
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The purpose of this work is to use biorelevant media to evaluate the robustness of a poorly water soluble weakly basic drug to variations along the gastrointestinal tract (GIT) after incorporation in liquisolid compacts and to assess the success of these models in predicting the in vivo performance. Liquisolid tablets were prepared using mosapride citrate as a model drug. A factorial design experiment was used to study the effect of three factors, namely: drug concentration at two levels (5% and 10%), carriers at three levels (avicel, mannitol and lactose) and powder excipients ratio (R) of the coating material at two levels (25 and 30). The in vitro dissolution media utilized were 0.1 N HCl, hypoacidic stomach model and a transfer model simulating the transfer from the stomach to the intestine. All compacts released above 95% of drug after 10 min in 0.1 N HCl. In the hypoacidic model, the compacts with R 30 were superior compared to R 25, where they released >90% of drug after 10 min compared to 80% for R 25. After the transfer of the optimum compacts from Simulated gastric fluid fast (SGFfast) to fasted state simulated intestinal fluid, slight turbidity appeared after 30 min, and the amount of drug dissolved slightly decreased from 96.91% to 90.59%. However, after the transfer from SGFfast to fed state simulated intestinal fluid, no turbidity or precipitation occurred throughout time of the test (60 min). In vivo pharmacokinetic study in human volunteers proved the success of the in vitro models with enhancement of the oral bioavailability (121.20%) compared to the commercial product.
Introduction
Dissolution testing has expanded substantially to address not only questions regarding quality control of dosage forms but also to screen formulations and as a means of identifying potential bioavailability problems as well as to assess the need for further bioequivalence studies. Hence, it is essential to run dissolution tests under conditions that closely resemble the key parameters of human gastrointestinal physiology. Biorelevant media have been developed to simulate conditions in the stomach and small intestine before and after meals (Reppas & Vertzoni, Citation2012).
The pH of the gastrointestinal fluids is of vital importance to the solubility of ionizable drugs. The solubility of weak acids and bases is dependent on the compound’s ionization constant and the pH of the local environment. Reduction of the pH below the pKa value for weak bases, for example, leads to an increase in solubility due to the contribution of the more soluble, ionized form of the drug. As for the specific case of the poorly soluble weak bases, one must not only consider solubility and dissolution but also the possibility of precipitation as the drug moves from the favorable pH conditions in the stomach to a less favorable pH environment in the small intestine (Ramenskaia et al., Citation2011; El-Hammadi & Awad, Citation2012). When the pH approaches, or even exceeds the pKa of the basic drug, its solubility undergoes a sharp decrease and the concentration of the drug in solution may in fact exceeds its solubility due to increase in the unionized form of the drug and decrease in the ionized soluble form (Ramenskaia et al., Citation2011; El-Hammadi & Awad, Citation2012). Thus, the question is does the drug stay in supersaturation solution or does it precipitate and therefore become no longer available for absorption?
Mosapride citrate (MC) is a gastroprokinetic agent that enhances gastrointestinal motility by stimulating the serotonin (5-HT4) receptor (Yoshikawa et al., Citation1998). It is used for the treatment of acid reflux (Ruth et al., Citation1998), irritable bowel syndrome and functional dyspepsia (Mizuta et al., Citation2006). According to the Biopharmaceutical Classification System, MC is classified as a class II drug with poor solubility and high permeability, which affects its absorption and hence its bioavailability. In addition, MC is an ionizable weak base and its solubility is pH dependant; decreasing with increase in the pH (Kumagai et al., Citation2005), which may result in high variability in oral bioavailability due to pH variations in the fluids of GIT.
Liquisolid technique is a promising technique for enhancing dissolution rate of poorly water soluble drugs. A “liquisolid system” refers to formulations formed by conversion of drug solution in non-volatile, non toxic solvents into dry, free-flowing and compressible powder mixtures by blending the solution with selected carriers and coating materials (Spireas & Sadu, Citation1998). Better bioavailability of poorly water-soluble drugs could be achieved when the drug is present in liquisolid formulations. This is due to the dissolving of the insoluble drug in an orally safe water-miscible solvent, which is then loaded by simple blending to a mixture of powders. By using liquisolid compacts, one can create in situ supersaturated solution from subsaturated drug solution incorporated in liquisolid dosage form. After oral administration, the subsaturated drug solution will mix with aqueous milieu in the GIT (in which the drug is less soluble), changing the vehicle composition and creating in situ freshly prepared supersaturated solution. This induced supersaturation can provide enhanced thermodynamic activity driving force for absorption.
Hence, the aim of this work was to improve solubility, dissolution and thus bioavailability of MC using liquisolid technique and to examine if this technique can minimize the influence of pH variations on its dissolution rate, as a model of a weakly basic poorly soluble drug, through using biorelevant media in its assessment like hypoacidic and transfer models. Liquisolid tablets of MC were prepared using glycerol formal as a non volatile solvent. The in vitro drug dissolution of liquisolid tablets, pure drug and market tablets were compared using different dissolution tests at different pH. Furthermore, a bioequivalence study was performed in human volunteers using the prepared tablets and the conventional commercial product.
Materials and methods
Materials
MC dihydrate was obtained from Hetero Pharmaceuticals, Hyderabad, India. Glycerol formal was obtained from S.P. Quimica, Riudoms, Spain. Propylene glycol, microcrystalline cellulose (Avicel PH 102), colloidal silicon dioxide (Aerosil 200) and lecithin were obtained from EGPI, Cario, Egypt. Poly ethylene glycol 400 and lactose D.C were obtained from Amryia Free Zone, Cario, Egypt. Mannitol was obtained from Quingdao Bright Moon, Jiaonan, China. Low substituted hydroxypropyl cellulose (L-HPC21) was obtained from Joinway, Shanghai, China. All other reagents, chemicals and solvents used were of analytical grade.
