCA–AZT incorporated in the NPs was more stable in the rat liver homogenates than the free prodrug and no influence of the Pluronic coating was observed.
Conclusions: Considering the different potential applications of nanoparticles coated and uncoated with Pluronic (brain and macrophage targeting, respectively), both of these nanoparticle systems could be useful in the therapies against HIV.
Introduction
Poly(d,l-lactide-co-glicolide) (PLGA) belongs to a family of biodegradable polymers approved by US FDA for the use of drug delivery (Makadia & Siegel, Citation2011) and widely used to prepare micro- and nanoparticles. PLGA is biocompatible and it is eliminated from the body through natural pathways, being degraded to the lactic and glycolic acids (Jalil, Citation1990; Johansen et al. Citation2000; Bala et al., Citation2004). In the field of biomedical applications, the submicron size of the nanoparticles (NPs) can allow their administration by a variety of routes, such as intravenous, oral, nasal, ocular and transdermal (Chen et al., Citation2012; Danhier et al., Citation2012; Tomoda et al., Citation2012). However, NPs can have some limitations due to their small size and large surface area that can limit drug loading and cause relative high burst release (Leo et al., Citation2006b; Nafee et al., Citation2009). Indeed, NP size greatly affect drug release rate. Generally, smaller NP size leads to a shorter average diffusion path of the matrix-entrapped drug molecules (Chorny et al., Citation2002) and can lead to particle–particle aggregation, making physical handling of NP difficult in liquid and dry forms (Contado et al., Citation2013). On the other hand, the increase in the particle size seems to be associated mainly with extended porosity of the polymeric matrix owing to the enhancement of the drug loading. This effect overcompensates the effect of the increasing diffusion pathways with increasing NP radius, resulting in increased drug release with increasing device dimension (Siepmann et al., Citation2004). Encapsulation studies in PLGA nanoparticulate systems are therefore of primary importance, in particular for those drugs whose therapeutic effect and site-specific targeting needs are strictly related.
In this article we report an encapsulation study in PLGA NP of a prodrug of zidovudine (AZT) obtained by conjugation with ursodeoxycholic acid (UDCA). AZT is currently employed in therapeutic protocols that use multiple drug combinations against HIV (De Clerk, Citation2009) but, unfortunately, it is substrate of active efflux transport (AET) systems that cause its efflux in the bloodstream from the membranes inducing, therefore, multidrug resistance (MDR) for intracellular therapies (Liang, Citation1987; Vyas et al., Citation2006) and the inability of the drug to reach efficacious concentrations in the CNS (Wong et al., Citation1993; Takasawa et al., Citation1997; Pardridge, Citation2003).
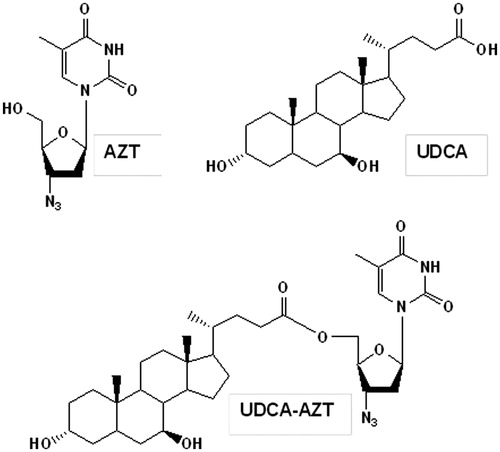
We have recently evidenced that the ester conjugation of AZT with UDCA allows obtaining a prodrug (UDCA–AZT; ) that is able to elude the AET systems (Dalpiaz et al., Citation2012). This prodrug appears, therefore, an interesting substrate for encapsulation studies in PLGA nanoparticulate systems potentially able to induce its delivery in specific sites of the body. Indeed, loaded UDCA–AZT NP may contribute to increase the AZT concentration in its therapeutic sites, where the delivered prodrug should not be effluxed by the AET systems.
Figure 1. Chemical formula of AZT, UDCA and their prodrug UDCA–AZT obtained by ester-conjugation.
In this article, we propose the formulation of UDCA–AZT-loaded NP by nanoprecipitation or by emulsion–solvent evaporation methods in presence of Pluronic F-68, a nonionic bifunctional triblock copolymer surfactant used primarily in pharmaceutical and cosmetic formulations as emulsifier (HLB = 29) under the trade name of Poloxamer 188, which has recently attracted a considerable interest as vehicle for diagnostic and therapeutic agents (Batrakov & Kabanov, Citation2008). Taking into account the potential targets of the prodrug UDCA–AZT, the NPs have been produced and characterized in both unpurified and purified forms. The unpurified form has been chosen taking into account that PLGA NP coated with Pluronic F-68 can induce long circulating proprieties and drug delivery in the brain (Gelperin et al., Citation2010). On the other hand, purified NP may be useful for the prodrug targeting in another activity site of AZT, i.e. macrophages (Owens III & Peppas, Citation2006), which when infected by HIV produces and harbors the virus for a long period (Carter & Ehrlich, Citation2008).
The size of the purified and unpurified NP has been evaluate by photon correlation spectroscopy (PCS) studies supported by sedimentation field flow fractionation (SdFFF) technique, whose employment appeared of great utility to identify the aggregation degree of the nanoparticulate systems. NPs were then characterized by evaluating their prodrug loading, their morphology and size and their ability to control the UDCA–AZT release. Finally, in the aim to verify the ability of the NP to stabilize the prodrug in physiological environments, we have considered that rat liver homogenates induce high instability of UDCA–AZT, causing its hydrolysis in few minutes (Dalpiaz et al., Citation2012); we have therefore employed rat liver homogenates to compare the hydrolysis rate of the prodrug in the free and encapsulated forms.
Materials and methods
Materials
The prodrug UDCA–AZT (solubility in water = 0.0030 ± 0.0001 mg/mL − 4.75 ± 0.19 10−6 M; solubility in the mixture water and methanol 70:30 v/v = 0.058 ± 0.002 mg/mL − 9.0 ± 0.3 10−5 M) was synthesized as previously described (Dalpiaz et al., Citation2012). PLGA 50:50, Purasorb® PDLG 5004 (Mw ∼30 000 Da) was kindly gifted by Purac Biochem (The Netherlands). Pluronic F-68 (polyethylene-polypropylene glycols), 7 n-propylxanthine (7 n-PX) were obtained from Sigma Aldrich (Milan, Italy). Methanol, acetonitrile, ethyl acetate and water were high-performance liquid chromatography (HPLC) grade from Merck (Darmstadt, Germany). The reversed-phase column (Hypersil BDS C-18 5U cartridge column, 150 mm × 4.6 mm i.d.) and the guard column (packed with Hypersil C-18 material) were obtained from Alltech Italia Srl BV (Milan, Italy). All other reagents and solvents were of analytical grade (Sigma-Aldrich).
