
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The sole objective of this work was to design successful dosage oral forms of diclofenac sodium (DiNa)-loaded solid lipid microparticles (SLM) based on solidified reverse micellar solution (SRMS). Hot homogenization technique was employed to prepare DicNa SLM using a mixture goat fat and Phospholipon® 90 G as lipid matrix and Tween®-80 as mobile surfactant. Characterization based on percentage yield, morphology, particle size, zeta potential, percentage encapsulation, pH and stability of SLMs were investigated. Anti-inflammatory, gastrointestinal tract (GIT) sparing effect and pharmacokinetics were carried out in rat model after oral administration. Results showed that the SLMs were spherical and smooth. The optimized formulation (SLM-4) had particle size of 79.40 ± 0.31 µm, polydispersity index of 0.633 ± 0.190, zeta potential of −63.20 ± 0.12 mV and encapsulation efficiency of 91.2 ± 0.1% with good stability after 8 months of storage. The DicNa SLM had sustained release effect with good anti-inflammatory activity. Higher and prolonged plasma DicNa concentration was shown by the SLM-4 compared to pure drug and a conventional sample. These studies demonstrate that DicNa-loaded SLM based on SRMS could be a promising oral formulation for enhanced bioavailability, pharmacologic activity and gastrointestinal sparing effect of the NSAID, DicNa.
Introduction
Diclofenac sodium (DicNa) is a non-steroidal anti-inflammatory drug (NSAID) used in the treatment of inflammation and degenerative disorder of the musculoskeletal system. It is used for the relief of pain due to inflammatory conditions such as rheumatoid arthritis, osteoarthritis, acute gout and following some surgical procedures (Wallace, Citation1997; Wallace et al., 1998). However, its low oral bioavailability, short plasma half-life and gastrointestinal tract (GIT) side effects limit its use in many patients with history of intestinal ulcer. It use is further limited by the multiple dosing required to attain effective plasma concentration. However, efforts have been made to address the challenges through the use of formulation approaches (Tunçay et al., Citation2000). Polymeric drug carrier delivery systems have been evaluated as a possible carrier (Momoh et al., Citation2013). However, the scarcity of safe polymers with regulatory approval and their high cost have limited the wide use of polymeric material in clinical medicine (Mukherjee et al., Citation2009). To overcome these limitations, lipids have been proposed as an alternative carrier. Lipid formulations such as solid lipid microparticles (SLMs) or nanoparticles, lipospheres and liposomes, have attracted attention among pharmaceutical formulators (Attama & Nkemnele, Citation2005). The most frequent role of lipid-based formulations has traditionally been used to improve the solubility of sparingly water soluble drugs. However, the spectrum of applications for lipid-based formulations has widened as the nature and type of active drugs under investigation vary. These include the protection of protect active compounds from biologic degradation or transformation, which in turn can lead to an enhancement of drug potency and reduction of drug toxicity by changing the biodistribution of the drug away from sensitive organs. This reduction in toxicity may allow higher drug dosing and forms the basis for the current success of several marketed lipid-based formulations (Attama & Nkemnele, Citation2005; Attama & Christel, 2008; Momoh & Esimone, 2012). Among the lipid formulation systems, SLMs have an added advantage in terms of controlled drug release and ease of formulation.
The aim of this study was to reduce the GI toxicity and ease of use of DicNa by formulating SLMs using the homolipid obtained from goat fat and semi-synthetic lipid (phospholipid). This fat from Capra hircus has been evaluated in the various drug formulations and its stability to rancidity and other physical change such as color, odor and taste has been evaluated (Attama et al., 2003).
Phospholipon® 90 G (P90G) is a purified, deoiled, and granulated soy lecithin with phosphatidylcholine content of at least 90%. It has been used in parenteral formulation (Attama & Müller-Goymann, Citation2006), and as a surface modifier for solid lipid nanoparticles (SLNs) (Jaspart et al., 2007), with resultant improvement in targeting and pharmacokinetics (Attama et al., 2008). Goat fat which is extracted from Capra hircus is cheap and available in Nigeria and with potential for drug delivery applications because of its crystal properties (Attama & Müller-Goymann, Citation2006). It consists mainly of triglycerides of C16, C18 and C18:1 fatty acids (Ulberth & Buchgraber, Citation2003).