Solubility studies
The saturation solubility of MC was done in different non-volatile solvents viz glycerol formal, PEG 400 and propylene glycol to select the best one for its solution in liquid medication. Excess amount of MC was suspended in each solvent in tightly closed screw cap vials, equilibrated in a shaking water bath at ambient temperature for 72 h then filtered using 0.45 µm Millipore filter and assayed spectrophotometrically at predetermined λmax. Three determinations were carried out to calculate the saturated solubility of MC.
Preparation of liquisolid systems
Different liquisolid formulations of MC with potency of 5 mg per unit dose were prepared adopting a full factorial design technique to study the effect of three factors on dissolution, namely: the drug concentration in the chosen solvent at two levels (5% and 10%), the carrier type at three levels (Avicel PH 102, mannitol and lactose D.C) and the R factor of the coating material (Aerosil 200) at two levels (25 and 30). Briefly, 5% or 10% w/w of the drug solution in the selected solvent were triturated with different carriers using a mortar and a pestle. Depending on the drug concentration in the liquid medication used, different loading factors (Lfs) were used according to capacity of each carrier. The wet masses were then converted into dry, flowable and compressible powder by incorporating the coating material (Aerosil 200) at powder excipients ratio (R) of 25 or 30. All powder systems were then passed through sieve, 500 µm. L-HPC21 (7%) used as a disintegrant was then added and mixed to all formulations. A typical design of the formulation characteristics of the prepared liquisolid systems are complied in .
Table 1. Composition and flow properties of the different prepared liquisolid systems.
Flow properties of liquisolid powders
The flow properties of the developed powders were evaluated using the following techniques:
Angle of repose. The angle of repose was measured according to the fixed funnel and cone method. A funnel with the end of its stem cut perpendicular to the axis of symmetry is fixed above a flat horizontal surface with its tip 10 cm high, H. The powders were allowed to flow through the funnel until a conical pile is formed with its apex reaching the tip of the funnel. The angle of repose was determined as follows:
where, θ is the angle of repose and R is calculated from the mean diameter of the powder cone.
Powders with angle of repose above 50° have unsatisfactory poor flow properties, 25°–40° reasonable flow potential, and minimum angles close to 25° represent very good flow properties (Staniforth, Citation2002).
Carr’s compressibility index. Five grams of each powder system were placed in a graduated cylinder and the volume occupied (bulk volume Vb) was recorded. The graduated cylinder was tapped several times till a constant volume was obtained and the volume of powder was then recorded (true or tapped volume Vt)
Carr’s index (CI%) was obtained from the bulk and tapped densities by the following equation (Carr, Citation1965):
where, ρbulk is the freely settled bulk density of the powder and ρtap is the tapped bulk density of the powder.
The smaller the value of the CI%, the superior the flow properties of the powder.
Hausner ratio (HR). HR was calculated according to the following equation (Hausner, Citation1967):
Compression of the liquisolid systems into tablets
The liquisolid powder mixtures were compressed into tablets containing 5 mg MC per tablet using a rotary compression machine (Karnavati, mini press II, Gujarat, India). Two punch sizes were used according to the unit tablet weight, where punch size 8 mm was used for powder weight <400 mg, and punch size 16 mm was used for powder weight >400 mg. The compression force applied was sufficient to produce acceptable tablet hardness.
Evaluation of the prepared liquisolid tablets
Tablets were assessed by performing quality control tests according to BP 2010 for weight variation, content uniformity, friability, in vitro disintegration and the crushing strength (hardness).
Dissolution studies
Dissolution in 0.1 N HCl. In vitro dissolution studies of the prepared MC tablets, conventional market tablet product and 5 mg pure drug were carried out in 500 ml 0.1 N HCl (pH 1.2) using USP Dissolution Tester (Pharmatest, Hainburg, Germany), Apparatus II (paddle) rotating at 75 rpm and maintained at 37 °C ± 0.5 °C. At predetermined time intervals (5, 10, 15, 30, 45 and 60 min), an aliquot of the dissolution media was withdrawn and analyzed spectrophotometrically for drug content after applying the second-order derivative. Withdrawn samples were replaced by fresh medium. All the determinations were carried out in triplicate.
Hypoacidic stomach model. Hypoacidic stomach model was applied for selected formulae to evaluate the robustness of MC formulation at high pH in stomach, which may be present in hypochlorohydric patients as a result of aging, some diseases as Helicobacter pylori infection, after some meals or drugs or at some people. The dissolution studies were carried out in 500 ml acetate buffer (pH 5) using USP Dissolution Tester, Apparatus II rotating at 75 rpm and maintained at 37 °C ± 0.5 °C as previously discussed (Dressman & Krämer, Citation2005). All the determinations were carried out in triplicate.
Transfer model. Transfer model was done to simulate MC transfer out of the stomach into the small intestine and to predict the possibility of its precipitation upon entry in the small intestine when it reaches pH near its pKa (6.5). The selected MC tablets, conventional market tablet product and 5 mg pure MC powder were completely dissolved in 100 ml of simulating gastric fluid SGFfast (donor phase) pH = 2. A tube of infusion set was then used to transfer the donor phase at a constant rate of 8.5 ml/min into a dissolution vessel of the USP Dissolution Tester, Apparatus II rotating at 75 rpm and maintained at 37 °C ± 0.5 °C, containing 500 ml of either fasted state simulated intestinal fluid (FaSSIF) or fed state simulated intestinal fluid (FeSSIF) pH = 6.5 as the acceptor phase. The pH was monitored in the acceptor phase along transfer of MC from donor phase to acceptor phase. After complete transfer of MC, an aliquot of the acceptor phase was withdrawn at 0, 5, 10, 20, 30, 45 and 60 min and analyzed spectrophotometrically for drug content after applying the second order derivative. Withdrawn samples were replaced by fresh medium from a separate vessel held at 37 °C ± 0.5 °C. All these determinations were carried out in triplicate.