UDCA–AZT-loaded NPs
NPs were prepared according to the nanoprecitation technique and single emulsion/solvent evaporation method. The method by Fessi et al. (Citation1989) was employed for the preparation of NPs by nanoprecipitation. Briefly, PLGA (100 mg) and UDCA–AZT (2.5 or 6.0 mg) were accurately weighted and dissolved in 4 mL of acetone. The organic phase was added under magnetic stirring to the aqueous phase (20 or 60 mL) containing 50 mg of Pluronic F-68. Particle precipitation occurred immediately. After 10 min, the organic solvent was removed under vacuum at 30 °C using a rotavapor (Mod. B-480, Büchi Laboratortechnick, Flawil, Switzerand). Empty NPs were prepared according to the procedure previously described, omitting the presence of the prodrug. When purified, the samples were recovered from suspension by gel-chromatography (Sepharose CL4B) or vacuum ultrafiltration.
Gel filtration chromatography was carried out using a Sepharose CL4B (Sigma Chemical) gel. The gel was filled into a column, avoiding bubbles and cracks. The column had a length of 50 cm, an inner diameter of 2 cm and contained ∼160 mL of gel. The NP samples were eluted using deionized water at a flow rate adjusted to 1.5 mL/min. The appearance of the NPs in the eluate was pointed out in ∼20 min using a turbidimeter (model DRT 15-CE, HF Scientific Inc., Fort Meyers, FL).
The vacuum ultrafiltration was performed by using a polycarbonate holder (SM 16510; Sartorius, Gottingen, Germany) equipped with a polypropylene filter (cutoff 0.2 µm; Pall Corporation, Ann Arbor, MI).
All samples were freeze-dried for 24 h at −55 °C at a pressure of 10−2 Torr (Lyovac GT2; Leybold-Heraeus, Hanau, Germany). The freeze-drying of the NPs ensured the elimination of any residual of the acetone that eventually remained in the particles.
The single emulsion/solvent evaporation method was carried out by dissolving either the PLGA (125 mg) and UDCA–AZT (2.5 mg) in 2 or 4 mL of dichloromethane. This organic solution was dispersed during 1 min by sonication at 25 Watts (XL 2000 Microson, Misonix, NY) into 10 or 30 mL of water containing 50 or 100 mg of Pluronic F-68. Then, the emulsion was kept at room temperature under stirring at 3000 rpm until the complete evaporation of the dichloromethane (2 h). Empty NPs were prepared according to the procedure previously described, omitting the presence of the prodrug. When purified, the resulting NPs were recovered by ultrafiltration, as described above. All samples were freeze-dried for 24 h. and report the preparation conditions of the NPs.
Table 1. Formulation parameters, size and physical–chemical characteristics of NPs obtained by nanoprecipitation technique.
Table 2. Formulation parameters, size and physical–chemical characteristics of NPs obtained by emulsion/solvent evaporation technique.
Determination of Pluronic F68 residuals
The amount of Pluronic F68 associated with NPs was determined by a colorimetric method based on the formation of a colored complex between two hydroxyl groups of Pluronic F68, Ba2+ and an iodine molecule (Childs, Citation1975). Briefly, 10 mg of lyophilized NPs were dissolved in 2 mL of dichloromethane. Then, 10 mL of deionized water was added and stirred until complete evaporation of the solvent (2 h). After the centrifugation of the water phase at 3500 g for 10 min (model 4235, A.L.C., Milan, Italy), 1 mL of a 5% w/v BaCl2 solution in HCl 0.1 M and 1 mL of a solution of I2/KI (0.05 M/0.15 M) were added to 4 mL of supernatant. Finally, the absorbance of the samples was measured spectophotometrically at 540 nm (WPA lighwave, Cambridge, UK) after 15 min incubation at room temperature. No absorption was observed when only PLGA polymer was used under identical condition.
Particle size measurement and morphological analysis
The shape and morphology of the samples were analyzed by scanning electron microscope (SEM) (XL-40 Philips, Amsterdam, The Netherlands). When necessary, before the analysis freeze-dried NPs were purified by vacuum ultrafiltration from Pluronic F-68 in order to avoid interference. Few drops of the concentrated reconstituted NP suspension were placed on an aluminum stub (TAAB Laboratories Equipment, Ltd, Berks, UK) using a double side sticky tab (TAAB Laboratories Equipment, Ltd) and, after drying, vacuum-coated with gold–palladium in an argon atmosphere for 60 s (Sputter Coater Emitech K550, Emitech LTD, Ashford, Kent, UK).
The size of the NPs was measured by PCS using a Zetasizer Nano ZS (Malvern, Worcs, UK). Each freeze-dried sample was diluted in deionized water until the appropriate concentration of particles was achieved to avoid multiscattering events. The obtained homogeneous suspension was examined to determine the volume mean diameter and the polydispersity and repeated three times for each sample. The data are the results of three independent experiments.
A colloid/particle fractionator SdFFF system (Model S101, Postnova Analytics, Landsberg, Germany) described in detail elsewhere (Contado & Argazzi, Citation2009, Citation2011), was also employed to determine the size distribution of the particles. Sample suspensions (0.1% w/v) were injected through a 50 µL Rheodyne loop valve. An HPLC Pump PN1121 (Postnova Analytics, Germany) was used to deliver the carrier. The outlet tube from the channel was connected to a UV/Vis detector Spectra SERIES UV100 (Thermo Separation Products) operating at a fixed wavelength of 254 nm. The SdFFF instrument was controlled by SPIN 1409 which was also used to acquire the fractrograms; the registered data were processed by FFF ANALYSIS; both were Windows compatible programs provided by Postnova Analytics (Germany) together with the instrument. The mobile phase was a 0.01% (w/v) solution of Triton X-100 in Milli-Q water (Millipore S.p.A., Vimodrone, Milan, Italy), flowing at 2 mL/min. The fractograms, i.e. the graphical results, were converted in particle size distribution (PSD) assuming a particle bulk density of 1.3 g/mL for all formulations.