Materials and methods
Materials
Phospholipon® 90 G (P90G) (Phospholipid GmbH, Köln, Germany), Tween®-80 (Merck, KGaA, Darmstadt, Germany), monobasic potassium phosphate, sodium hydroxide and concentrated hydrochloric acid (British Drug House (BDH), Poole UK) are used. Homolipid was obtained from a batch processed in our laboratory. Diclofenac sodium was a gift from AC Drugs Ltd, Enugu, Nigeria. Other reagents were of analytical grade and were used without further purification.
Preparation of the lipid matrices
The lipid matrices used in the SLM formulation consisting of 80% mixture of goat fat and 20% P90G were prepared by fusion. Briefly, the lipid components were melted together at 70 °C in a steam bath and stirred until solidification to obtain solidified reverse micellar solution (SRMS).
Formulation of SLM
SLMs containing the drug and the drug free (unloaded) were formulated by containing the following; 5 g lipid matrix was melted at 70 °C, and the aqueous phase containing 2% Tween-80 and 0.005% sorbic acid at the same temperature was added to the molten lipid matrix with gentle stirring using a magnetic stirrer (SR 1UM 52188; Remi Laboratory Instruments, Mumbai, Maharashtra, India). The mixture was further dispersed with a homogenizer (Ultra-Turrax® T18 digital; IKA® Werke GmBH & Co. KG, Staufen, Germany) at 8000 rpm for 5 min to produce the hot primary emulsion. The SLMs suspension obtained after cooling at room temperature was then lyophilized using a freeze dryer (Amsco/Finn-Aqua® Lyovac GT3, Hurth, Germany) to obtain water-free SLMs. By adding increasing concentrations of DicNa (25, 50, 75 and 100 mg) to each of the 5% w/w of lipid matrix and following the above described procedure, DicNa-loaded SLMs (SLM-1 to SLM-4) were obtained (). A batch of SLMs containing no drug (SLM-0), which served as negative control was similarly formulated.
Table 1. Quantities of material used for SLM formulation.
Characterization of SLMs
Determination of recovery rate of SLM
The water-free SLMs from each lipid matrix were weighed to obtain the yield of SLMs formulated per batch. The percentage (%) yield was then calculated using the formula:
(1)
(1)
where W1 is the weight of the SLMs formulated (g), W2 the weight of the drug added (g) and W3 the weight of the lipid and other additives (g).
pH analysis
The pH of the different batches of the SLMs formulations including those of the control were measured using a pH meter (Digital pH Meter; Labtech, Tamilnadu, India). This was repeated at time intervals of 24 h, 4 weeks, 16 weeks and 8 months. All the evaluations were done in triplicate using the dispersed SLMs.
Particle size analysis and determination of zeta potential
The particle size of the SLMs was determined by photon correlation spectroscopy using a Zetasizer Nano (ZEN 3600; Malvern Instruments, Great- Malvern , UK). For particle size analysis, the samples were measured after necessary dilutions using highly double distilled water. Measurements were made at 25 °C at a light-scattering detection angle of 90°. The mean particle size, polydispersity index (PDI) and zeta potential were determined. The zeta potential was similarly determined using a Zetasizer by phase analysis light scattering.
Particle morphology
The morphology of the particles of the SLMs was determined by computerized image analysis on a photomicroscope (Helmut Hund® GmBH, Wetzlar, Germany) within 1 week of preparation. Photomicrographs of the particles were also obtained.
Encapsulation efficiency
The encapsulation efficiency (EE%) of the prepared SLMs was assessed using a Vivaspin® 6 concentrator (50,000 MWCO; Vivascience, Hannover, Germany). SLMs dispersion (5.0 ml) was added to the centrifuge tube properly capped and centrifuged for 25 min at 5000 rpm. The filtrate was diluted (1:3) with distilled water and analyzed using a spectrophotometer (Jenway 6504; Jenway Scientific Equipment, Essex, UK) at 275 nm. The amount of drug encapsulated in the microparticles was calculated by reference to a standard Beer’s plot for DicNa to obtain the EE% using the formula:
(2)
(2)
In vitro drug release studies
Drug release studies from the SLMs were performed using a USP XXII rotating paddle apparatus (Erweka GmBH, Heusenstamm, Germany). Freshly prepared phosphate-buffered saline (PBS pH 7.2) maintained at 37 °C was used as the dissolution medium. A polycarbonate dialysis membrane (MWCO 5000–8000; Spectrum Laboratories, Breda, The Netherlands) pre-treated by soaking in the dissolution medium for 24 h prior to the commencement of each release experiment was used as release barrier. The choice of molecular weight was based on the earlier worked by Attama et al. (Citation2009), and that the higher the molecular weight, the higher the pore. However, release of drug from formulation depends on the formulation parameters, not the dialysis membrane use as the medium. In each case, 0.1 g of the SLMs was placed in the dialysis membrane containing 5.0 ml of the dissolution medium, securely tied with a thermo-resistant thread and then immersed in the dissolution medium under agitation provided by the paddle at 100 rpm. Aliquots of the release medium (5.0 ml) were withdrawn at predetermined time intervals, filtered through 0.45 μm filter, appropriately diluted, and the absorbance measured at 275 nm using a spectrophotometer (Jenway 6405). The volume of the dissolution medium was kept constant by replacing it with 5.0 ml of fresh medium after each withdrawal to maintain sink condition. The amount of DicNa released at each time interval was determined with reference to standard Beer’s plot. This test was carried out in triplicate for all the batches.
