
Abstract
Bone is the primary site of skeletal metastasis in prostate cancer (PCa). Atelocollagen (ATE)-mediated siRNA delivery system can be used to silence endogenous genes involved in PCa metastatic tumor cell growth. However, we hope that the delivery system can target PCa cells to reduce damage to the bone tissue and improve the therapeutic effect. RNA aptamer (APT) A10-3.2 has been used as a ligand to target PCa cells that express prostate-specific membrane antigen (PSMA). APT was investigated as a PSMA-targeting ligand in the design of an ATE-based microRNA (miRNA; miR-15a and miR-16-1) vector to PCa bone metastasis. To observe the targeted delivery and transfection efficiency of ATE–APT in PSMA-overexpressing cells, luciferase activity and biodistribution of nanoparticles in Balb/c mice was analyzed. The anticancer effect of nanoparticles in vivo was investigated using the survival times of human PCa bone metastasis mice model. Luciferase assays of pGL-3 expression against PC3 (PSMA−) and LNCaP (PSMA+) cells showed that the transfection efficiency of the synthesized DNA/ATE–APT complex was higher than that of the DNA/ATE complex. The anticancer efficacy of miRNA/ATE–APT was superior to those of other treatments in vivo. This PSMA-targeted system may prove useful in widening the therapeutic window and allow for selective killing of PCa cells in bone metastatic foci.
Introduction
Autopsy showed that more than 90% patients who died from prostate cancer (PCa) had bone metastasis, indicating that bone is the primary site of skeletal metastasis in PCa (Keller & Brown, Citation2004; Siegel et al., Citation2012). Skeletal metastasis may cause serious complications including bone pain, impaired mobility, pathological fracture, spinal cord compression and symptomatic hypercalcemia. Various therapeutic approaches have been explored in animal studies and clinical trials, offering some hope for better outcomes in the treatment of PCa. But unfortunately, most of these approaches have produced modest results as a whole (Takeshita et al., Citation2010).
MicroRNA (miRNA) or siRNA has been widely used for the treatment of PCa. Recent reports showed that miR-15a and miR-16-1 acted as tumor suppressor genes in PCa and associate with the expression of BCL2, CCND1 and WNT3A, thus promoting survival, proliferation and invasion in advanced PCa cells (Sherr, Citation1996; Dhanasekaran et al., Citation2001; Clevers, Citation2006; Bonci et al., Citation2008). These miR-15a and miR-16-1 are coded on chromosome 13q14. Dong et al. (Reed, Citation1998; Dong et al., Citation2001) reported that initiation and metastasis of high-grade and high-stage PCa were attributed to the loss of heterogeneity at 13q14, suggesting that miR-15a and/or miR-16 could be a novel therapeutic target against PCa metastasis. But the key to the successful use of the therapeutic strategy depends on how to deliver miRNA to target cells safely and effectively (Lu et al., Citation2006).
Atelocollagen (ATE) is a highly purified pepsin-treated type I collagen from the calf dermis. Collagen is a fibrous protein in the connective tissue that plays an important role in the maintenance of the morphology of tissues and organs (Sano et al., Citation2003). Recent studies (Ochiya et al., Citation1999; Honma et al., Citation2001) have demonstrated that ATE complexed with DNA or RNA molecules was efficiently transduced into mammalian cells and allowed long-term gene expression (Ochiya et al., Citation1999; Honma et al., Citation2001). In addition, ATE displays low-toxicity and low-immunogenicity when it is transplanted in vivo (Ochiya et al., Citation2001; Takeshita et al., Citation2005). It was reported that an ATE-mediated siRNA delivery system could be used to silence endogenous genes involved in PCa metastatic tumor cell growth (Minakuchi et al., Citation2004). Although the ATE-mediated siRNA delivery system has proved to be an efficient bone-targeting system, whether it could target PCa cells to reduce damage to the bone tissue and improve the therapeutic effect is unclear.
Aptamers (APTs), RNA or DNA oligonucleotides (Ellington & Szostak, Citation1990; Tuerk & Gold, Citation1990), are ligands that can bind target antigens with high affinity and specificity (Wu et al., Citation2009) by folding into unique three-dimensional conformations (Brody & Gold, Citation2000; Ishida et al., Citation2003). A10 (Lupold et al., Citation2002) is a RNA APT that can bind to the extracellular domain of the prostate-specific membrane antigen (PSMA) that is commonly found on the cell surface of certain PCa cells. Its capability of active binding and uptake by targeted cancer cells in vitro has been demonstrated (Farokhzad et al., Citation2004; Cheng et al., Citation2007; Dhar et al., Citation2008; Tong et al., Citation2010). However, A10 has some deficits such as difficult chemical synthesis, low stability and low activity in vivo, which limit its effect on tumor-specific treatment. To optimize A10, the second-generation APT A10-3.2 has been synthesized recently (Dassie et al., Citation2009). A10-3.2 is obtained through truncating A10 from 71 nt to 39 nt. Compared with A10, A10-3.2 is easy to be synthesized chemically and has enhanced silencing activity and specificity. Its in vivo kinetics can be optimized by modification.