HPLC analysis
The quantification of the prodrug UDCA−AZT and its potential hydrolysis product AZT was performed by HPLC. The chromatographic apparatus consisted of a modular system (model LC-10 AD VD pump and model SPD-10A VP variable wavelength UV−Vis detector; Shimadzu, Kyoto, Japan) and an injection valve with 20 μL sample loop (model 7725; Rheodyne, IDEX, Torrance, CA). Separation was performed at room temperature on a reverse phase column Hypersil BDS C-18, 5U, equipped with a guard column packed with the same Hypersil material. Data acquisition and processing were accomplished with a personal computer using Class-VP software (Shimadzu). The detector was set at 260 nm. The mobile phase consisted of a mixture of water and methanol regulated for the analysis of both AZT and UDCA–AZT by a gradient profile programed as follows: 10 min isocratic elution at H2O/MeOH ratio 80:20 (v/v); 1 min linear gradient elution from the water/MeOH ratio 80:20 (v/v) to 25:75 (v/v); 10 min isocratically at the H2O/MeOH ratio 25:75 (v/v); after each cycle the column was conditioned back to the initial conditions for 10 min. The flow rate was 1 mL/min. The xanthine derivative 7-n-PX was employed as internal standard for human rat liver homogenates (see below). The retention times for 7-n-PX, AZT and the prodrug UDCA−AZT were 6.5, 8.4 and 19.6 min, respectively. For the analysis of UDCA–AZT alone the elution was isocratic with a mixture H2O/MeOH 20:80 (v/v). In this case the retention time of UDCA–AZT was 4.8 min.
The chromatographic precision for each compound was evaluated by repeated analysis (n = 6) of the same sample (25 μM). The calibration curves of peak areas versus concentration were generated in the range 0.5–50 μM for AZT dissolved in aqueous phase and UDCA−AZT dissolved in methanol or a mixture of aqueous phase and methanol (70:30 v/v ratio).
UDCA–AZT content in the NPs
The NPs (∼1 mg) were accurately weighed using a high-precision analytical balance (d = 0.01 mg; Model CP 225D; Sartorius), and dissolved into 1 mL of dichloromethane. The samples were reduced to dryness under a nitrogen stream, then 1 mL of methanol was added and, after centrifugation at 12 000 g for 10 min, 10 μL were injected into the HPLC system. All the values were obtained as the mean of three independent incubation experiments. Preliminary experiments indicated the absence of interferences by the components of PLGA NPs on the chromatograms. The actual drug loading (%, w/w) was calculated from the weight of the NPs and the amount of drug incorporated.
In vitro UDCA–AZT release studies from NPs
An accurately weighted amount of NPs (15 mg of samples NP1-LO1P, NP2- LO1, NP2-LO2P, EM1-LO1P and EM2-LO1 or 7.5 mg of the sample NP2-LO2, all containing ∼0.12 mg of the prodrug UDCA–AZT) was added to 15 mL of a mixture of water and methanol (70:30 v/v) and immediately dispersed by sonication (30 s, at 25 W; XL 2000 Microson, Misonix, Farmingdale, NY). The samples were maintained at 37 °C and stirred mechanically (100 rpm) during the release experiments. Aliquots (250 µL) were withdrawn at fixed time intervals and filtered upon centrifugation at 13 000 g for 12 min using Microcon filter devices (YM 30; Millipore Corporation, Bedford, MA). The withdrawn volume was immediately substituted with same amount of the mixture of water and methanol (70:30 v/v). The filtered solution (10 µL) was injected into the HPLC apparatus for the evaluation of UDCA–AZT contents. Preliminary experiments indicated that concentrations of UDCA–AZT in the mixture of water and methanol (70:30 v/v) were not significantly altered by the microcon filtration process.
The dissolution studies of the prodrug were performed by adding 0.15 mg of UDCA–AZT to 15 mg of the mixture of water and methanol (70:30 v/v). Aliquots (150 µL) were withdrawn at fixed time intervals and 10 µl of filtered samples (0.45 µm) were injected into the HPLC system for UDCA–AZT detection. The dissolution experiments were performed in the absence and in the presence of 0.5% Pluronic-F68 in the mixture of water and methanol (70:30 v/v). All the values obtained are the mean of three independent experiments.
Preparation of rat liver homogenates
The livers of male Wistar rats were immediately isolated after their decapitation, washed with ice-cold saline solution and homogenized in 4 volumes (w/v) of Tris–HCl (50 mM, pH 7.4, 4 °C) with the employment of a Potter−Elvehjem apparatus. The supernatant obtained after centrifugation (2000 g for 10 min at 4 °C) was decanted off and stored at −80 °C before its employment for kinetic studies. The total protein concentration in the tissue homogenate was determined using the Lowry procedure (Lowry et al., Citation1951) and resulted in 29.4 ± 1.1 μg protein per microliter.
Kinetic analysis of UDCA–AZT in rat liver homogenates
The prodrug UDCA−AZT or its potential hydrolysis product AZT were incubated at 37 °C in 3 mL of rat liver homogenates, resulting in a final concentration of 30 μM obtained by adding 3 μL of 10−2 M stock solution in DMSO for each milliliter incubated. During the experiment, the samples were shaken continuously and gently in an oscillating water bath. At regular time intervals, 100 μL of samples were withdrawn and immediately quenched in 200 μL of ethanol (4 °C); 100 μL of internal standard (30 μM 7-n-PX) was then added. After centrifugation at 13 000 g for 10 min, 300 μL aliquots were reduced to dryness under a nitrogen stream, then 150 μL of the water and methanol mixture (70:30 v/v) was added and, after centrifugation for 10 min at 12 000 g, 10 μL was injected into the HPLC system. The same procedure was adopted in the presence of unloaded NPs (Sample NP2-UN1, 1.66 mg/mL), with the only difference that 50 µL of dichloromethane was added to the withdrawn samples before the extraction procedure. All the values were obtained as the mean of three independent incubation experiments. A preliminary analysis performed on blank rat liver homogenate samples showed that its components did not interfere with the AZT, UDCA–AZT and 7-n-PX retention times.
The accuracy of the analytical method was determined by recovery experiments, comparing the peak areas of extracted rat liver homogenate samples at 4 °C (n = 6) with those obtained by injection of an equivalent concentration of the analytes dissolved in their mobile phase. For all compounds analyzed, the calibration curves were constructed by employing eight different concentrations in rat liver homogenates at 4 °C ranging from 0.5 to 50 μM and expressed as peak area ratios of the compounds and the internal standard versus concentration.
For stability studies of the nanoencapsulated UDCA–AZT, 3 mL of rat liver homogenate was spiked at 37 °C with loaded NPs (samples NP2-LO1, NP2-LO2, NP2-LO2P) resulting in a final concentration of about 30 µM for encapsulated UDCA–AZT. At 30 min of incubation, 1 mL of dichloromethane was added and 250 µL of samples were withdrawn and quenched in 400 µL of ice-cold ethanol added with 200 µl of 30 µM 7-n-PX as internal standard. After 10 min of centrifugation at 13 000 g, 600 µL aliquots were reduced to dryness under a nitrogen stream. About 300 µL of the water and methanol mixture (70:30 v/v) was added and, after centrifugation, 10 mL was injected into the HPLC system. The accuracy of the method was determined as described above. All the values obtained are the mean of four independent experiments.