In vivo studies
Animals
Albino Wistar rats of either sex weighing ∼190–200 g were housed in a 12:12 h light–dark cycle and were maintained at a temperature of 22 °C. All animal experimental procedures were reviewed and approved by the Committee for Animal Research of the Department of Pharmaceutics, University of Nigeria, Nsukka, Nigeria, and were in compliance with the Federation of European Laboratory Animal Science Association and the European Community Council Directive of November 24, 1986 (86/609/EEC).
Assessment of anti-inflammatory activity
Anti-inflammatory activity was evaluated using carrageenan-induced rat paw edema model (Momoh & Adikwu, Citation2008). Male albino rats (190–200 g) were divided into groups of six of five rats per group. Group I served as control group and received normal saline, group II received pure DicNa (5 mg/kg) dispersed in distilled water (2 ml) and groups III through VI received 5 mg/kg of the test formulations SLM-1, SLM-2, SLM-3 and SLM-4, respectively. The formulations were prepared as homogenous suspensions in 2 ml normal saline (0.9% NaCl) and administered to the rats orally. One hour after administration of the formulations, each rat received a subplanter injection of 0.1 ml of 0.2% w/w γ-carrageenan solution in the right hind paws. The percentage paw edema and % inhibition were calculated using Equations (3) and (4) as described by Delporte et al. (Citation2005).
(3)
(3)
Here, Po is the initial paw volume before carrageenan injection and Pt is the final paw volume after carrageenan injection,
(4)
(4)
where Ic is the edema rate of control group and It is the edema rate of the treated group.
Evaluation of GI side effect as a function of ulcerogenic effect
The SLMs were tested for GI sparing effect as a function of ulcer protection in rat model. DicNa-loaded SLMs ulcers were determined using a method described by Chung-Chin et al. (2006). The studies were carried out on healthy male Wistar rats of an average weight of 210 g after acclimatization as previously described. The rats were divided into five groups (n = 5). SLM-0 to SLM-4 were administered to groups 1, 2, 3, 4 and 5, respectively, at a dose of 50 mg/kg per oral for 3 days. Prior to the test, the rats were fasted for 8 h but were allowed free access to food and water. They were sacrificed 8 h after the administration of the last dose of the formulation. The sacrificed rats were subjected to midline abdominal incision according to our previous work (Momoh & Adikwu, Citation2008). The abdomen was opened and stomach removed after ligating both the esophageal and pylorus ends. Incision was made in the stomach along its greater curvature; mucosal surface was exposed and washed with normal saline. It was then stretched and pinned on a cork board. The mucosal surface was then examined under a microscope fitted with a 4× binocular magnifier for erosions and ulcerations. Ulcer index was calculated using an established method (Rajashree et al., Citation2011).
Pharmacokinetics study
The bioavailability of DicNa from the SLMs in the plasma was evaluated in 15 Albino rats of an average weight of 180 g. They were divided into three groups of five animals each. Based on the in vitro release, SLM-4 was used for this assessment. It was suspended in 3 ml of distilled water and then orally administered to rats in group I (5 mg/kg of body weight). Pure DicNa (PS) and a marketed formulation (MKT-S) were similarly administered to rats in groups II and III, respectively, at the same dose of level for comparison. The dose of SLM used here was based on the results of the in vitro studies and the dose used in the anti-inflammatory evaluation which was indicative of good bioavailabilty. At predetermined time intervals of 0, 0.5, 1, 2, 4, 8, 10, 12, 16, 20, 24, 48 and 72 h post-administration, blood samples were collected from the pre-orbital sinus into a clean test tube. The samples were then centrifuged at 9000 rpm for 45 min, the supernatant collected and stored at 28 °C. They were analyzed spectrophotometrically (Jenway 6405) at a wavelength of 275 nm. The concentration of DicNa was determined using a standard curve.