Differential scanning calorimetry
Differential scanning calorimetry (DSC) was performed using Shimadzu differential scanning calorimeter DC-60, in order to assess the thermal behavior of MC, Avicel PH 102, Aerosil 200, their physical mixture in the ratio 1:1:1 and the crushed liquisolid tablets prepared. About 5 mg of the sample was sealed in the aluminum pans and heated at the rate of 10 °C/min, covering a temperature range of 0–250 °C under nitrogen atmosphere of flow rate 100 ml/min.
X-ray diffraction (XRD)
X-ray diffraction experiments were performed on MC, Avicel 102, Aerosil 200, their physical mixture in the ratio 1:1:1 and the crushed liquisolid tablets prepared using X-ray diffractometer. The patterns of the samples were recorded with area detector operating at a voltage of 40 kV and current of 20 mA using CuKa radiation (k = 0.154 nm) and X-ray tube (PW3373/00CuLFF). X-ray diffractograms were obtained at scanning rate of 0.3°/min, and the scanning scope of 2θ was from 3 to 60 at room temperature.
Stability study
Tablets of selected formulations were stored at 40 °C ± 2 °C and 75% ± 5% relative humidity for 3 and 6 months to undergo an accelerated stability study (ICH, Citation2003). Stored tablets were examined visually for any changes in every week. Moreover, the dissolution profiles using 500 ml 0.1 N HCl was repeated after the storage period as previously discussed for the fresh tablets. Dissolution profiles of the fresh and stored tablets were compared using the similarity factor (ƒ2).
Pharmacokinetic study in healthy volunteers
Subject selection
Six healthy human male volunteers with age range between 20 and 40 years were selected for the study. The selected subjects had no history of drug or alcohol abuse as well as no chronic disease. The volunteers were instructed not to take any drug for two weeks before and during the study period. This research was performed according to the international clinical research guidelines, outlined in the Declaration of Helsinki resolved in Helsinki in 1964 and amended in Seoul, South Korea, October 2008. The protocol of the study was approved by the institutional review board of Genuine Research Center, Cairo, Egypt.
Study design and drug administration
Randomized, single dose, two-way crossover open-label study was performed using the prepared LS-4 tablets and the commercial market product. Following an overnight fast of at least 10 h in all phases, subjects swallowed the oral tablets with a cup of water. No additional water or fluids were permitted till two hours after the dose. A standard meal was provided at approximately four hours after drug administration. Meal plans were identical in the two periods and were served approximately at the same time. Subjects were housed post dosing until the 12-h blood draw. Seven days were allowed between the two doses as a washout period.
Sample collection
Blood samples were collected in glass tubes before administration of the formulations and at 0.25, 0.50, 0.75, 1.00, 1.50, 2.00, 2.50, 3.00, 4.00, 5.00, 6.00, 8.00 and 12.00 h after drug administration. All samples were collected, and plasma was immediately separated from the blood cells by centrifugation at 3000 rpm for 10 min and stored frozen at −20 °C until analysis.
Sample preparation
All frozen human plasma samples were thawed at ambient temperature. For each sample, an amount of 0.5 ml was placed in 7 ml glass tubes, and 100 µl of Internal standard (IS) solution was added to each and vortexed for 1 min. Four milliliter methyl-t-butyl-ether was then added and samples were then vortexed for 2 min. The tubes were then centrifuged for 10 min at 4000 rpm (1790 g). The upper organic phases were then evaporated to dryness using centrifugal vacuum concentrator Vacufuge 5301 at 40 °C. Dry residues were then reconstituted in 200 µl of mobile phase, vortexed for 1 min and 20 µl was injected using the autosampler.
LC-MS/MS assay of MC in human plasma
Plasma samples were analyzed for MC using a validated LC–MS/MS method. Twenty microliter aliquots of the processed samples were injected on a Luna C18 (Phenomenex, Torrance, CA) (50 × 4.0 nm), 5 µm particle size using a Shimadzu Prominence (Shimadzu, Japan) series LC system with degasser (DGU-20A3), solvent delivery unit (LC-20AB) and an auto-sampler (SIL-20AC). The isocratic mobile phase was formed of 70% acetonitrile and 30% 0.05 M ammonium acetate that was delivered at a flow rate of 1.0 ml/min into the mass spectrometer’s electrospray ionization chamber. Quantitation was done by MS/MS detection in positive ion mode for both MC and Itopride (Internal standard), using a MDS Sciex (Foster City, CA) API-3200 mass spectrometer, equipped with a Turbo ion spray TM interface at 500 °C. The ion spray voltage was adjusted at 5500 V and the common parameters, nebulizer gas, curtain gas, auxillary gas and collision gas were adjusted at 40, 10, 40 and 7 psi, respectively. The compound parameters, declustering potential, collision energy, entrance potential and collision exit potential were 45, 31, 5.5 and 6 V for MC and 30, 23, 6 and 4 V for IS, respectively. The ions were detected in the multiple reactions monitoring mode, monitoring the transition of the m/z 421.9 precursor ions to the m/z 198.1 for MC and m/z 358.7 precursor ion to the m/z 72.0 for IS. Quadrupoles Q1 and Q3 were set on unit resolution. The analytical data were processed by Analyst software (version 1.6).
Pharmacokinetic and statistical analysis
Plasma concentration–time data of MC was analyzed for each subject by non-compartmental pharmacokinetic models using Kinetica® software (version 4.4.1). The peak plasma concentrations (Cmax) and the time of their occurrence (Tmax) were directly obtained from the concentration–time data. The area under the plasma concentration–time curve (AUC0–12) from time zero to last measured concentration was calculated according to the linear trapezoidal rule. The elimination rate constant (λz) was estimated by linear regression of the terminal part of the ln(concentration)–time curve, and the elimination half life was calculated. The relative bioavailability of MC tablets compared to the commercial product was calculated according to the following equation:
Data analysis
All data were expressed as mean ± SD. All the results and the presented data in the study were compared using one-way analysis of variance followed by the least significant difference test or Student t-test using StatView software, version 5, USA.