Results and discussion
Characterization of the NPs: size, ζ potential and morphology
The formulation parameters and results of the physical–chemical characterization of the particles obtained by nanoprecipitation are reported in . The batch NP1 is the only one subjected to the purification by vacuum filtration before freeze-drying; the NP2 and NP3 batches were obtained by increasing the volume of the aqueous phase (NP2) and by omitting the surfactant (NP3) with respect to the formulation conditions adopted for batch NP1. The sample NP2-LO2P was purified by gel filtration.
The analysis showed that all the samples had a negative ζ potential value, according to the polymer features, characterized by the presence of carboxylic groups deprotonated at our experimental conditions. The ζ potential values of the NPs obtained by nanoprecipitation ranged between −20 and −30 mV, except that in the not purified sample NP2-LO2 (ζ potential = −13.6 mV), obtained with a higher amount of prodrug.
The presence of Pluronic F68 did not seem to influence the ζ potential of NPs as no significant changes were observed between the purified and unpurified samples.
In general, the nanoparticulate systems allow obtaining relatively stable suspensions when their ζ potential is higher than 30 mV as absolute value (Hunter, Citation1989). The samples reported in do not seem to reach this condition and the analysis of their size by PCS appears to confirm this aspect. Indeed, the size values indicate that the samples are characterized by entities with a mean diameter between 500 and 800 nm if we exclude the sample NP1-LO1. These values seem to represent samples highly polydispersed, i.e. the powders which could contain particles of different sizes or more, probably aggregates. The highest degree of aggregation was observed in the case of batch NP1-LO1, likely due to the combined effect of the prodrug dispersed in a relatively small amount of water. In fact, in all other cases the aggregation was more circumscribed. A possible reason is connected with the increase of the aqueous phase, which favors the prodrug dissolution by reducing its precipitation and its ability to act as a nucleus of aggregation for NPs.
The presence of the surfactant coating the unpurified samples did not seem relevant in reducing the aggregation of the NPs. Indeed, the purified and unpurified samples of batch NP2 and the batch NP3, prepared under the same conditions than NP2 but in the absence of Pluronic, showed similar aggregation degrees.
A different approach to size the NPs is offered by the SdFFF, a separation technique able to sort micro- and nanoparticles according to their effective mass. This method detects the particles through UV measurements (absorption and/or scattering) only after their separation, on the contrary of batch measurements given by the PCS technique. The complementarities of the two measurement methods might be well understood by observing , where the PSD plots of some selected samples are reported. The separation conditions were selected in order to highlight only the sizes comprehended between 200 and 2000 nm, neglecting on purpose larger dimensions. Samples NP1-UN1P and NP1-LO1P are classified by the PCS as very polydispersed (PDI values 0.5 and 0.8, respectively) with average sizes of 527 and 1214 nm (); the SdFFF PSD plot (A) show that the NP1-UN1 contains aggregates of roughly 650 nm, forming a single population spanning between 300 nm and up to 1 µm, whereas the NP1-LO1 contains either aggregates of sizes ranging from 250 to 800 nm, but also agglomerates of larger dimensions which are not fractionated under the selected experimental conditions but which exit immediately from the channel and that appear in the PSD plot in the region of the small sizes (<250 nm). The SdFFF measurements confirm therefore that the prodrug encapsulation in this formulation promotes the particle aggregation.
Figure 2. Particle mass distributions of the samples. (A) NP1-UN1P and NP1-LO1P; (B) NP2-UN2 and NP2-LO2 and (C) EM1-UN1P and EM1-LO1P. The conversion from the fractograms was achieved by setting a particle density of 1.3 g/mL for all samples. The fractionations were obtained under programed field conditions by using a 0.01% w/v solution of Triton X-100 as mobile phase, flowing at 2 mL/min.
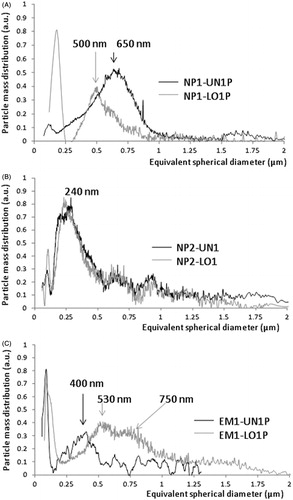
On the contrary, the encapsulation of the prodrug on the formulations of batch NP2 does not determine significant changes in the PSD plots of the samples as highlighted by the plot B, where the two representative sample profiles (UN1 – black and LO1 – gray) appear perfectly superposable, with a main peak centered at about 240–260 nm. The similar behavior could be insight also by the similar ζ potential values (26–27 mV). The secondary populations at 600 nm and 900 nm are of minor intensity and negligible compared to the main one. The PCS measurements ascribe to these samples instead, a certain polydispersity (PDI values ∼0.6 or 0.4) and sizes around 710 nm for the UN1 sample and ∼580 nm for the LO1 sample. Therefore, the SdFFF results indicate that the polydispersity values obtained by PCS are mainly due to different degrees of aggregation, occurring among particles of about 240 nm. This phenomenon seems to be favored in a restricted volume, such as a couvette, whereas it is hampered by the spatial separation in the SdFFF channel.
reports the characteristics of the particles obtained by the emulsion/solvent evaporation method. In general, ζ potential values of the unloaded NPs obtained by emulsion/solvent evaporation method appear lower than those of particles obtained by nanoprecipitation, probably due to a different rearrangement of the polymer in this type of preparation. The presence of the prodrug in the EM1 batch provoked a relatively high aggregation of the NPs, as observed previously also in the case of batch NP1. The effect of aggregation in the presence of drug disappeared as the external aqueous phase was increased probably owing to enhancement of the prodrug dissolution.
The PSDs of the EM1 samples (, plot C), chosen as example for the emulsion formulation method, confirm the presence of a single population of about 400 nm for the sample EM1-UN1 and the aggregation of the particles when the prodrug is present (EM1-LO1).
reports, as representative, the SEM microphotography related to samples NP1-LO1P, NP2LO2P and EM1-UN1P. These images evidence quite spherical and well-formed NPs characterized by a significant polydispersity mainly due to aggregation phenomena. These morphological characteristics are in good agreement with the size data obtained by PCS and SdFFF techniques.
Figure 3. SEM micrographs of the samples NP1 -LO1P (A), NP2-LO2P (B), obtained by nanoprecipitation and EM 1-UN1 (C), obtained by emulsion–solvent evaporation method.