In this study, we used APT A10-3.2 as a new ligand to conjugate with ATE to carry miR-15a and miR-16-1 in an attempt to establish an efficient PSMA-targeting system (miRNA/ATE–APT) to improve the therapeutic effect on PCa. A PSMA-positive PCa cell line LNCaP (PSMA+) was used as a model cell line, and a PSMA-negative PCa cell line PC3 (PSMA–) was used as a control to determine the specificity of the delivery system. The characteristics and anticancer effect of miRNA/ATE–APT were investigated in vitro. An in vivo mice model for human PCa bone metastasis was used to determine the specificity of the delivery system in vivo.
Materials and methods
Materials
Non-human primates were used in this research. The animal study proposal was approved by the Institutional Animal Care and Use Committee (IACUC) of the Second Military Medical University with the permit number: SMMU IACUC2012-022. All Balb/c mice experimental procedures were performed in accordance with the Regulations for the Administration of Affairs Concerning Experimental Animals approved by the State Council of People's Republic of China.
Materials used in this study were as follows: ATE, a highly purified type I collagen of the calf dermis with pepsin treatment (Koken Co., Ltd., Tokyo, Japan); sulfosuccinimidyl-4-(N-maleimidomethyl) cyclohexane-1-carboxylate (sulfo-SMCC, Pierce, Rockford, IL); plasmid pEGFP-N2 (Clontech, Palo Alto, CA) and a pGL3-control vector (Promega, Madison, WI) purified from bacteria using a Qiagen plasmid mega kit (Qiagen GmbH, Hilden, Germany); system and reporter lysis buffer (Promega); anti-PSMA APT (sequence 5′-GGGAGGACGAUGCGGAUCAGCCAUGUUUACGUCACUCCU-(CH2)6-S-S-(CH2)6-OH-3′ with 2′-fluoro pyrimidines), FAM-labeled anti-PSMA APT (FAM–APT) and nuclease-free water (RiboBio, Guangzhou, China); miRNA-15a (sequence 5′-UAGCAGCACAUAAUGGUUUGUG-3′ with 2′-fluoro pyrimidines); miRNA-16-1 (sequence 5′-UAGCAGCACGUAAAUAUUGGCG-3′ with 2′-fluoro pyrimidines), negative control-miRNA (NC-miRNA, sequence 5′-UUCUCCGAACGUGUCACGUUTT-3′ with 2′-fluoropyrimidines) and Cy3-labeled negative control-miRNA (Cy3-NC-miRNA, GenePharma, Shanghai, China); and PC3 and LNCaP cell lines (American Type Culture Collection, Manassas, VA). Male Balb/c mice (4–5 weeks old) weighing 20–25 g were provided by the Department of Experimental Animals of the Second Military Medical University (Shanghai, China) and maintained under standard housing conditions. All animal experiments were carried out in accordance with the guidelines evaluated and approved by the ethics committee of the Second Military Medical University. All the other materials were reagent grade and obtained from commercial sources.
Synthesis of ATE–APT
To prepare 3′-SH-modified APT (SH-APT), 0.1 µmol OH-(CH2)6-S-S-(CH2)6-APT was dissolved in 2.5 mL 100 mM Dithiothreitol (DTT) (pH 8.0) and incubated at room temperature for 30 min. The entire sample was loaded onto a Glen Gel-Pak™ 2.5 (Glen Research, Sterling, VA) desalting column, equilibrated with 25 mL 50 mM sodium phosphate (pH 6.0), allowed to drip through and eluted with 2.5 mL sodium phosphate (pH 6.0). Finally, the purified conjugation of SH-APT was collected (Wu et al., Citation2011).
To prepare Maleimide-activated (MAL-activated) ATE, solutions of ATE concentration 80 μg ml−1 were prepared by serial dilution. The ATE was reacted with a 50-fold molar excess of Sulfo-SMCC for 2 h at room temperature (RT) according to the manufacturer's instructions. Excess crosslinker was removed using a desalting column equilibrated with conjugation buffer. Finally, the purified conjugation of MAL–ATE was collected.
To conjugate the APT with ATE, MAL-activated ATE (80 µg/ml) was dissolved and reacted with SH–APT (20 µg/ml) in nuclease-free phosphate-buffered solution (pH 7.0) for 12 h at 4 °C, and the resulting product ATE–APT was purified by ultrafiltration using a 20 kDa molecular weight cutoff membrane to remove unreacted APT and suspended in nuclease-free water. The anti-PSMA APT-conjugated ATE (ATE–APT) was stored at 4 °C. The corresponding conjugate FAM-labeled ATE–APT (ATE–APT–FAM) was synthesized as described previously.