Stability studies
The SLM formulations (unlyophilized) were assessed for stability using particles size, PDI, zeta potential and EE% after storage for 8 months as parameters. The method used in this stability study was based on the International Conference on Harmonization (ICH) Guidelines of Stability Studies & Accelerated Stability Studies (Yogeshwar & Vandana, Citation2009).
In vitro release was also carried out and compared with that of newly prepared SLM, in addition to the physical examinations such as color change and presence of odor.
Determination of injectability
To evaluate the ability of the formulations to pass through injection needle gauge, injectibility test was carried out on the unlyophilized SLM. A 5 ml of each formulation was gently pushed through a 5-ml plastic disposable syringe (MENO-JECT; Shandong, China) through hypodermic needles of various sizes 18, 22, 23 25 and 27 needle gauge. The testing was started with the smallest needle (27 gauge) and continued progressively until all the content of sample was delivered. The injectebility was recorded as the needle that allowed the passage of the complete sample.
Results and discussion
Percentage recovery of SLMs
The percentage recovery of the SLMs recovered was high () ranging from 93 to 97%, with the values increasing with in increased DicNa concentration. This is an indication of the suitability of this method of preparation.
Table 2. Properties of the SLMs formulation.
The pH studies
The pH could be an index for stability evaluation. It determines the possibility of product degradation, which can affect the stability as well as the therapeutic value of the formulation. In these studies, we evaluated the pH stability of the different batches of the SLMs periodically (24 h, 1 week and 8 weeks after formulation). It was observed from the results () that, the pH change decrease from the values obtained 24 h after the formulation in all the bathes of SLMs. SLM-1 had a range of pH of 7.81–6.71; SLM-2, SLM-3 and SLM-4 had their pH in the range of 7.98–6.91, 8.8–9.76 and 6.81–6.76 for the loaded SLMs. The unloaded SLMs also showed similar decrease with a range of 8.51–7.13. The pure sample of DicNa at 1% w/w aqueous solution of showed a pH of 7.41 as compared to the literature from the manufacturer for 10% w/w aqueous solution of DicNa that showed a pH of 7.48–7.4.
Table 3. Anti-inflammatory activity and time-resolved pH analysis.
The pH of the different batches of SLMs was measured periodically after preparation to ascertain the variation of pH with time, which could be a function of degradation of the active pharmaceutical ingredient (API) or excipients. Formulation stability may be affected by degradation of excipients or API with storage through generation of unfavorable pH (increase or decrease) or protonation in the functional group (Attama et al., Citation2009). It is necessary to know the pH of maximum of stability of an API or its stability profile and utilize that in designing a suitable formulation for the API. This will inform the formulation scientist whether to include a stabilizer or not in the new formulation. There was a gradual decrease in pH values of the formulations both in loaded and unloaded formulation. Generally, the pH of DicNa in solution was about pH of 7.4. Shortly after the formulation, the pH was slightly higher than the pure sample but overtime it showed a gradual reduction. This result is not peculiar to the loaded formulation alone, but also to the unloaded formulation, an indication that the carriers may have undergone certain degradation whereby free fatty acids may be release and may be implicated in the fall to acidic pH.
Morphologic studies
The SLMs () were smooth and relatively spherical shape indicating that the homogenization resulted in uniform and well-formed particles. However, there was a small aggregation noticed in SLM-1 and SLM-4, which on gently shaken for redispersing easily. In drug delivery system based on SLMs preparation, the shape of the particles and particle sizes determine to some extent the capability of the drug to be released from the formulation as well as the amount of the drug encapsulated in the lipid core.
Figure 1. Photomicrographs of SLM-0 (0 mg DicNa) SLM-1 (25 mg DicNa), SLM-2 (50 mg DicNa), SLM-3 (75 mg DicNa) and SLM-4 (100 mg DicNa) after 2 weeks of preparation. Bar represents 65.0 μm.

Particle size analysis and zeta potential
The result of the particle size analysis carried out on the SLMs expressed as volume distribution is presented in and . All the formulations exhibited a narrow size distribution () with PDI in the range of 0.611–0.617 (). The results indicate that while the particle sizes were generally within the same range, they varied with the amount of drug incorporated in the SLMs.
Figure 2. Particle-size distribution of formulations a SLM-1 to SLM-4 containing 25, 50, 75 and 100 mg of diclofenac sodium, respectively. SLM-0 is the unloaded formulation.