Characterization of the NPs: UDCA–AZT loading
The polymer PLGA employed for the formulation of the NPs was chosen with a molecular weight (∼30 kDa) suitable for the encapsulation of drugs with relatively low molecular weight (Dalpiaz et al., Citation2009). In the aim to verify the loading effects, the different formulations proposed here keep constant the type of polymer (PLGA) and surfactant (Pluronic F68) and differ for the weight ratios between prodrug, Pluronic F-68 and PLGA or for the different volumes of organic and aqueous solvents which were used, as reported in and . The actual UDCA–AZT loading (percent w/w) were also reported in and for the NPs obtained by nanoprecipitation and emulsification techniques, respectively. The loading values have been obtained by HPLC analysis with isocratic elution at H2O/MeOH ratio 20:80 (v/v), as previously described (Dalpiaz et al., Citation2012). The chromatographic precision for UDCA−AZT dissolved in MeOH or in a H2O/MeOH mixture 70:30 (v/v) was represented by relative standard deviation (RSD) values ranging between 0.95% and 0.96%. The calibration curves of the prodrug were linear over the range of 0.5–50 μM (n = 8, r > 0.998, p < 0.0001). The employment of methanol as co-solvent was necessary to increase the low solubility of the prodrug in water. In particular, the solubility value of UDCA−AZT in water was 0.0030 ± 0.0001 mg/mL (4.75 ± 0.19 × 10−6 M), whereas in the mixture of water and methanol (70:30 v/v), it was 0.058 ± 0.002 mg/mL (9.0 ± 0.3 × 10−5 M) (Dalpiaz et al., Citation2012). For this reason, also the release studies of UDCA–AZT from the NPs were performed in the same water/methanol mixture (see below).
The data showed in the Tables indicate that the prodrug content in the nanoparticulate powders obtained by nanoprecipitation or by emulsion/solvent evaporation was not modified by the vacuum-ultrafiltration purification process. This is a very important data that shows how the process of purification in the case of a very slightly water soluble drug as the prodrug UDCA–AZT does not modify the drug loading, but changes only the amount of surfactant present in the final product. Indeed, as reported in and , the Pluronic residual was dependent on the purification methods applied. The unpurified particles showed a Pluronic residual of 33% w/w, while the purified particles showed a residual of 0.5% or 0.9% w/w for vacuum ultrafiltration and gel filtration, respectively. Therefore, the elimination of the Pluronic was poorly affected by the method of purification, as demonstrated in a previous work (Leo, Citation2006a). On the other hand, the sample NP2-LO2P, purified by gel filtration, showed a reduced prodrug content with respect to its parent unpurified sample NP2-LO2. In this case the purification process, able to perform a separation of solid particles on the basis of their size, appeared able to separate the solid particles of the prodrug not solubilized and adsorbed on the surface of the NP. The absence of surfactant during the preparation appeared detrimental for the UDCA–AZT loading as evident from the batch NP3 in which only traces of prodrug were found (0.051% w/w), notwithstanding the sample was unpurified. The lack of drug loading in this batch is probably due to its high drug lipophilicity that, in the absence of Pluronic, determines the complete drug adhesion on the walls of the glassware and the absolute lack of drug recovery. On the other hand, in the presence of surfactant, drug loading was of about 0.8% w/w both in the samples unpurified and purified, irrespective of the amount of aqueous phase used in the preparation (NP1 and NP2 batches). Increasing the theoretical content of the drug (NP2-LO2) an increase of its actual loading was observed (until 1.53% w/w) without current increase of the particles’ size. After gel filtration, the prodrug content appeared slightly >0.80% being eliminated the most part of unloaded drug.
The samples obtained by simple emulsion/solvent evaporation method () showed prodrug contents around 0.8% w/w. The formulation conditions for the sample EM1 (high Pluronic F-68 amounts and small water phase volumes) appeared to improve the prodrug presence in the nanoparticulate powder. Indeed the prodrug percentage found for the sample EM1-LO1 was the same as detected for the sample EM2-LO1, that was not purified and was prepared using different organic/water phase ratio and a lower Pluronic F68 amount. Therefore, these parameters seem to not influence the recovery of the prodrug in nanoparticulate powder.
In vitro N6-cyclopentyladenosine release studies from NPs
The release studies of the prodrug from the loaded nanoparticulate samples have been performed in a mixture of water/methanol 70:30 (v/v) with the aim to induce the solubility of UDCA–AZT in water, which is very poor (0.0030 ± 0.0001 mg/mL – 4.75 ± 0.19 ċ 10−6 M). Therefore, in order to create sink conditions, the release studies were performed in the presence of 30% methanol in the release medium, with the aim of optimizing both the prodrug solubility and HPLC detection for the release studies. Even if these conditions do not mimic the physiological conditions, we considered our release studies as a way of comparison of the ability of the NPs to control the release of the prodrug. reports the release profiles of UDCA–AZT from the loaded NPs. The release patterns are compared with those of the dissolution during time of the raw powder of the prodrug in the water/methanol mixture 70:30 (v/v), both in the absence and in presence of 0.5% Pluronic F-68. Also in this case, the amounts of prodrug to be solubilized were chosen in manner to obtain, after complete dissolution, a concentration of prodrug corresponding to the 15% of its saturation value. It can be observed that the dissolution rate of UDCA–AZT in the absence of surfactant appeared very low. Indeed, after eight hours of incubation, <30% of the total raw powder amount was solubilized. On the other hand, the presence of the surfactant in the incubation medium allows to obtain a complete dissolution of the prodrug within 1.5 h. The presence of the surfactant seems, therefore, to play an important role in reducing the interfacial tension between the solid prodrug and the liquid phase, allowing a complete wetting of the raw powder of the prodrug. It is interesting to observe in that the sample NP1-LO1P showed a release pattern characterized by a high-burst effect (>80%), followed by a release of the prodrug that was completed within one hour. On the whole, the release rate of UDCA–AZT from all samples appeared higher than the dissolution rate of the prodrug in the presence of Pluronic F-68. This phenomenon has been attributed to the size of the particles in the µm or nm ranges, that seemed to induce a great increase of surface contact of the solid prodrug with the incubation medium, allowing therefore to increase the dissolution rate with respect to the raw powder. However, as NP1-LO1P sample is larger than the others with a low drug loading, we would expect a slower drug diffusion rate. The relatively fast release of UDCA–AZT from sample NP1-LO1P suggests that the prodrug amount encapsulated in the interior structure of the NPs is very poor. As a consequence, the formulation conditions chosen for sample NP1-LO1P, appear detrimental for an efficient encapsulation of the prodrug in the NPs. The low amount of organic phase employed during formulation seems useful to induce adsorption phenomena of UDCA–AZT to the NPs, but not their loading. On the other hand, the formulation conditions employed for sample NP2-LO1 optimized the prodrug encapsulation in the PLGA NP. Indeed, following a burst effect of ∼60%, probably due to the presence of the prodrug adsorbed on the surface of NP that has not been removed by purification, the release of UDCA–AZT appeared completed within six hours, with a release rate significantly lower than the dissolution rate of the prodrug in the presence of surfactant. It is important to remark that, despite the sample NP2-LO1 has a smaller size and has not been purified, its burst effect appeared the lowest among the burst effect values of the samples reported in . This finding can be explained assuming that the formulation parameters allow to obtain a low porous and high compact matrix which leaded to a reduction in the drug dissolution rate despite the reduction of the particle size. As a consequence, the enhancement of the volume of the water phase with respect to the formulation conditions chosen for the sample NP1-LO1, appears to have favored the encapsulation of the prodrug in the NP2-LO1 NPs, probably by increasing the efficacy of the nanoprecipitation of the polymer when it was desolvated from acetone by action of the aqueous “non-solvent”. Finally, keeping constant the formulation conditions of the samples NP2 and increasing the initial amount of drug (theoretical loading), we obtained NPs characterized by a lower ability to control the release of UDCA–AZT. Indeed, the samples NP2-LO2 and NP2-LO2P showed burst effects values about the 74% and 60%, respectively, then the prodrug release was completed within six hours, but with a rate in the first phases higher than that showed by the sample NP2-LO1. The increase of the initial prodrug amount seems therefore to have induced the creation of a porous structure in the polymeric matrix of the NPs, with a consequent increase of the release rate of UDCA–AZT. Considering the samples NP2-LO2 and NP2-LO2P, we can notice how the presence of Pluronic does not substantially modify the kinetics of drug release from NPs nor the extent of the burst effect which is instead only influenced by the preparation parameters adopted.