Preparation of miRNA/ATE–APT complexes
The miRNAs and ATE complexes were prepared as follows. An equal volume of ATE (in PBS at pH 7.4) and miRNAs solution were combined and mixed by rotation at 4 °C for 20 min. The complex was then kept at 4 °C for 16 h before use. The final concentration of ATE in vitro and in vivo was 0.008% and 0.5%, respectively. The corresponding conjugate complexes Cy3–NC-miRNA/ATE, NC-miRNA/ATE–APT–FAM and Cy3–NC-miRNA/ATE–APT–FAM were prepared as described previously. The size and zeta potential of the complexes formed in phosphate-buffered saline (10 mM, pH 7.4) were measured in triplicate using a Zetasizer Nano-ZS90 (Malvern Instruments Ltd., Worcestershire, UK).
Characterization of miRNA/ATE–APT complexes
The characteristics of miRNA/ATE–APT complexes were determined by flow cytometry analysis (FCM). A dispersion of Cy3–NC-miRNA/ATE, NC-miRNA/ATE–APT–FAM and Cy3–NC-miRNA/ATE–APT–FAM was introduced directly into the flow cytometer and analyzed using a FAC Scan flow cytometer (Becton Dickinson, San Jose, CA). NC-miRNA/ATE was also analyzed as a control.
Stability of miRNA/ATE–APT complex
An aliquot of 5 µg of NC-miRNA and ATE–APT or cationic Lipofectamine 2000 (Invitrogen, Carlsbad, CA) complexes were incubated in the presence of 0.1 mg/ml RNase A (NipponGene, Tokyo, Japan) for 0, 5, 15, 30, 45 and 60 min at 37 °C. The solutions were extracted with phenol and phenol/chloroform/isoamyl alcohol (25:24:1). The NC-miRNA was precipitated with ethanol and agarose gel electrophoresed (3.5%) and visualized by ethidium bromide staining (Minakuchi et al., Citation2004).
Efficiency of gene expression in PC3 and LNCaP
PC3 and LNCaP cell lines were cultured in RPMI 1640 medium (Gibco, Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% antibiotics at 37 °C in a humidified atmosphere with 5% CO2. PC3 and LNCaP cells were seeded into 24-well plates (Corning-Coaster, Tokyo, Japan) at a density of 3 × 104 cells per well and incubated at 37 °C for 24 h (to reach 70% confluence) prior to transfection. The DNA/ATE, DNA/ATE–APT or DNA/Lipo 2000 complexes (3 μg DNA/well) were added to the cells in FBS-free medium, and the mixture was incubated at 37 °C for 1 h according to the method described previously (Visai et al., Citation2000; Honma et al., Citation2001). After 48 h, fluorescence images of the gene expression products (green fluorescent protein) were acquired and photographed under a fluorescence microscope. The efficiency of gene transfer was quantitatively determined by luciferase activity using the pGL3-control vector. After 48-h transfection, the activity of the gene expression product, luciferase, was quantified using a luciferase assay system (Promega) following the manufacturer's instructions. Briefly, cells were first lysed in 150 μL cell culture lysis reagent for 30 min at room temperature, the luciferase activity was measured by a luminometer (Promega E6080), and finally the protein content was measured using a micro-bicinchoninic acid protein assay kit.
Western blot analysis
To assess the downregulation of Bcl-2, Cyclin D1 and Wnt3a gene in LNCaP cells, cells were transfected with miRNA (miRNA-15a:miRNA-16-1 = 1:1)/ATE–APT complexes and negative control-miRNA (NC-miRNA)/ATE–APT complexes, harvested at 24, 48, and 72 h post-transfection and lysed in cold Radio-Immunoprecipitation Assay (RIPA) buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1 mM EGTA and 0.25% sodium deoxycholate). The lysate was then incubated for 15 min at 4 °C and removed by centrifugation at 12 000 rpm for 15 min. The supernatant was analyzed for protein concentrations using the Bradford assay (Bio-Rad, Hercules, CA). An equal amount (10 mg) of protein was subjected to electrophoresis on sodium dodecyl sulfate polyacrylamide gels and then transferred to polyvinylidene fluoride membrane (Millipore, Billerica, MA). The blotted membrane was immunostained with antibodies specific to anti-Bcl-2 (BioWorld, Visalia, CA), anti-cyclin D1 (BioWorld) and anti-Wnt3a antibodies (R&D Systems Inc., Minneapolis, MN). Blots were developed by the enhanced chemiluminescence reagents (Amersham Pharmacia, Buckinghamshire, UK) and visualized by the Gene-Genius Imaging System (Syngene, Frederick, MD).