Results and discussion
Solubility studies
The average solubility of MC in PEG 400, propylene glycol and glycerol formal were 15, 175 and 197 mg/ml, respectively. The results showed that the solvating power of glycerol formal for MC was significantly higher than the other solvents used (p < 0.0001). Therefore, it was chosen as the liquid vehicle for the preparation of the liquisolid systems.
Flow properties of liquisolid powders
The flow properties of the liquisolid compacts are listed in . The results shows that formulae prepared using Avicel PH 102 (LS-1, LS-2, LS-3 and LS-4) were superior in their flowability shown in their lowest Carr’s index, lowest angle of repose and best HR. This was due to the good flowability of Avicel PH 102 and its good loading capacity to glycerol formal. Moreover, the presence of Aerosil particles, which were adsorbed onto host powder particles smoothing out its surface, thus decreasing both friction and mechanical interlocking of particles during flow. On the other hand, shows that formulae prepared using lactose and mannitol almost had the same flow properties, which is lower than those prepared by Aerosil.
Evaluation of the prepared liquisolid tablets
lists the quality control tests of the prepared liquisolid tablets. The liquisolid tablets compiled with the BP requirements of the uniformity of tablet weight and drug content. All the liquisolid tablets had acceptable friability with a percentage loss in tablet weights <1%. However, formulae prepared using Avicel PH 102 as a carrier were superior in comparison with others prepared using mannitol or lactose D.C. This might be due to the binding property of Avicel PH 102 as well as its high compressibility.
Table 2. Quality control tests of the liquisolid tablets.
Formulation should have optimum tablet hardness without applying excessive compression force, while at the same time, assuring rapid tablet disintegration and drug dissolution. Formulae prepared using Avicel (LS-1, LS-2, LS-3 and LS-4) were more hard than those prepared using lactose and mannitol (p < 0.01). That reveals the good compressibility of Avicel containing formulae.
The hardness of Avicel containing formulae might be due to the nature of the microcrystalline cellulose particles. The particles are held together by hydrogen bonds, and they are deformed plastically when compressed forming a strong compact due to the large number of surfaces brought in contact during the plastic deformation as well as the strength of the hydrogen bonds formed (Shangraw, Citation1989). Furthermore, presence of Aerosil 200 is responsible for the hardness in all formulations (Augsburger & Shangraw, Citation1966). It showed that as the level of Aerosil 200 in the formulation increased, the hardness of the tablets increased.
In vitro disintegration test results shows that most of the liquisolid tablets had acceptable disintegration time which is <5 min. However, LS-1 and LS-3 formulae needed more than 5 min to disintegrate, and this might be due to the presence of Avicel PH 102, which has high compressibility that in turn produce hard tablets on compression. It is worthy to note that although LS-2 and LS-4 tablets were also prepared using Avicel PH 102, yet they have lower disintegration time compared to LS-1 and LS-3, this might be due to the lower punch size used in their compression (8 mm) compared to 16 nm in case of LS-1 and LS-3.
Dissolution studies
Dissolution in 0.1 N HCl
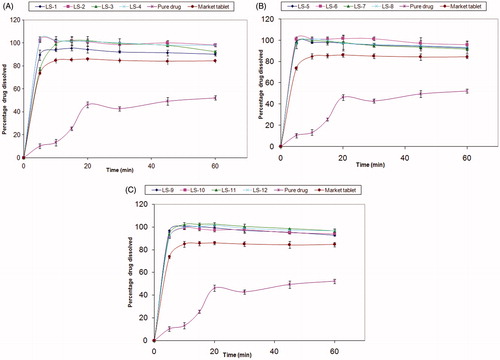
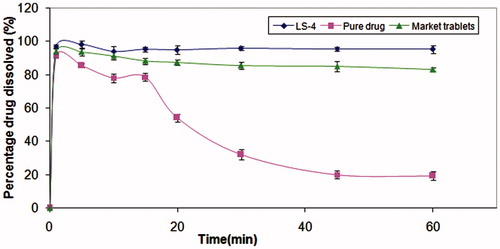
shows the dissolution profiles of MC from the liquisolid tablets, pure drug and conventional market tablets in pH 1.2. In vitro dissolution profiles of MC reveal the great dissolution pattern enhancement in both rate and extent attained by the liquisolid technique compared to the pure drug and the conventional market tablets, where the average percent of MC dissolved after 10 min from liquisolid tablets was >90%, while pure drug and conventional market tablets released only 13.33 and 84.9%, respectively. Moreover, almost 100% of dissolution occurred within 60 min from all liquisolid tablets, while pure drug and conventional market tablets released only 52.3% and 84.7% within the same time, respectively.
Figure 1. Dissolution profile of tablets in 500 ml 0.1 N HCl. (A) Avicel formulae, (B) mannitol formulae and (C) lactose formulae.