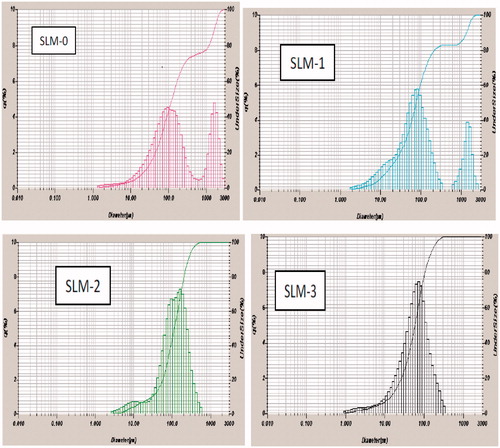
SLM-0 which contained no drug had the least particle size, while for the loaded formulations, SLM-1 had the highest particle size (86.0 µm) and SLM-4 had the least particles size (79.0 µm). Particle size and size distribution are very important characteristics for the evaluation of the stability of disperse systems and also influence the penetration mechanism of drugs into the biologic system (Souto et al., Citation2004). The particle size depends largely on the reliability of the production technology and other production steps such as degree of homogenization and pressure. Excipients, drug solubility, polymorphic nature of the lipid and the degree of particle size growth are also strong determinants. Generally, the particle sizes depend on the quantity of the drug incorporated into the lipid matrix.
Zeta potential is the potential difference between the dispersion medium and the stationary layer of fluid attached to the dispersed particle as they appear in electrical double layer. The zeta potential for the formulations ranged from −43.0 to −64.7 mV as shown in (). Large negative or large positive zeta potential is required for colloidal dispersion stability. The general dividing line between stable and unstable suspension is generally taken as either +30 or −30 mV (Momoh et al., 2010). For molecules and particles that are small enough, a high zeta potential will confer the stability by resisting aggregation (He et al., Citation2010). Based on the values obtained, the SLMs formulated with the two lipids are expected to be stable. The result of the particles size analysis after 8 months of storage () supports this as there was no aggregation or flocculation.
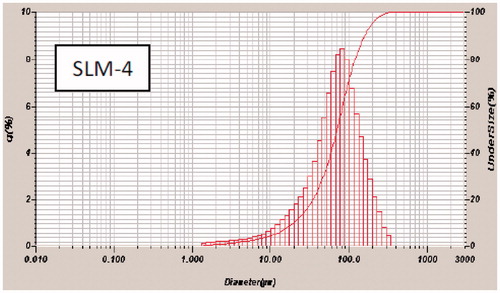
In vitro release studies
The in vitro release profiles of DicNa in phosphate buffer indicate good release of diclofenac from all batches of the formulation as shown in . The release of the DicNa from the SLMs shows an initial fast release of between 55 and 70% in 2 h. This could have been due to the proportion of drug trapped on the surface of the carrier during the production process (Huang & Brazel, Citation2001; Emmanuel et al., 2012). Inspection of reveals that size was a major determinant of the release profile, and drug initial release rate decreased with increasing particle size. This is consistent with Fick’s law of diffusion which attributes this decrease in drug release rate to an increase of diffusion pathways (reduced surface area to volume ratio for large spheres). For instance, in SLM-1 with highest particle size (88.00 ± 0.10 µm) gave a maximum release of 82%, while SLM-4 with the least particle size (79.00 ± 1.10 m) gave a maximum release of 96%. This trend may be attributed to the amount of drug encapsulated in the batches. Increase in the drug concentration had no effect on the particle size. However, it automatically led to an increase in the quantity of drug released. The result here is in agreement with earlier report, which indicates that the release of the active ingredient from a formulation increases with decrease in the particle size (Attama et al., 2010). Regarding their sustaining potentials, all the batches of the SLMs had good sustaining properties. However, SLM-4 formulations produced better sustained controlled release properties compared with SLM-1, SLM-2 and SLM-3. At initial state of the release, SLM-4 released <30% of the drug within the first 1 h, which is one of the criteria for sustained release formulation, after which a particular amount of the encapsulated drug was released and maintained unlike in SLM-1, SLM-2 and SLM-3.
Figure 3. In vitro release profile of DicNa from SLM formulated in phosphate buffer (pH 7.4) system. SLM-1 to SLM-4 contains 25, 50, 75 and 100 mg of DicNa, respectively.