In vitro anticancer effect
The antiproliferation effect of miRNA/ATE–APT on LNCaP cells was evaluated by a cell counting kit-8 (CCK-8, Dojindo, Kumamoto, Japan). LNCaP cells and PC3 cells were seeded in 96-microwell plates at a density of 1 × 104 cells per well and incubated at 37 °C for 24 h. Cells were treated with various concentrations (0.02–50 µM) of miRNA (miRNA-15-a/miRNA-16-1 = 1)/ATE–APT, NC-miRNA/ATE–APT and miRNA (miRNA-15-a/miRNA-16-1 = 1)/ATE for 72 h, followed by addition of 10 μL CCK-8 to each well and incubation for another 2 h. The absorbance of each well was determined with a microplate reader (Thermo Scientific, Rockford, IL), and a standard curve was drawn according to the absorbance and cell number. Each sample was tested in triplicate. The antiproliferation effect was expressed as the IC50 value, concentration that inhibits cell growth by 50% as compared with nontreated controls. IC50 value was calculated according to the equation: y = bottom × (top–bottom)/[1 + 10 (logIC50 − x) × Hill slope], where bottom and top are the minimum and maximum y-axis values, respectively, of a plateau in the curve, and Hill slope is the steepness of the curve using GraphPad 5.0 PRISM® (GraphPad Software Inc., San Diego, CA).
Establishment of the mice model of human PCa bone metastasis
Balb/c mice in each group were anesthetized, and the left leg was cleaned with 70% ethanol. PCa cells were aspirated into a 25-μl syringe fitted with a 25-gauge needle. The needle was inserted through the cortex of the anterior tuberosity of the tibia, 3–5 mm below the diaphysis of the tibia and 10 μl cell suspension was injected. After four weeks, the mice model of human PCa bone metastasis was established (Fisher et al., Citation2002).
In vivo anticancer effect
The anticancer effect of nanoparticles in vivo was investigated using the survival times of human PCa bone metastasis mice model. The mice were randomly divided into four groups and treated with 100 μl of NaCl (0.9%), miRNA (miRNA-15-a/miRNA-16-1 = 25 μg/25 μg)/ATE–APT, NC-miRNA/ATE–APT and miRNA (miRNA-15-a/miRNA-16-1 = 25 μg/25 μg)/ATE days 1, 3, 5 and 7 after establishment. The body weight was monitored every other day, and the death was recorded as occurring on the following day (Xin et al., Citation2010).
Statistical analysis
The data were presented as mean ± standard deviation. Statistically significant differences were determined using two-sample t-test and analysis of variance, with p < 0.05 as the significance level.
Results and discussion
Characterization of miRNA/ATE–APT complexes
ATE exhibits a stick-like structure, 300 nm in length, 1.5 nm in diameter and 300 kDa in molecular weight (Mu et al., Citation2009). It shows neither antigenicity nor toxicity because it is free from antigenic telopeptides with pepsin treatment. ATE is a liquid at 4 °C and a gel at 37 °C when it is concentrated at 0.5%. This biochemical feature of ATE contributes to controlled release of miRNA directly injected into the tumor. We used 0.8% and 0.05% ATE for local and systemic delivery of miRNA. The complex with miRNA formed a nano-sized particle miRNA/ATE 197 ± 7.4 nm (mean ± SD; n = 6; ). However, compared with miRNA/ATE, miRNA/ATE–APT was about 25 nm increased (), presumably owing to the presence of APT on the nanoparticles surface. APT was coupled to ATE, triggering the receptor-mediated mechanism to increase the accumulation of miRNA/ATE–APT in PCa.
Table 1. Size and zeta potential of the nanoparticles (n = 6).
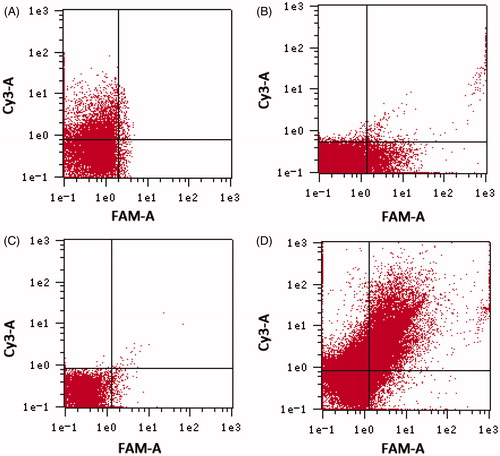
Meanwhile, the binding of APT to ATE was confirmed by FCM. As shown in , non-APT modified Cy3-NC-miRNA/ATE only showed red fluorescence and NC-miRNA/ATE–APT–FAM showed green fluorescence. In contrast, Cy3–NC-miRNA/ATE–APT–FAM showed both red and green fluorescences, suggesting that ATE conjugated APT. Sulfo-SMCC is a commonly used heterobifunctional cross-linker containing N-hydroxysuccinimide (NHS) esters and maleimide groups. The NHS ester groups can covalently bind with sulfhydryl-containing molecules, and the maleimide groups allow covalent conjugation of amino-containing molecules. The maleimide groups of Sulfo-SMCC can react with amino groups of ATE to form amide bonds, resulting in maleimide-activated ATE (Xin et al., Citation2010). MAL–ATE could covalently bind to sulfhydryl groups on SH–APT. This strategy adopts a chemical combination method that uses Sulfo-SMCC to directly cross-link APT onto ATE.