This enhanced drug dissolution patterns from liquisolid systems might be attributed to several mechanisms. First, the increased surface area of drug available for release, where the liquisolid tablets contain a solution of MC in glycerol formal solvent adsorbed on the powder carrier. Hence, the drug is available in a solubilized, molecularly dispersed state, which in turn extremely increases surface of the drug available for dissolution. In contrast, conventional MC tablets are just exposing micronized drug particles (Spireas & Sadu, Citation1998; Javadzadeh et al., Citation2007a). Second, it is expected that the solubility of the MC might be increased with liquisolid systems, but the small amount of glycerol formal in a liquisolid compact is not sufficient to increase the overall solubility of the MC in the dissolution medium. However, at the solid–liquid interface between the liquisolid primary particle and the release medium, it is possible that in this microenvironment, the amount of glycerol formal diffusing out of a single liquisolid particle together with the MC molecules might be sufficient to increase the solubility of MC if the liquid vehicle acts as a co-solvent. Such an increase in solubility will result in a larger drug concentration gradient thereby increasing the dissolution rate (Spireas & Sadu, Citation1998; Javadzadeh et al., Citation2007b). Third, improvement in the wetting of the liquid–solid primary particles might have enhanced dissolution. Another explanation to this dissolution enhancement is the increase in thermodynamic activity of MC through reaching a supersaturation state. Using liquisolid compacts, one can create in situ supersaturated solution from subsaturated drug solution incorporated in liquisolid dosage form. After oral administration, the subsaturated drug solution will mix with aqueous milieu in the GIT (in which the drug is less soluble), changing the vehicle composition and creating in situ freshly prepared supersaturated solution. This induced supersaturation can provide enhanced thermodynamic activity driving force for dissolution (Moser et al., Citation2001).
For better comparison, Q5 (percent of MC dissolved within 5 min) from liquisolid compacts was investigated using factorial design to study the effect of the drug concentration, the carrier type and the R factor of the coating material.
The drug concentration had a significant effect on Q5 (p < 0.001). shows that at high drug concentration (10%), Q5 was significantly increased compared to the low drug concentration (5%) with values of 97.49% and 92.03%, respectively. This might be due to the higher amount of solvent used in case of formulae with low drug concentration (5%), which required more carrier and coating material to maintain the good flow and compressibility of powder and thus entrapping the drug within these excipients causing a significant retardation of drug release.
Figure 2. Plots for the main effect of the different factors on Q5. (A) Drug concentration, (B) carrier type and (C) R factor of the coating material.
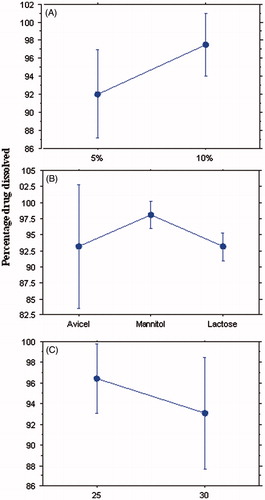
shows that the type of diluent affected significantly the release of the drug in the first five minutes (p < 0.001). Tablets prepared using mannitol gave the highest MC dissolved after 5 min with Q5 value of 98.06% compared to 93.12% and 93.11% in case of Avicel and lactose, respectively. This might be due to the low disintegration time of the mannitol tablets as a result of their low hardness, moreover being a water soluble diluent, it dissolved rapidly and hence enhanced dissolution. The lower Q5 values in the formulations prepared by Avicel might be due to the higher hardness values of these formulations as a result of the good compressibility of Avicel. However, these tablets did not take more than 10 min to disintegrate and release >90% of the drug. It was shown that Avicel PH 102 had more Lf (0.25) in comparison with lactose D.C and mannitol (0.2). Furthermore, Avicel PH 102 has superior physical and compressibility properties, which appeared through tabletting with higher hardness values. Moreover, it has a disintegrating property.
shows that the release of MC from the liquisolid formulations did not differ significantly by changing R factor from 25 to 30 (p > 0.001). It is worthy to note that Spireas et al. recommended R-value not less than 20 in case of immediate release solid dosage form (Spireas et al., Citation1999). They revealed that liquisolid compacts with lower R-values (carrier:coating ratio) contain smaller amounts of carrier powder, and larger amounts of fine drug-loaded silica particles, and the ratios of the amounts of their liquid medication per powder are higher. However, liquisolid compacts with higher R values contain low liquid/powder ratios, high presence of carrier and low presence of silica. This could be directly related to deaggregation, enhanced wicking and disintegration, and thus an increase in the R-value results in an enhanced release rate. While, the liquisolid tablets with low R-values have poor dissolution (Spireas et al., Citation1998). In such cases, even though drug diffusion out of the primary particles may be rapid, oversaturation might occur resulting in local precipitation/recrystallization of the drug and thus decreased release rate (Spireas et al., Citation1999; Javadzadeh et al., Citation2007a). Moreover, as silica is a hydrophobic material, high amounts of it can cause retardation of drug release.
Based on the above results of the effect of the drug concentration, the carrier type and the R factor of the coating material on Q5, combinations of liquisolid compacts prepared using 10% drug concentration, Avicel and both tried R values (25 and 30) viz LS-2 and LS-4 were subjected for further studies.
Hypoacidic stomach model
Dissolution of poorly soluble, weakly basic drug as MC is very hard to simulate owing to the variability in gastric conditions. The pH has a great influence on solubility; hence any modest change in pH can create a change in solubility. To test the robustness of the formulation to variations in gastric pH, dissolution should be obtained not only in pH 1.2 but also in a model that reflects the conditions in the hypochlorhydric stomach as a result of aging, some diseases as H. pylori infection, after some meals or due to using some drugs. Acetate buffer adjusted to pH 5 and having a very low buffer capacity is a good choice for this model, since hypochlorhydria exists by reduction in HCl secretion rather than the addition of buffer species (Dressman & Krämer, Citation2005).
The discrimination with respect to robustness of the release of the liquisolid tablets (LS-2 and LS-4) against conventional market tablets using the higher pH medium is shown in . Conventional market tablets showed a significant decrease in the rate of dissolution in acetate buffer, where the amount of drug dissolved decreased from 84.9% in 0.1 N HCl to 55.6% after 10 min; however, only a slight decrease has occurred from 84.57% in 0.1 N HCl to 82.3% in acetate buffer after 60 min. As for the liquisolid tablets, it was observed that the percentage of drug dissolved from LS-2 decreased from 98.27% in 0.1 N HCl to 87.84% in acetate buffer after 60 min, and, in case of LS-4 only a slight decrease has occurred from 98.24% in 0.1 N HCl to 93.6% in acetate buffer keeping their dissolution rate without significant decrease along time of dissolution.