Figure 4. In vitro release of UDCA–AZT from PLGA NP, obtained by nanoprecipitation (A) or emulsion/solvent evaporation (B) methods. The release profiles are compared with those of the raw UDCA–AZT powder dissolution during time in the absence or in the presence of 0.5% Pluronic F-68. Results are the means of three independent experiments.
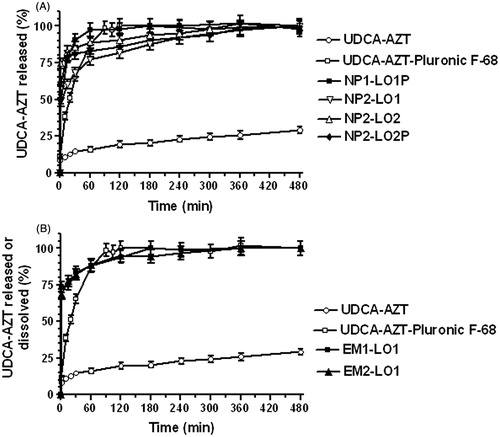
reports the release profiles of UDCA–AZT from the NPs obtained by simple emulsion/solvent evaporation. Both the samples showed a poor ability to control the release of the prodrug, with a burst effect of about 75%; the 90% of UDCA–AZT was released within one hour and the release was completed within three or four hours. As a consequence, the formulation conditions chosen for EM1 and EM2 batches appear inappropriate for the encapsulation of the prodrug in the PLGA NPs. Moreover, the inability to control the release of the prodrug was showed by other samples, obtained at the same conditions of the samples EM1-LO1P and EM2-LO1, but by increasing the prodrug amounts during preparation (6.0 mg). In particular, despite a significant increase of loading, the prodrug release was characterized by burst effects ranging from 85% to 90%, with a complete release of the drug within two hours. It is probable that UDCA–AZT is able to reach the aqueous phase where Pluronic F-68 is dissolved, during the solidification phase of polymer. Only a rapid solidification phenomenon, such as that allowed by nanoprecipitation seems to consent the UDCA–AZT encapsulation in the PLGA NP. We have previously verified that a moderately water-soluble drug, the N6-cyclopentyladenosine (CPA), can easily diffuse in aqueous phase during the preparation of PLGA NPs, both by nanoprecipitation and single or double emulsion/solvent evaporation methods. Using such methods, either a poor grade of CPA encapsulation or an unsatisfied control of its release was achieved (Leo et al., Citation2006b). We have therefore demonstrated that the lipophilic prodrug 5′-octanoyl-CPA (Dalpiaz et al., Citation2001) can be encapsulated in PLA NP and be able to control its release. These satisfactory results were obtained by the nanoprecipitation technique (Dalpiaz et al., Citation2005; Leo et al., Citation2006a). It is important to remark that CPA is an adenosine derivative, whereas AZT is a timidine derivative. Both these derivatives are moderately water-soluble and an increase of their lipophilicity by a prodrug synthesis allows to obtain their encapsulation in polymeric NPs, with results optimized by the nanoprecipitation procedure.
Hydrolysis studies of free and encapsulated UDCA−AZT in rat liver homogenates
On the basis of the encapsulation and release results, the samples NP2-LO1, NP2-LO2 and NP2-LO2P appear potentially useful for the targeting of the prodrug UDCA–AZT in specific sites of the body where the antiviral activity of zidovudine is required. In particular, the unpurified and Pluronic coated samples NP2-LO1 and NP2-LO2 appear potentially suitable for the prodrug targeting in the CNS. Indeed, it has been demonstrated that PLGA NPs coated with Pluronic F-68 can induce drug delivery in the brain (Gelperin et al., Citation2010). The AZT targeting in CNS should be of great importance because HIV is able to easily reach the brain where it induces neurological disorders ranging from mild cognitive impairments to the severe AIDS dementia complex (Davis et al., Citation1992; Gray et al., Citation1996). Even if anti-HIV agents can be efficacious in periphery, they are in general unable to reach the CNS and that therefore becomes a sanctuary of the virus from which the periphery can be continuously reinfected (Kolson & Gonzales Sacrano, 2000). Moreover, the purified sample NP2-LO2P appears potentially useful for the prodrug targeting in the macrophages, that when infected by HIV produces and harbor the virus for a long period (Carter & Ehrlich, Citation2008). As a consequence, the opsonization and the following phagocytosis of the purified NPs by the macrophages of the mononuclear phagocytic system (MPS) (Owens III & Peppas, Citation2006) can constitute an efficacious way to target UDCA–AZT in an important therapeutic site of AZT.
In the aim to evaluate the ability of the NPs to protect and stabilize the prodrug in physiologic environments, we have compared the hydrolysis rate of UDCA–AZT in rat liver homogenates in the free and encapsulated forms. We have chosen the rat liver homogenates because the hydrolysis rate of the prodrug is relatively fast (Dalpiaz et al., Citation2012). The average recovery ± SD of UDCA–AZT and AZT from rat liver homogenates were 83.2 ± 2.9% and 85.5% ± 3.4%, respectively. The concentrations of these compounds were therefore referred to as peak area ratio with respect to their internal standard 7-n-PX. The precision of the method based on peak area ratio was represented by RSD values ranging between 1.1% and 1.3%. The calibration curves referred to AZT and its prodrug UDCA–AZT incubated in rat liver homogenates were linear over the range 0.5–50 μM (n = 8, r > 0.989, p < 0.0001).