Figure 1. Determination of APT on the surface of nanoparticles by flow cytometry. (A) Cy3–NC-miRNA/ATE; (B) NC-miRNA/ATE–APT–FAM; (C) NC-miRNA/ATE–APT; and (D) Cy3–NC-miRNA/ATE–APT–FAM.
APT, aptamer and ATE, atelocollagen.
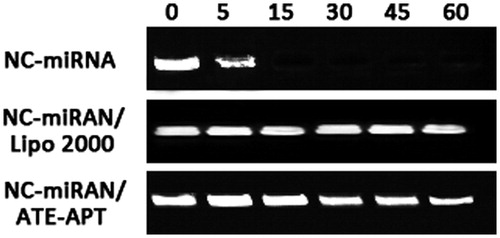
In addition, to examine whether ATE–APT blocks degradation of siRNA from nuclease, naked NC-miRNA, miRNA/Lipofectamine 2000 complex and NC-miRNA/ATE–APT complex were incubated in the presence of RNase (0.1 mg/ml) for 0, 5, 15, 30, 45 and 60 min at 37 °C followed by agarose gel electrophoresis. The results indicated that the NC-miRNA/ATE–APT complex showed partial resistance to degradation of miRNA in the presence of nuclease ().
Figure 2. ATE–APT blocks degradation of siRNA from nuclease, naked NC-miRNA, miRNA/Lipofectamine 2000 complex and NC-miRNA/ATE–APT complex were incubated in the presence of RNase (0.1 mg/ml) for 0, 5, 15, 30, 45 and 60 min at 37 °C and then agarose gel electrophoresed. The presence of miRNA was revealed by ethidium bromide staining. APT, aptamer; ATE, atelocollagen; and NC, negative control.
Efficiency of gene expression in PC3 (PSMA−) and LNCaP (PSMA+)
ATE not only plays an important role in protection against degradation by nucleases and sustained release of miRNA or siRNA but also acts as a positive transporter of nucleases into cells (Xin et al., Citation2010). ATE with positive electric charge interacts with negative-charged nucleic acid electrostatically to form a RNA/ATE or NDA/ATE complex. The shape of this complex can be controlled, depending on condition adjustment (Sano et al., Citation2003).
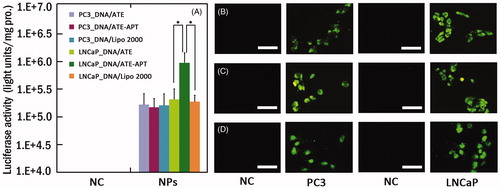
To observe the targeted delivery and transfection efficiency of ATE–APT in PSMA-overexpressing cells, LNCaP cells and PC3 cells were treated with DNA/ATE, DNA/ATE–APT or DNA/Lipo 2000 complexes using 3 µg pEGFP () or pGL3-control vector (). The result of luciferase activity assay indicated that DNA/ATE–APT complexes induced high gene expression in LNCaP cells compared with DNA/ATE complexes, while the gene expression was rarely observed in PC3 cells transfected by DNA/ATE–APT complexes. As shown in , the luciferase activity of ATE–APT (1.074601 × 106 units/mg protein) was 5.4-fold higher than that of ATE (1.93712 × 105 units/mg protein) in LNCaP cells, but no difference was observed in PC3 cells. Compared to DNA/Lipo 2000, DNA/ATE group displayed no significant (p > 0.05) transfection. When compared to DNA/Lipo 2000, DNA/ATE–APT group (p < 0.01) significantly enhanced the transfection efficiency. This might be attributed to the targeting head group APT, which could enhance the transfection efficiency by selectively binding to PSMA-overexpressing cells through the APT-mediated approach. Therefore, when the ATE delivered system to the bone tissue, the APT could mediate the system to target PCa cells at the second step. In this way, the damage to the bone tissue could be reduced, and the therapeutic effect could be improved.
Figure 3. (A) Luciferase activity analysis (n = 4, error bars represent the standard deviation) and (B–D) fluorescence microscopy images (scale bar 50 µm) after transfection of pGL-3/ATE (B), pEGFP/ATE–APT (C) or pEGFP/Lipo 2000 (D) against PC3 and LNCaP cells, respectively. *p < 0.05. APT, aptamer; ATE, atelocollagen; NC, negative control; NPs, nanoparticles; and Lipo 2000, Lipofectamine 2000.