Anti-inflammatory studies
All the SLM formulations produced anti-inflammatory activity as evidenced by the inhibition of edema ranging from 55.9 to 85.2% (). All the formulation had better inhibition than unformulated drug. This is an indication that the formulation process did not adversely affect the drug but rather improved its bioavailability. The observed anti-inflammatory activity of the loaded formulations may be due to the lipid matrix blending with the physiologic system and subsequently anchored to the wall of the GIT for better absorption. The presence of lipids in the GIT has been reported to induce secretion of gastric lipases, pancreatic lipases and co-lipases (Zhang & Zhou, Citation2009). Since gastric lipases hydrolyze up to 25% of the acyl chains of the lipids (Zhang & Zhou, Citation2009), the relatively high absorption (compared to the unformulated drug, PS) of the lipid-based formulations could be due to increased drug absorption due to the breakdown of the fatty acid chain. Endogenous and formulation-derived phospholipids are hydrolized by phospholipase A2 resulting in a free fatty acid and lysophophatidylcholine (Varshosaz et al., Citation2009). In addition, the secretion of biliary lipids, bile salts and cholesterol is stimulated, yielding the formation of various colloidal structures, including mixed micelles or unilamellar and multi-lamellar vesicles, which incorporate or associate to solubilize the drug. All of these physiologic changes resulted in the high bioavailability of the drug.
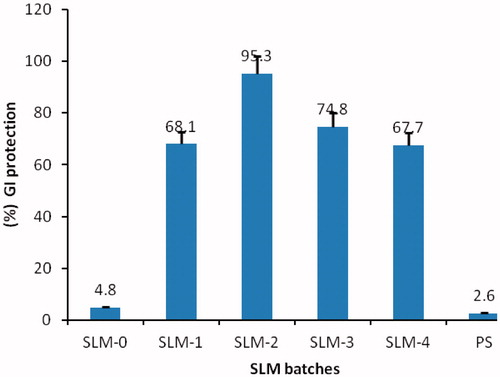
GI sparing effect
The mean ulcer index and the GI sparing or protection effects of the formulations are shown in and . The SLMs formulations were appreciably better tolerated than the unformulated DicNa (PS) with lower ulcer indices and higher GI protection. The formulation with the highest drug concentration (SLM-4) was the least protective of the formulations. The significant reduction in the ulcerogenic potentials of the NSAID could be due to activity of the phospholipid in the formulations which might have coated the gastric mucosa thus protecting it from irritation. Study have shown that pre-associating NSAID with phospholipid before administration resulted in remarkable protection of the GIT from ulceration compared to the un-associated NSAID administered in the same dose (Lichtenberger et al., Citation1994; Lichtenberger, Citation1995; Reithmeier et al., 2001). The mechanism for the observed effect is not clear. However, Lichtenberger et al. (Citation1995) have attributed this effect to the interaction of the core lipid and the phospholipid to improve the mucous gel layer of the GIT thereby preventing the direct action of GIT acid on the mucosa.
Figure 4. Mean ulcer index (±SD) of SLM-0 to SLM-4 containing 0, 25, 50, 75 and 100 mg of DicNa, respectively. PS represent the pure sample (diclofenac sodium) as a positive control.
Figure 5. Percentage GI protection of SLM-0 to SLM-4 containing 0, 25, 50, 75 and 100 mg of DicNa, respectively. PS represent the pure drug sample as a positive control.
Pharmacokinetics of DicNa after oral administration of the drug to rats
shows the mean plasma concentration–time profiles of the drug after oral administration of DicNa-loaded SLM at a dose of 50 mg/kg to rats in Group I, PS and MKT-S to rats in Group II and III, while some pharmacokinetic parameters are listed in . Drug absorption from the rat GIT was rapid with DicNa detected in plasma within the first few minutes after administration in all the groups of rats. The plasma concentration was higher in the PS compared with MKT-S (conventional sample) and the SLM. This is because of the unformulated drug as PS would go into solution faster thus making the drug available for transport across the GI epithelial membrane. After 12 h, the AUC of SLM-4 was 1456.30 ± 1.21 µgh/ml which was significantly greater (p < 0.05) than the controls (PS = 412.10 ± 1.13 and MKT-S = 724.23 ± 1.10) () which could be due to the slower clearance of the SLM. SLM had higher bioavailability than MKT-S as indicated by their Cmax () although it took the same length time (2 h) for the two to attain maximum concentration. The T1/2 of SLM was about twice than those of other preparation (). This may be due to lipid as well as surface modification in the formulation, which will form bond with the drug. This would affect factor such as the diffusion of the particles through the mucosa wall and the epithelia interaction which can affect the plasma concentration (Luo et al., Citation2006; Yuan et al., Citation2007), resulting in delayed drug clearance.