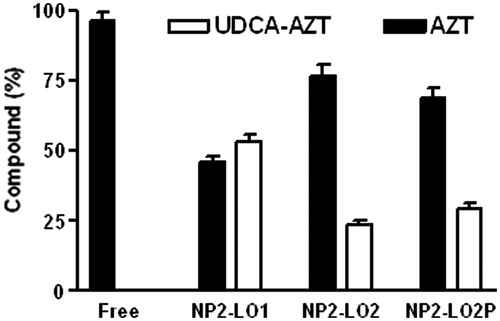
As reported in , UDCA–AZT was degraded in rat liver homogenates, following a first order kinetic with a half-life of 2.78 ± 0.15 min. This value appears in good agreement with that obtained by previous studies on UDCA–AZT pharmacokinetics in same incubation media, where it was demonstrated that the prodrug was hydrolyzed allowing the release of AZT (Dalpiaz et al., Citation2012). This process was completed within 30 min, suggesting the need to increase the half-life value of UDCA–AZT in the case of potential in vivo administration. The stability studies have been performed first by evaluating the effect of the presence of the unloaded NPs on the hydrolysis of UDCA–AZT by rat liver homogenates. As reported in , the degradation curve of UDCA–AZT appears superimposable with that of its degradation in the presence of the sample, i.e. the unloaded and unpurified NPs of the batch NP2. In particular, the half-life of UDCA–AZT in the presence of the unloaded NPs was 2.50 ± 0.15. This value was not significantly different from the half-life value of UDCA–AZT alone indicating that the presence of the coating of Pluronic on NPs does not change the half-life value of the prodrug. These data are in good agreement with those obtained by previous pharmacokinetic studies of different drugs and prodrugs in the presence of unloaded PLA, PLGA or solid lipid micro and NPs (Dalpiaz et al., Citation2001, Citation2002, Citation2005, Citation2008, Citation2009, Citation2010; Leo et al., Citation2006a,Citationb). The amounts of AZT and UDCA–AZT detected after 30 min of incubation of the free or encapsulated prodrug in rat liver homogenates are reported in . All the values are reported as the percentage of the overall amount of incubated prodrug. It can be observed that after this time period the free prodrug appeared totally hydrolyzed, with the 100% of AZT released and the total absence of UDCA–AZT. On the other hand, the incubation of the sample NP2-LO1 allowed saving more than 50% of the UDCA–AZT incubated for 30 min in rat liver homogenates. Indeed, lower amounts of UDCA–AZT were hydrolyzed with respect to the free prodrug, as confirmed by the release of the 48% of AZT. The samples NP2-LO2 and NP2-LO2P have also contributed to save the prodrug from a complete hydrolysis, even if their efficacy appeared lower with respect to the sample NP2-LO1. In this case the amounts of UDCA–AZT saved ranged between 24% and 30% with corresponding release of AZT ranging from 69% to 76%. These results indicate that the ability of the PLGA NP to stabilize the prodrug in rat liver homogenates is greatly related to their ability to control the release of encapsulated UDCA–AZT. Interestingly, poor differences were found between the samples NP2-LO2 and NP2-LO2P, confirming that the presence of Pluronic F-68 on the surface of the NPs do not alter the hydrolysis rate of the encapsulated prodrug.
Figure 5. Degradation profiles of the free prodrug UDCA−AZT in rat liver homogenates in the absence or in the presence of unloaded nanoparticulate systems obtained by nanoprecipitation. All the values are reported as the percentage of the overall amount of incubated prodrug. Data are reported as the mean ± SD of four independent experiments.
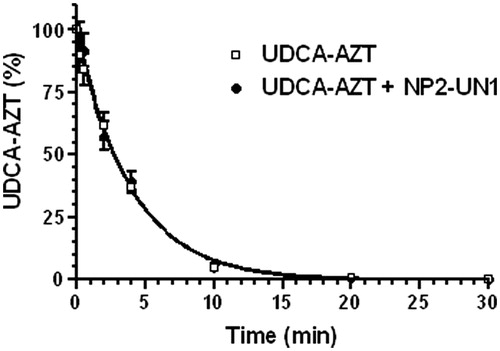
Figure 6. Degradation in rat liver homogenates of free or encapsulated UDCA–AZT in PLGA NP. All UDCA–AZT and AZT values are reported as the percentage of the overall amount of incubated prodrug. Data are reported as the mean ± SD of four independent experiments.
Conclusions
We have demonstrated that PLGA NPs obtained by nanoprecipitation in the presence of Pluronic-F68 as surfactant, are able to encapsulate and control the release of a prodrug of zidovudine (AZT), obtained by its conjugation with UDCA. The encapsulated prodrug UDCA–AZT shows in physiologic fluids, an increased stability with respect to the free form. Despite these interesting aspects, the loading of the formulations appeared relatively low, whereas the PDI, burst and release values were relatively high. Therefore, this type of device can be considered useful for in vitro study about opsonization and the following phagocytosis by macrophages. The ability of the described nanoparticles to increase the dissolution rate of the prodrug, with respect to its raw solid form, may allow to obtain an appropriate delivery of the prodrug for studies that can elucidate its effect in solubilized form in the macrophages, where an anti-HIV action should be needed in the case of AIDS syndrome.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this work.
This work was supported by a grant from Ministero dell’Istruzione, dell’Università e della Ricerca (PRIN 2012).