Figure 3. Dissolution profile of tablets in 500 ml 0.1 N HCl and acetate buffer.
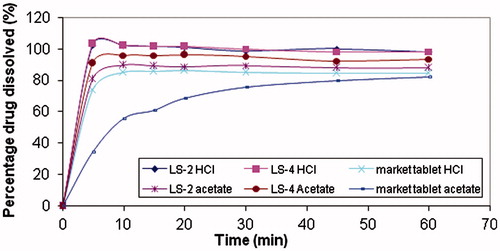
The insignificant decrease in dissolution in the hypoacidic model may be explained by the increase in the solubility of the MC, through usage of liquisolid technique as discussed previously. In fact, the relatively small amount of glycerol formal in a liquisolid compact is not sufficient to increase the overall solubility of MC in the aqueous dissolution medium. However, at the solid–liquid interface between an individual liquisolid primary particle and the release medium, it is possible that in this microenvironment, the amount of glycerol formal diffusing out of a single liquisolid particle together with the MC molecules might be sufficient to increase the solubility of MC if the liquid acts as a co-solvent (Spireas & Sadu, Citation1998; Javadzadeh et al., Citation2007a). The superiority of LS-4 compared to LS-2 might be due to its lower amount of silica, which could be due to enhanced wicking, disintegration and deaggregation. Hence, LS-4 was selected for further studies.
Transfer model
When the drug moves from the stomach into the small intestine through the pylorus, it will meet rapid changing environment including pancreatic and bile secretions, which contain different enzymes and surface active bile components, and increase in pH, so, simulation of conditions in the GIT is crucial to predict the in vivo behavior of drug formulation to decrease the number of human studies required to assess a drug with suitable performance in both fast and fed state.
The pH of GIT fluids is of central importance in determining the solubility of ionizable drugs. For the specific case of poorly soluble weak bases like MC, one must not only consider solubility and dissolution but also the possibility of precipitation as the drug moves from the favorable pH conditions in the stomach to a less favorable pH environment in the small intestine. As the pH approaches or even exceeds the pKa of the basic drug, its solubility undergoes a sharp decrease and the concentration of the drug may in fact exceed the solubility (Kostewicz et al., Citation2004). The question is does the drug stay in supersaturated solution till complete absorption occurs or does it precipitate and become no longer available for absorption. A Transfer model that simulates drug transfer out of the stomach into the intestine gives good prediction of the in vivo behavior. In this model, a solution of the drug dissolved in the simulated gastric fluid is allowed to transfer into simulated intestinal fluid at a suitable rate of (0.5–9 ml/min) under paddle rotation. Drug precipitation in the acceptor medium is examined via concentration time measurements (Kostewicz et al., Citation2004). This is performed under both fasting and fed conditions.
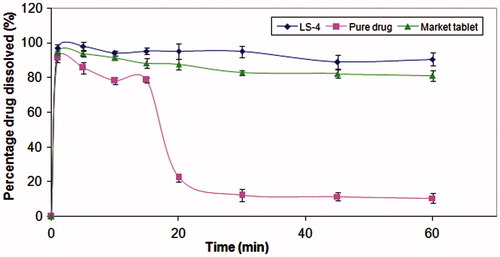
Upon transfer of pure drug dissolved in simulated gastric fluid SGFfast pH = 2 to FaSSIF at a constant rate of 8.5 ml/min, slight turbidity was observed after 15 min, which turned into a white color turbidity in all the vessel after 20 min. Moreover, shows that when pure drug was transferred from acidic to alkaline pH in FaSSIF, the amount of dissolved drug decreased from 91.3% to 10.5% after 60 min. MC is a poorly soluble weak basic drug, which when moves from the favorable pH conditions in the stomach to a less favorable pH environment in the small intestine that is almost equal to, or even exceeds, the pKa of the base (6.5), its solubility undergoes a sharp decrease and the concentration of the drug in the solution may, in fact, exceed the solubility and hence turbidity occurs in the vessel (Kostewicz et al., Citation2004). On the other hand, the conventional market tablets showed slight turbidity in the vessels and the amount of MC dissolved after their transfer from simulated gastric fluid SGFfast pH = 2 to FaSSIF decreased from 93.87% to 81.11% (). Concerning the liquisolid tablet (LS-4), no precipitation or turbidity occurred in the first 30 min; however, slight turbidity appeared after 30 min. After complete transfer, a slight decrease in the amount of drug dissolved from 96.91% to 90.59% have occurred with the amount of the drug dissolved still be more than 90% till 60 min, which is considered a suitable period for complete absorption of the drug ().
Figure 4. Concentration of MC after the transfer of pure drug, conventional market tablets and LS-4, from SGFfast to FaSSIF at a constant rate of 8.5 ml/min.
After the transfer of LS-4 from SGFfast pH = 2 to FeSSIF, no turbidity or precipitation occurred throughout time of the test (60 min). Moreover, there was nearly no decrease in the amount of drug dissolved (). However, the turbidity was still obvious in case of the conventional market tablets, and a milky appearance was shown in case of pure drug after 15 min as pH 5 was still near Mosapride pKa 6.5. These results revealed that liquisolid technique improved solubility and dissolution of MC greatly through increasing its thermodynamic activity and made them nearly pH independent, which appeared in preventing its precipitation along the changes in pH of transfer model from 2 to 6.5.
Figure 5. Concentration of MC after the transfer of pure drug, conventional market tablets and LS-4, from SGFfast to FeSSIF at a constant rate of 8.5 ml/min.