Figure 6. Plasma concentration versus time plotting of DicNa after oral administration of SLM-4, PS and MKT-S within 18 h (mean ± SD, n = 3). DicNa = diclofenac sodium, SLM-4 = batch 4 of the diclofenac sodium-loaded SLMs, PS = pure drug and MKT-S = market sample of diclofenac sodium.
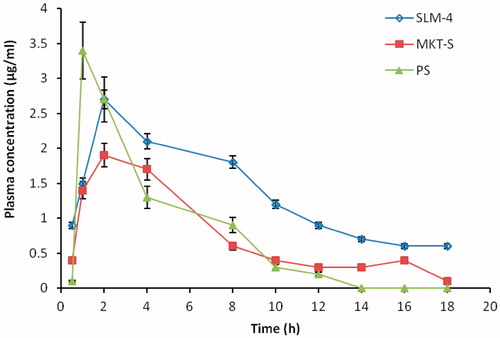
Table 4. Pharmacokinetic parameters of SLM-4, PS and MKT-S after oral administration to Albino rats (mean ± SD, n = 3).
In addition adhesion of the lipid to the GIT wall which will also improve the bioavailability of the drug through increased residence time. The P90G which is a heterolipid has a long chain of fatty acid and the hydrophobic nature of the lipid would also increase the lymphatic uptake of the formulations.
Stability studies
After 8 months of storage at 5–10 °C, zeta potential and PDI and particle sizes (PS) showed no significant (p > 0.05) changes. They were in the range of −54 to −57 ± 0.06 mV, 0.59 to 0.61 ± 0.01 and 84 to 86.1 ± 0.11 µg, respectively. The data for the % EE are shown in . The water-free DicNa SLM demonstrated physical and chemical stability for at least 8-month storage at 4–10 °C. There was no significant (p > 0.05) change in the particle size after 8 month of the preparation.
Physical (visual) observations showed no changes in the color or odor formation within the period of the storage. The physical stability of these formulations may be attributed to the P90G which is a core part of the lipid and further strengthened by Tween®-80 since they can be used as stabilizers and surface modifier to prevent increase in particle size of SLNs (Attama & Müller-Goymann, Citation2006).
Conclusion
Stable SLMs based on SRMS were formulated with good stability and bioavailability properties in comparison to a conventional formulation. The SLMs were found to have sustained release properties which could be exploited to reduce the frequency of dosing. The SLMs had significantly lower GI toxicity in terms of ulcer protection. All of these improve patient tolerability and compliance.
Acknowledgements
The authors wish to thank Phospholipid GmbH, Köln, Germany for the generous gift of Phospholipon® 90 G and AC Drugs Ltd, Enugu, Nigeria for the gift of diclofenac used in this study.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Attama AA, Christel CM. (2008). Effect of beeswax modification on the lipid matrix and solid lipid nanoparticle crystallinity. Colloids and Surfaces A: Physicochem Eng Aspects 315:189–95
- Attama AA, Müller-Goymann CC. (2006). A critical study of physically structured lipid matrices composed of a homolipid from Capra hircus and theobroma oil. Int J Pharm 322:67–78
- Attama AA, Nkemnele MO. (2005). In vitro evaluation of drug release from self micro-emulsifying drug delivery systems using a biodegradable homolipid from Capra hircus. Int J Pharm 304:4–10
- Attama AA, Nzekwe IT, Nnamani PO. (2003). The use of solid self-emulsifying systems in the delivery of diclofenac. Int J Pharm 262:23–8
- Attama AA, Reichl S, Müller-Goyman CC. (2008). Diclofenac sodium delivery to the eye: in vitro evaluation of novel solid lipid nanoparticles formulation using human cornea construct. Int J Pharm 355:307–13
- Attama AA, Okafor CE, Builders PF, Okorie O. (2009). Formulation and in vitro evaluation of a PEGylated microscopic lipospheres delivery system for ceftriaxone sodium. Drug Deliv 16:448–57
- Chung-Chin M, Santos JL, Oliveira EV, et al. (2009). Synthesis, ex-vivo and in vitro hydrolysis study of an indoline derivative designed as anti-inflammatory with reduced gastric ulceration properties. Molecules 14:3187–97
- Delporte C, Backhouse N, Erazo S, et al. (2005). Analgesic and anti-inflammatory properties of Proustia pyrifolia. J Ethnopharmacol 99:119–24
- Emmanuel CU, Franklin CK, John DO, et al. (2012). Preparation of novel solid lipid microparticles loaded with gentamicin and its evaluation in vitro and in vivo. J Microencap 29:296–307