References
- Bala I, Hariharan S, Kumar MN. (2004). PLGA nanoparticles in drug delivery: the state of the art. Crit Rev Ther Drug Carrier Syst 21:387–422
- Batrakov EV, Kabanov AB. (2008). Pluronic block copolymers: evolution of drug delivery concept from inert nanocarriers to biological response modifiers. J Contr Rel 130:98–106
- Carter CA, Ehrlich LS. (2008). Cell biology of HIV-1. Infection of macrophages. Annu Rev Microbiol 62:425–33
- Chen J, Zhang C, Liu Q, et al. (2012). Solanum tuberosum lectin-conjugated PLGA nanoparticles for nose-to-brain delivery: in vivo and in vitro evaluations. J Drug Target 20:174–84
- Childs CE. (1975). Determination of polyoxyethylen glycol in gammaglobulin solutions. Microchem J 20:190–2
- Chorny M, Fishbein I, Danenberg HD, Golomb G. (2002). Lipophilic drug loaded nanospheres prepared by nanoprecipitation: effect of formulation variables on size, drug recovery and release kinetics. J Contr Rel 83:389–400
- Contado C, Argazzi R. (2009). Size sorting of citrate reduced gold nanoparticles by sedimentation field-flow fractionation. J Chromatogr A 1216:9088–98
- Contado C, Argazzi R. (2011). Sedimentation field flow fractionation and flow field flow fractionation as tools for studying the aging effects of WO3 colloids for photoelectrochemical uses”. J Chromatogr A 1218:4179–87
- Contado C, Vighi E, Dalpiaz A, Leo E. (2013). Influence of secondary preparative parameters and aging effects on PLGA particle size distribution: a sedimentation field flow fractionation investigation. Anal Bioanal Chem 405:703–11
- Dalpiaz A, Cacciari B, Mezzena M, et al. (2010). Solid lipid microparticles for the stability enhancement of a dopamine prodrug. J Pharm Sci 99:4730–7
- Dalpiaz A, Leo E, Vitali F, et al. (2005). Development and characterization of biodegradable nanoparticles as delivery systems of antiischemic adenosine derivatives. Biomaterials 26:1299–306
- Dalpiaz A, Mezzena M, Scatturin A, Scalia S. (2008). Solid lipid microparticles for the stability enhancement of the polar drug N(6)-cyclopentyladenosine. Int J Pharm 355:81–6
- Dalpiaz A, Scatturin A, Menegatti E, et al. (2001). Synthesis and study of 5′-ester prodrugs of N6-cyclopentyladenosine, a selective A1 receptor agonist. Pharm Res 18:531–6
- Dalpiaz A, Scatturin A, Pavan B, et al. (2002). Poly (lactic acid) microspheres for the sustained release of antiischemic agents. Int J Pharm 242:115–20
- Dalpiaz A, Vighi E, Pavan B, Leo E. (2009). Fabrication via a nonaqueous nanoprecipitation method, characterization and in vitro biological behaviour of N6-cyclopentyladenosine-loaded nanoparticles. J Pharm Sci 98:4272–84
- Dalpiaz A, Paganetto G, Pavan B, et al. (2012). Zidovudine and ursodeoxycholic acid conjugation: design of a new prodrug potentially able to bypass the active efflux transport systems of the central nervous system. Mol Pharmaceutics 9:957–68
- Danhier F, Ansorena E, Silva JM, et al. (2012). PLGA-based nanoparticles: an overview of biomedical applications. J Contr Rel 161:505–22
- Davis LE, Hjelle BL, Miller VE, et al. (1992). Early viral brain invasion in iatrogenic human immunodeficiency virus infection. Neurology 42:1736–9
- De Clerk E. (2009). Anti-HIV drugs: 25 compounds approved within 25 years after the discovery of HIV. Int J Antimicrob Agents 33:307−20
- Fessi H, Puisieux F, Devissaguet JP, et al. (1989). Nanocapsule formation by interfacial polymer deposition following solvent displacement. Int J Pharm 55:R1–4
- Gelperin S, Maksimenko O, Khalansky A, et al. (2010). Drug delivery to the brain using surfactant-coated poly(lactide-co-glycolide) nanoparticles: influence of the formulation parameters. Eur J Pharm Biopharm 74:157–63
- Gray F, Scaravilli F, Everall I, et al. (1996). Neuropathology of early HIV-1 infection. Brain Pathol 6:1–15
- Hunter RJ. (1989). Foundations of colloid science, vol I. Oxford, New York: Oxford University Press
- Jalil R. (1990). Biodegradable poly(lactic acid) and poly(lactide-co-glycolide) polymers in sustained drug delivery. Drug Dev Ind Pharm 16:2353–67
- Johansen P, Men Y, Merkle HP, Gander B. (2000). Revisiting PLA/PLGA microspheres: an analysis of their potential in parenteral vaccination. Eur J Pharm Biopharm 50:129–46
- Kolson DL, Gonzales Sacrano F. (2000). HIV and HIV dementia. J Clin Invest 106:11--13
- Leo E, Contado C, Bortolotti F, et al. (2006a). Nanoparticle formulation may affect the stabilization of an antischemic prodrug. Int J Pharm 307:103–13
- Leo E, Scatturin A, Vighi E, Dalpiaz A. (2006b). Polymeric nanoparticles as drug controlled release systems: a new formulation strategy for drugs with small or large molecular weight. J Nanosci Nanotechnol 6:3070–9
- Liang V. (1987). Multidrug resistance P-glycoprotein expression. Ann NY Acad Sci 507:7–8
- Lobato M, et al. (2012). Cannabinoid derivate-loaded PLGA nanocarriers for oral administration: formulation, characterization, and cytotoxicity studies. Int J Nanomedicine 7:5793--806
- Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. (1951). Protein measurement with the folin phenol reagent. J Biol Chem 193:265–75
- Makadia HK, Siegel SJ. (2011). Poly lactic-co-glycolic acid (PLGA) as biodegradable controlled drug delivery carrier. Polymers 3:1377–97
- Nafee N, Schneider M, Schaefer UF, Lehr CM. (2009). Relevance of the colloidal stability of chitosan/PLGA nanoparticles on their cytotoxicity profile. Int J Pharm 38:130–9
- Owens DE III, Peppas NA. (2006). Opsonization, biodisribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm 307:93–102
- Pardridge WM. (2003). Blood-brain barrier drug targeting: the future of brain drug development. Mol Interventions 3:90–105
- Siepmann J, Faisant N, Akiki J, et al. (2004). Effect of the size of biodegradable microparticles on drug release: experiment and theory. J Control Release 96:123–34
- Takasawa K, Terasaki T, Suzuki H, Sugiyama Y. (1997). In vivo evidence for carrier-mediated efflux transport of 3′-Azido-3′- deoxythymidine and 2′,3′-dideoxyinosine across the blood-brain barrier via a probenecid sensitive transport system. J Pharmacol Exp Ther 281:369–75
- Tomoda K, Watanabe A, Suzuki K, et al. (2012). Enhanced transdermal permeability of estradiol using combination of PLGA nanoparticles system and iontophoresis. Colloids Surf B Biointerfaces 97:84–9
- Vyas SP, Subhedar R, Jain S. (2006). Development and characterization of emulsomes for sustained and targeted delivery of an antiviral agent to liver. J Pharm Pharmacol 58:321–6
- Wong SL, Van Belle K, Sawchuk RJ. (1993). Distributional transport kinetics of zidovudine between plasma and brain extracellular Fluid/cerebrospinal fluid in the rabbit: investigation of the inhibitory effect of probenecid utilizing microdialysis. J Pharmacol Exp Ther 264:899–909