Better results as shown in ( and ) occurred after the transfer from simulated gastric fluid SGFfast pH = 2 to FeSSIF compared to FaSSIF as the FeSSIF pH is slightly lower than that in the fasted state and the amount of sodium taurocholate and lecithin were higher in fed state in comparison with fasted state, which worked as surfactants and increased wettability of MC. Generally, the fed-state intestinal pH is slightly lower than that in the fasted state (Dressman et al., Citation1990), and the volume of the lumenal contents increases to about 1 ± 1.5 L (Fordtran & Locklear, Citation1966). Furthermore, bile is secreted in response to food arrival in the small intestine, so the concentrations of amphiphilic bile salts and phosphatidylcholine are elevated. The decrease in pH together with the increased volume and the bile input would all be favorable to solubility and dissolution of weak bases. Hence, we can conclude that taking medication containing MC in fed state is more recommended than in fasted state.
In summary, use of biorelevant media to determine solubility in the upper gut combined with assessment of formulation with respect to robustness and ability to protect drug from precipitation is key to an efficient development process for weakly basic poorly soluble drug and it will lead to better prediction of in vivo behavior of other different drugs (Dressman & Krämer, Citation2005).
From the dissolution studies, LS-4 formula was considered the best formula among all prepared liquisolid systems and it was selected for further stability and bioequivalence studies.
Differential scanning calorimetry
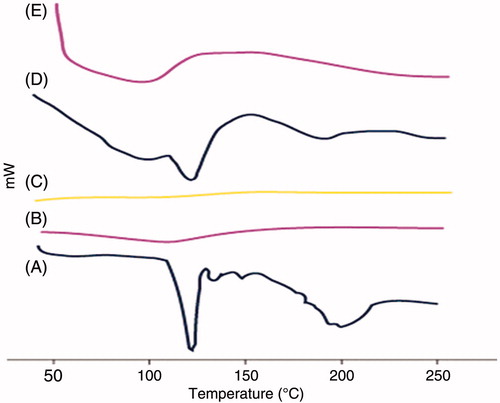
DSC thermograms for MC, Avicel PH 102, Aerosil 200, physical mixture and crushed liquisolid tablet LS-4 are shown in . MC peaks are clear in its DSC thermogram demonstrating a sharp characteristic endothermic peak at 114.27 °C corresponding to its melting temperature; such sharp endothermic peak signifies that MC used was in pure crystalline state. The thermogram of Avicel PH 102 displayed a short broad endothermic peak at 110.34 C that might correspond to the volatilization of adsorbed water and decomposition of the crystalline cellulosic material. Aerosil 200 did not show any sharp peaks; indicating that the coating substance was in an amorphous state. Physical mixture showed the same peak of MC in the same area with some shortening, which indicates the absence of interaction between MC and the used excipients. On the other hand, the liquisolid system thermogram displayed complete disappearance of the characteristic peak of MC indicating the formation of drug solution in the liquisolid powdered system.
Figure 6. DSC thermograms of (A) MC, (B) Avicel PH 102, (C) Aerosil 200, (D) physical mixture and (E) liquisolid system.
X-ray diffraction (XRD)
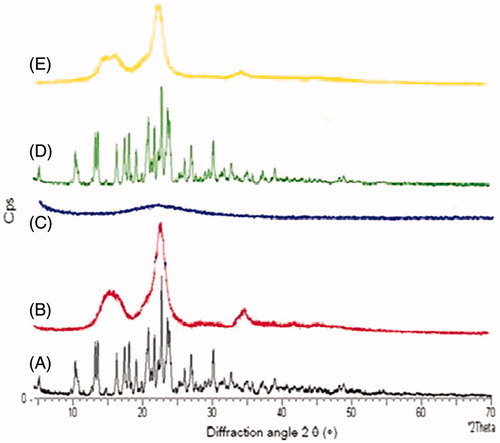
XRD patterns for MC, Avicel PH 102, Aerosil 200, physical mixture and crushed liquisolid tablet LS-4 are shown in . The XRD results were in good agreement with the thermal analysis data, where X-ray diffraction pattern revealed that MC was clearly in crystalline state. The crystalline nature of the drug was demonstrated by the characteristic XRD pattern with peaks appearing at 2θ values 13.19, 16.28, 17.46, 18.08, 20.87 and 22.76. MC characteristic peaks were observed in the physical mixture, indicating no change in its crystalline nature during the physical mixing. On the other hand, the diffraction pattern of the liquisolid powder showed only one sharp diffraction peak at 2θ angle 22.5 due to Avicel PH 102, indicating its crystalline state. Such absence of MC peaks in the liquisolid X-ray diffractogram indicates that drug was changed from crystalline to amorphous form, such lack of crystallinity in the liquisolid system indicates MC solubilization in the liquid vehicle. This amorphization or solubilization of MC in the liquisolid system may contribute to the consequent improvement in the apparent solubility, dissolution rate and therefore the bioavailability of MC.
Figure 7. X-ray diffractograms of (A) pure MC, (B) Avicel PH 102, (C) Aerosil 200, (D) physical mixture and (E) crushed liquisolid tablet.
Stability studies
There was no visual change in the color or the appearance of LS-4 during storage time. There was no significant change in the dissolution of the aged tablets after the accelerated stability study compared to the freshly prepared ones (Figure not shown). Similarity factor of the two release profiles against fresh tablets was 84.812 and 68.07 after three and six months, respectively. This means that aging has no effect on dissolution behavior of MC liquisolid tablets. Based on the results of the storage conditions, LS-4 tablets were selected for further in vivo studies.
Pharmacokinetic study in healthy volunteers
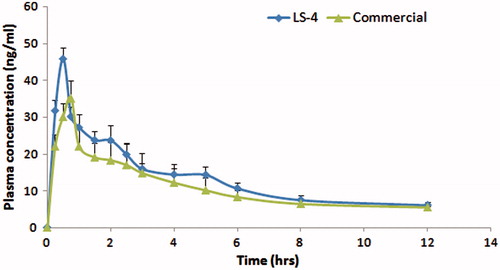
The mean plasma concentrations of commercial MC (Mosapride®) and liquisolid (LS-4) formulations after oral administration to human volunteers are illustrated in . MC commercial tablets gave plasma concentration of 30.16, 35.15 and 22.09 ng/ml at 0.5, 0.75 and 1 h, respectively. On the other hand, LS-4 gave plasma concentration of 31.77, 45.86 and 30.16 ng/ml at 0.25, 0.5 and 0.75 h, respectively.