- He C, Hu Y, Yin L, et al. (2010). Effects of particle size and surface charge on cellular uptake and biodistribution of polymeric nanoparticles. Biomaterials 31:3657–66
- Huang X, Brazel CS. (2001). On the importance and mechanisms of burst release in matrix controlled drug delivery systems. J Control Rel 73:121–36
- Jaspart S, Bertholet P, Piel G, et al. (2007). Solid lipid microparticles as sustained release system for pulmonary drug delivery. Eur J Pharm Biopharm 65:47–56
- Lichtenberger LM. (1995). The hydrophobic barrier of gastrointestinal mucus. Annu Rev Physiol 57:565–83
- Lichtenberger LM, Wang ZM, Romero JJ, Barreto JC. (1994). Certain NSAIDs chemically associate with surface phospholipids: insight into the mechanism and reversal of NSAID-induced gastric injury. Gastroenterology 106:154–8
- Lichtenberger LM, Wang ZM, Romero JJ, et al. (1995). Non-steroidal anti-inflammatory drugs (NSAIDs) associate with zwitterionic phospholipids: insight into the mechanism and reversal of NSAID induced gastrointestinal injury. Nat Med 1:54–8
- Luo YF, Chen DW, Ren LX, et al. (2006). Solid lipid nanoparticles for enhancing vinpocetine’s oral bioavailability. J Control Rel 114:53–9
- Momoh MA, Adikwu MU. (2008). Evaluation of oral insulin delivery using bovine mucin in non-diabetic rats. J Pharm Res 7:5–9
- Momoh MA, Adikwu MU, Ibezim EC. (2010). Formulation and evaluation the bioadhesive properties of drug delivery system based on PEGylated mucin matrices. Asian Pac J Trop Med 4:412–20
- Momoh AM, Esimone CO. (2012). Phospholipon® 90H (P90H)-based PEGylated microscopic lipospheres delivery system for gentamicin: an antibiotic evaluation. Asian Pac J Trop Biomed 2012:889–94
- Momoh MA, Kenechukwu FC, Attama AA. (2013). Formulation and evaluation of novel solid lipid microparticles as a sustained release system for the delivery of metformin hydrochloride. Drug Deliv 20:102–11
- Mukherjee S, Ray S, Thakur RS. (2009). Solid lipid nanoparticles: a modern formulation approach in drug delivery system. Indian J Pharm Sci 71:349–58
- Rajashree SC, Harinath NM, Ashok VB. (2011). Synthesis, characterization and evaluation of analgesic and anti-inflammatory activities of some novel indoles. Trop J Pharm Res 10:463–73
- Reithmeier HJ, Herrmann N, Gopferich A. (2001). Development and characterisation of lipid microparticles as a drug carrier for somatostatin. Int J Pharm 218:133–43
- Souto EB, Wissing SA, Barbosa CM, Müller RH. (2004). Development of a controlled release formulation based on SLN and NLCcfor topical clotrimazole delivery. Int J Pharm 278:71– 7
- Tunçay M, Caliş S, Kaş HS, et al. (2000). In vitro and in vivo evaluation of diclofenac sodium loaded albumin microspheres. J Microencap 2:145–55
- Ulberth F, Buchgraber M. (2003). Analytical platforms to assess the authenticity of cocoa butter. Eur J Lipid Sci Technol 105:32–42
- Varshosaz J, Minayian M, Moazen E. (2009). Enhancement of oral bioavailability of pentoxifylline by solid lipid nanoparticles. J Liposom Res 20:231–9
- Wallace JL. (1997). Nonsteroidal anti-inflammatory drugs and gastroenteropathy: the second hundred years. Gastroenterology 112:1000–16
- Wallace JL, Bak A, McKnight W, et al. (1998). Cyclooxygenase 1 contributes to inflammatory responses in rats and mice: implications for gastrointestinal toxicity. Gastroenterology 115:101–9
- Yogeshwar GB, Vandana BP. (2009). Microemulsion-based vaginal gel of clotrimazole: formulation, in vitro evaluation, and stability studies. AAPS Pharm Sci Tech 10:476–81
- Yuan H, Chen J, Du YZ, et al. (2007). Studies on oral absorption of stearic acid SLN by a novel fluorometric method. Colloids Surf B Biointerfaces 58:157–64
- Zhang Z, Lv H, Zhou J. (2009). Novel solid lipid nanoparticles as carriers for oral administration of insulin. Pharmazie 64:574–8