Western blot analysis
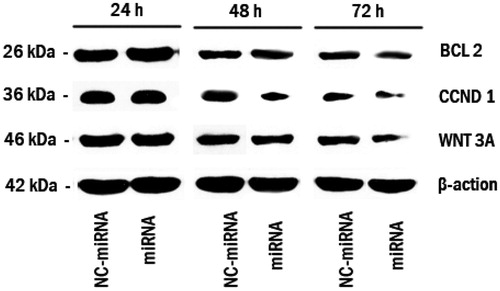
shows the results of Western blot of Bcl-2, Cyclin D1 and Wnt3a as the direct target of miRNA-15a and miRNA-16-1 determined at 24, 48 and 72 h after transfection. Their expressions began decreasing 48 h after miRNA transfection and decreased significantly after 72 h, especially for Wnt3a. However, this phenomenon was rarely observed in NC-miRNA transfection. Thus, downregulation of Bcl-2, Cyclin D1 and Wnt3a by the delivery of miRNA (whose functions are directly associated with cancer progression and invasion) (Tong et al., Citation2010) is expected to increase cell apoptosis. It has been demonstrated that ATE–APT can carry miRNA effectively into cells to exert the inhibitory effect, and therefore the use of miR-15a and miR-16-1 in the treatment of PCa bone metastasis may become a reality.
Figure 4. Bcl-2, Cyclin D1 and Wnt3a knocks down following miRNA/ATE–APT complexes delivery to LNCaP cells. Cells were treated with miRNA/ATE–APT (miRNA) and NC-miRNA/ATE–APT (NC-miRNA) complexes and processed for immunoblotting with anti-Bcl-2, anti-Cyclin D1 and anti-Wnt3a antibodies at 24, 48 and 72 h post-transfection. β-actin was a loading control. APT, aptamer; ATE, atelocollagen; and NC, negative control.
In vitro anticancer effect
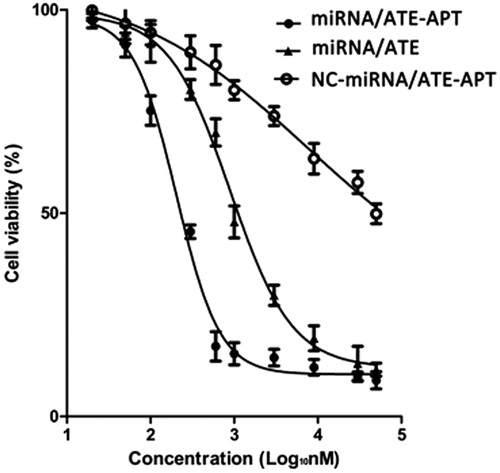
To evaluate the anticancer potential of miRNA/ATE–APT, we performed a series of in vitro cytotoxicity assays using the CCK8 assay to determine the IC50 value. LNCaP cells were treated with miRNA/ATE–APT, NC-miRNA/ATE–APT and miRNA/ATE complexes (0.02–50 µM). The results showed that cell growth was inhibited in a dose-dependent manner (). The calculated IC50 value was 0.204 µM (0.15–0.27 µM), 7.66 µM (4.29–13.7 µM) and 0.896 µM (0.69–1.16 µM), respectively. Therefore, miRNA/ATE–APT had an approximately 4.4-fold lower IC50 value than miRNA/ATE. It could be concluded from the efficiency of gene expression in LNCaP as described earlier that ATE–APT could raise the gene transfection rate in LNCaP, whereby we may infer that ATE–APT could carry more miRNA into LNCaP. In addition, the results of Western blot analysis showed that the more miRNA was carried into LNCaP, the further Bcl-2, Cyclin D1 and Wnt3a were down regulated, which explains why miRNA/ATE–APT had a lower IC50 value. It is therefore reasonable to believe that miRNA/ATE–APT could become a novel nanotargeted drug delivery system for the treatment of skeletal metastasis in PCa.
Figure 5. Viability of LNCaP cells treated with miRNA/ATE–APT, NC-miRNA/ATE–APT and miRNA/ATE complexes (n = 3, error bars represent the standard deviation). APT, aptamer; ATE, atelocollagen; and NC, negative control.
In vivo anticancer effect
The anticancer effect of miRNA/ATE–APT in close comparison with those of miRNA/ATE, NC-miRNA/ATE–APT and NaCl was demonstrated by the survival time of human PCa bone metastasis mice model in . We can see that the anticancer efficacy of miRNA/ATE–APT was superior to those of other treatments. As shown in , the mean survival time of NaCl group, NC-miRNA/ATE–APT group, miRNA/ATE group and miRNA/ATE–APT group were 27.2, 28.2, 38 and 57 d, respectively. Compared to NaCl, NC-miRNA/ATE–APT group displayed no significant prolongation in the survival time (p > 0.05) through log-rank analysis. When compared to NaCl, NC-miRNA/ATE–APT and miRNA/ATE, miRNA/ATE–APT group (p < 0.01) significantly prolonged the survival time. These results implied that ATE–APT was non toxic, and the anticancer efficacy of miRNA/ATE–APT was superior to those of other treatments. What's more, miRNA/ATE–ATE group resulted in significant prolongation in the survival time compared to miRNA/ATE group. It indicates that after the miRNA/ATE–APT was transplanted in vivo, ATE could take the system to the bone tissue. Then, the APT could help the system to identify the PCa cells. In this way, the damage to the bone tissue could be reduced, and the therapeutic effect could be improved. In a word, the anticancer efficacy of miRNA/ATE–APT was superior to miRNA/ATE for human PCa bone metastasis mice model. These results might be explained by the increased local concentration of APT in the tumor tissue, since accumulation of ATE in bone was favored by its efficient bone-targeting property.