Figure 8. Mean plasma concentrations time profiles of MC from different formulae after oral administration to human volunteers.
The comparative pharmacokinetic parameters after oral administration of MC formulae are reported in . The mean values for the maximum plasma concentration (Cmax) were 35.38 ± 4.52 and 45.86 ± 3 ng/ml after the oral administration of the commercial product and LS-4 to the volunteers, respectively. In addition, the mean values for time to peak plasma concentration (Tmax) were 0.71 ± 0.1 and 0.5 ± 0 h, while the mean values for the area under the plasma concentration-time curve AUC(0–12) were 133.77 ± 22.79 and 162.12 ± 9.62 ngh/ml, for the two aforementioned formulae, respectively. Furthermore, the two formulations exhibited mean residence time of 6.25 ± 0.06 and 5.81 ± 0.63 for commercial product and LS-4, respectively. Liquisolid compacts enhanced the oral bioavailability of MC with a relative oral bioavailability of 121.2% compared to the commercial product.
Table 3. Pharmacokinetic parameters of MC after oral administration of LS-4 and the commercial product.
Conclusion
The use of biorelevant media to evaluate the robustness of poorly soluble weak bases to variations along the GIT after incorporation in liquisolid compacts proved to be an efficient development process that was successful in predicting the in vivo behavior.
Declaration of interest
The authors report no conflict of interest.
References
- Augsburger LL, Shangraw RF. (1966). Effect of glidants in tableting. J Pharm Sci 55:418–23
- Carr RL. (1965). Evaluating flow properties of solids. Chem Eng 18:163–8
- Dressman JB, Berardi RR, Dermentzoglou LC, et al. (1990). Upper gastrointestinal (GI) pH in young, healthy men and women. Pharm Res 7:756–61
- Dressman J, Kramer J. (2005). Pharmaceutical dissolution testing. : Boca Raton, FL, USA. 213–19
- El-Hammadi M, Awad N. (2012). Investigating the use of liquisolid compacts technique to minimize the influence of pH variations on loratadine release. AAPS PharmSciTech 13:53–8
- Fordtran JS, Locklear TW. (1966). Ionic constituents and osmolality of gastric and small-intestinal fluids after eating. Am J Dig Dis 11:503–21
- Hausner H. (1967). Friction conditions in a mass of metal powder. Int J Powder Metall 3:7–13
- International Conference on Harmonisation (ICH). (2003). Stability testing of new drug substances and products Q1A(R2). Available from: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q1A_R2/Step4/Q1A_R2__Guideline.pdf
- Javadzadeh Y, Jafari-Navimipour B, Nokhodchi A. (2007a). Liquisolid technique for dissolution rate enhancement of a high dose water-insoluble drug (carbamazepine). Int J Pharm 341:26–34
- Javadzadeh Y, Siahi MR, Asnaashari S, Nokhodchi A. (2007b). An investigation of physicochemical properties of piroxicam liquisolid compacts. Pharm Dev Technol 12:337–43
- Kostewicz ES, Wunderlich M, Brauns U, et al. (2004). Predicting the precipitation of poorly soluble weak bases upon entry in the small intestine. J Pharm Pharmacol 56:43–51
- Kumagai Y, Fujita T, Ozaki M, et al. (2005). Pharmacokinetics ofmosapride citrate in patients withgastrectomy. Clin Pharmacol Ther 77:51–3
- Mizuta Y, Shikuwa S, Isomoto H, et al. (2006). Recent insights into digestive motility in functional dyspepsia. J Gastroenterol 41:1025–40
- Moser K, Kriwet K, Froehlich C, et al. (2001). Permeation enhancement of a highly lipophilic drug using supersaturated systems. J Pharm Sci 90:607–16
- Ramenskaia GV, Shokhin IE, Savchenko AI, Volkova EA. (2011). The dissolution test in biorelevant media as a prognostic tool for modeling of drug behavior in vivo. Biomed Khim 57:482–9
- Reppas C, Vertzoni M. (2012). Biorelevant in-vitro performance testing of orally administered dosage forms. J Pharm Pharmacol 64:919–30
- Ruth M, Hamelin B, Rohss K, Lundell L. (1998). The effect of mosapride, a novel prokinetic, on acid reflux variables in patients with gastro-oesophageal reflux disease. Aliment Pharmacol Ther 12:35–40
- Shangraw RF. (1989). Compressed tablets by direct compression. 2nd ed. In: Lieberman HA, Lachman L, Schwartz JB, eds. Pharmaceutical dosage forms tablets. vol. 1. , Marcel Dekker Inc., 195–220
- Spireas S, Sadu S, Grover R. (1998). In vitro release evaluation of hydrocortisone liquisolid tablets. J Pharm Sci 87:867–72
- Spireas S, Wang T, Grover R. (1999). Effect of powder substrate on the dissolution properties of methyclothiazide liquisolid compacts. Drug Dev Ind Pharm 25:163–8
- Spireas S, Sadu S. (1998). Enhancement of prednisolone dissolution properties using liquisolid compacts. Int J Pharm 166:177–88
- Staniforth JN. (2002). Powder flow. In: Aulton ME, ed. Pharmaceutics: the science of dosage form design. : Churchill Livingstone, 205–8
- Yoshikawa T, Yoshida N, Mine Y, Hosoki K. (1998). Affinity of mosapride citrate, a new gastroprokinetic agent, for 5-HT4 receptors in guinea pig ileum. Jpn J Pharmacol 77:53–9