Figure 6. Kaplan–Meier survival curve of human PCa bone metastasis mice model. APT, aptamer; ATE, atelocollagen; and NC, negative control.
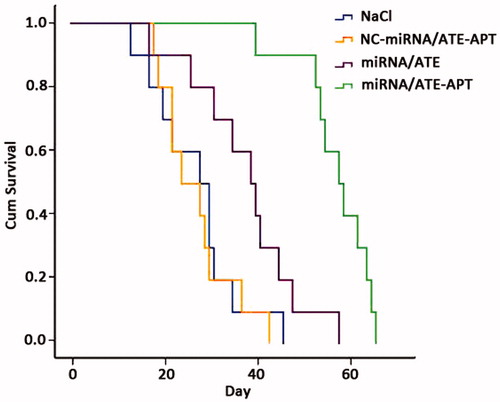
Table 2. In vivo effects of nanoparticles on human PCa bone metastasis mice model.
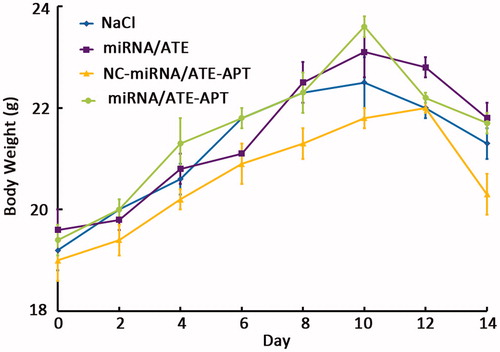
In order to evaluate the safety of these nanoparticles, body weight was monitored as a marker of overall toxicity. The change in body weight was recorded in 14 d because the first mouse in NaCl group died on day 14 after implantation. From , we can see the mice treated with miRNA/ATE–APT, miRNA/ATE and NC-miRNA/ATE–APT gaining weight during 10 d just like that treated with NaCl. These results implied that miRNA/ATE–APT was safe. On the contrary, there was weight loss 12 d after treatment. The cause of body weight loss is the tumor grew rapidly in bone after 12 d. So the miRNA/ATE–APT could be an effective drug delivery system for PCa bone metastasis treatment.
Figure 7. Change in body weight of animals as a function of time in human PCa bone metastasis mice model (n = 10). APT, aptamer; ATE, atelocollagen; and NC, negative control.
Conclusion
In this study, we successfully constructed an efficient target gene delivery system mediated by the second-generation APT (ATE–APT), which could synergistically induce selective cell death of PCa cells by loading miR-15a and miR-16-1. ATE–APT could effectively deliver miRNA to PCa cells that overexpress PSMA, resulting in tumoricidal efficacy as verified by cell viability assays and the survival time of human PCa bone metastasis mice model. It should be emphasized that because ATE–APT uses the second-generation APT A10-3.2, synthesis of the system becomes much easier and biological targeting becomes even stronger. In addition, use of ATE decreases toxicity of the system and endows it with better biocompatibility. Using miRNA provides multiple-target inhibition on PCa and enhances the anticancer effect of the system. Therefore, this research may prove to be a significant contribution to the use of nanocarrier-based targeted delivery of miRNAs for the treatment of skeletal metastasis in PCa.
Declaration of interest
The authors report no conflicts of interest in this work.
We acknowledge the financial support from the National Natural Science Foundation of China (no. 81302212), Medical Science Foundation of Sanya (no.YW1310 and YW1311) and Key scientific and technological project of Hainan (no. ZDXM20110036).
References
- Bonci D, Coppola V, Musumeci M, et al. (2008). The miR-15a-miR-16-1 cluster controls prostate cancer by targeting multiple oncogenic activities. Nat Med 14:1271–7
- Brody EN, Gold L. (2000). Aptamers as therapeutic and diagnostic agents. J Biotechnol 74:5–13
- Cheng J, Teply BA, Sherifi I, et al. (2007). Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials 28:869–76
- Clevers H. (2006). Wnt/beta-catenin signaling in development and disease. Cell 127:469–80
- Dassie JP, Liu XY, Thomas GS, et al. (2009). Systemic administration of optimized aptamer-siRNA chimeras promotes regression of PSMA-expressing tumors. Nat Biotechnol 27:839–49
- Dhanasekaran SM, Barrette TR, Ghosh D, et al. (2001). Delineation of prognostic biomarkers in prostate cancer. Nature 412:822–6
- Dhar S, Gu FX, Langer R, et al. (2008). Targeted delivery of cisplatin to prostate cancer cells by aptamer functionalized Pt(IV) prodrug-PLGA-PEG nanoparticles. Proc Natl Acad Sci U S A 105:17356–61
- Dong JT, Boyd JC, Frierson HF Jr. (2001). Loss of heterozygosity at 13q14 and 13q21 in high grade, high stage prostate cancer. Prostate 49:166–71
- Ellington AD, Szostak JW. (1990). In vitro selection of RNA molecules that bind specific ligands. Nature 346:818–22
- Farokhzad OC, Jon S, Khademhosseini A, et al. (2004). Nanoparticle-aptamer bioconjugates: a new approach for targeting prostate cancer cells. Cancer Res 64:7668–72
- Fisher JL, Schmitt JF, Howard ML, et al. (2002). An in vivo model of prostate carcinoma growth and invasion in bone. Cell Tissue Res 307:337–45
- Honma K, Ochiya T, Nagahara S, et al. (2001). Atelocollagen-based gene transfer in cells allows high-throughput screening of gene functions. Biochem Biophys Res Commun 289:1075–81
- Ishida S, Usui T, Yamashiro K, et al. (2003). VEGF164-mediated inflammation is required for pathological, but not physiological, ischemia-induced retinal neovascularization. J Exp Med 198:483–9
- Keller ET, Brown J. (2004). Prostate cancer bone metastases promote both osteolytic and osteoblastic activity. J Cell Biochem 91:718–29
- Lu W, Takahashi H, Furusato M, et al. (2006). Allelotyping analysis at chromosome 13q of high-grade prostatic intraepithelial neoplasia and clinically insignificant and significant prostate cancers. Prostate 66:405–12
- Lupold SE, Hicke BJ, Lin Y, Coffey DS. (2002). Identification and characterization of nuclease-stabilized RNA molecules that bind human prostate cancer cells via the prostate-specific membrane antigen. Cancer Res 62:4029–33
- Minakuchi Y, Takeshita F, Kosaka N, et al. (2004). Atelocollagen-mediated synthetic small interfering RNA delivery for effective gene silencing in vitro and in vivo. Nucleic Acids Res 32:e109
- Mu P, Nagahara S, Makita N, et al. (2009). Systemic delivery of siRNA specific to tumor mediated by atelocollagen: combined therapy using siRNA targeting Bcl-xL and cisplatin against prostate cancer. Int J Cancer 125:2978–90
- Ochiya T, Nagahara S, Sano A, et al. (2001). Biomaterials for gene delivery: atelocollagen-mediated controlled release of molecular medicines. Curr Gene Ther 1:31–52
- Ochiya T, Takahama Y, Nagahara S, et al. (1999). New delivery system for plasmid DNA in vivo using atelocollagen as a carrier material: the Minipellet Nat Med 5:707–10
- Reed JC. (1998). Molecular biology of chronic lymphocytic leukemia. Semin Oncol 25:11–8
- Sano A, Maeda M, Nagahara S, et al. (2003). Atelocollagen for protein and gene delivery. Adv Drug Deliv Rev 55:1651–77
- Sherr CJ. (1996). Cancer cell cycles. Science 274:1672–7
- Siegel R, Naishadham D, Jemal A. (2012). Cancer statistics. CA Cancer J Clin 62:10–29
- Takeshita F, Minakuchi F, Nagahara S, et al. (2005). Efficient delivery of small interfering RNA to bone-metastatic tumors by using atelocollagen in vivo. Proc Natl Acad Sci U S A 102:12177–82
- Takeshita F, Patrawala L, Osaki M, et al. (2010). Systemic delivery of synthetic microRNA-16 inhibits the growth of metastatic prostate tumors via downregulation of multiple cell-cycle genes. Mol Ther 18:181–7
- Tong R, Yala L, Fan TM, Cheng J. (2010). The formulation of aptamer-coated paclitaxel-polylactide nanoconjugates and their targeting to cancer cells. Biomaterials. 31:3043–53
- Tuerk C, Gold L. (1990). Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 249:505–10
- Visai L, Xu Y, Casolini F, et al. (2000). Monoclonal antibodies to CNA, a collagen-binding microbial surface component recognizing adhesive matrix molecules, detach Staphylococcus aureus from a collagen substrate. J Biol Chem 275:39837–45
- Wu F, Wuensch SA, Azadniv M, et al. (2009). Galactosylated LDL nanoparticles: a novel targeting delivery system to deliver antigen to macrophages and enhance antigen specific T cell responses. Mol Pharm 6:1506–17
- Wu X, Ding B, Gao J, et al. (2011). Second-generation aptamer-conjugated PSMA-targeted delivery system for prostate cancer therapy. Int J Nanomedicine 6:1747–56
- Xin H, Chen L, Gu J, et al. (2010). Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ɛ-caprolactone) nanoparticles: in vitro and in vivo evaluation. Int J Pharm 402:238–47