
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The aim of this study was to prepare levofloxacin-loaded chitosan microspheres and to evaluate their in vitro and in vivo characteristics. Glutaraldehyde-crosslinked microspheres were prepared using a spray-drying method, and characterized in terms of the morphological examination, particle size distribution, entrapment efficiency, drug loading and in vitro release. Pharmacokinetics and colon biodistribution studies were used to evaluate that microspheres have more advantage than the conventional formulations. The surface morphology of the freeze-dried microspheres were smooth, discrete with a regular spherical to near-spherical shape. Size of the microspheres after freeze-drying was 4.96 ± 0.76 μm and well-distributed. The zeta potential of microspheres was −29.3 ± 2.1 mV. An average drug loading of 9.3 ± 0.4% and encapsulation efficiency of 81.1 ± 4.7% of levofloxacin microspheres were obtained with the optimized preparation parameters. The cumulative release rate of levofloxacin microspheres was followed by a sustained release and fitted for classic Higuchi kinetic model. In vivo studies showed that chitosan microspheres are thought to have the potential to maintain levofloxacin concentration within target ranges for a long time, decreasing side effects caused by concentration fluctuation, ensuring the efficiency of treatment and improving patient compliance by reducing dosing frequency. It also does not cause any harmful or toxic effect in colon and rectum as evaluated by histopathologic studies.
Introduction
Preoperative bowel preparation has traditionally been the essential preparatory step to reduce postoperative infectious complications. The clinical practice guidelines address oral and parenteral prophylactic antibiotics are the recommendations (Townsend et al., Citation2012). Numerous studies of oral antibiotic bowel preparation before colorectal resection have been shown to reduce surgical site infections (Lewis, Citation2002; Nelson et al., Citation2009; Bellows et al., Citation2011). The risk of infection after operation can shorten the patient’s hospitalization and reduce the readmission rate. The total treatment expenditure could be reduced greatly as well (Dimick et al., Citation2004; Cannon et al., Citation2012; Kelly et al., Citation2012).
Levofloxacin (LFX) is a broad-spectrum antibiotic of the fluoroquinolone drug class. It is rapidly and completely absorbed after oral administration (McGregor et al., Citation2008). Peak plasma concentration (Cmax) is usually attained 1–2 h after oral dosing. The plasma concentration profile of LFX after intravenous administration is comparable in area under the concentration–time curve (AUC) to that observed for oral tablets when equal doses are administered. LFX is excreted largely (87%) as unchanged drug in the urine. The mean terminal plasma elimination half-life (t1/2) of LFX ranges from ∼6 to 8 h (Giordano et al., Citation2007). It follows that oral LFX agents most frequently used for bowel preparation are inexpensive and likely cost-effective, considering the high costs of infectious complications after colectomy. However, there is obvious deficiency in the oral LFX bowel preparation. For example, after the oral intake of LFX, drugs are normally digested first in the stomach and small intestine and then absorbed into the blood. This can keep the plasma concentration in a certain level. But some localized tissue like colon and rectal has only limited antibacterial activity because of the low concentration in this part.
For a large majority of pharmaceutical formulations in contemporary use, their “specificity” toward appropriate sites of a disease is not based on their ability to accumulate selectively in the target organ or tissue. Usually, they are more or less evenly distributed within the body. Moreover, to reach the target area, the drug has to cross different biologic barriers like organs, cells, and even intracellular compartments, where it can cause undesirable side reactions, or be partially inactivated. The best solution to this issue is drug targeting (Shixuan et al., Citation2009).
Chitosan is a natural polysaccharide derived from chitin by alkaline deacetylation, and it is the second most abundant biopolymer after cellulose (Ravi Kumar, Citation2000). Chitosan exhibits several favorable biologic properties that make it an interesting polymer for use in pharmaceutical formulations, as demonstrated by the number of scientific reports published. It is a mucoadhesive and biodegradable polymer that possesses penetration enhancing properties and an adequate toxicity profile (Dodane et al., Citation1999; Sogias et al., Citation2008). It is popularly used as colon-specific drug delivery vehicle for colon site infections. This is due to chitosan can be selectively digested by colonic microflora to induce site-specific drug release (Zhou et al., Citation2014).
Prolonged colonic matrix retention and consistent local colonic drug exposure are more easily achieved using particulate system. Exploring alternative routes of administration like microspheres to develop a targeted drug delivery system and to act locally on the organ of infection will improve the therapeutic efficacy, reduce side effects and thereby provide patient compliance (Delgado et al., Citation2000; Dhanaraj et al., Citation2001).
In the present study, the development of a chitosan microspheres delivery system for LFX was the aim. In order to develop the system, LFX glutaraldehyde-crosslinked chitosan microspheres were prepared using the spray-drying method. The systems were characterized for surface morphology and size distribution. Technologic studies were performed to evaluate drug-loading coefficient, entrapment efficiency and in vitro release. Moreover, pharmacokinetics and biodistribution studies of LFX-loaded chitosan microspheres were investigated.
Materials and methods
Materials
LFX was purchased by Canbo Pharma Co., Ltd. (Hubei, China). LFX tablets were obtained from Eashu Pharma Co., Ltd. (Guangdong, China). Medium molecular weight chitosan (deacetylation ratio of 75–85%), acetic acid, glutaraldehyde and dehydrated alcohol were purchased from Sinopharm, Shanghai, China. Norfloxacin (≥98.00% purity), used as internal standard, were purchased from National Institute for Food and Drug Control (Beijing, China). All other materials or solvent were of reagent or analytical grade.
The experiments were performed on rats weighing 220 ± 15 g. The animals were kept in cages in a room at a temperature of 25 ± 2 °C, with a 12:12 light–dark cycle. Food and water were freely available. All experiments were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals as adopted by the China National Institutes of Health (Shanghai, China).
Preparation of the microspheres
Chitosan microspheres containing LFX were prepared via the modified spray-drying method (Cai et al., Citation2011). Chitosan (900 mg) was dissolved in aqueous acetic acid solution (100 mL, 1% v/v) and filtered through a nylon cloth to remove insolubles. Then, LFX was dispersed directly into the chitosan solution to a final concentration of 25% (w/w of chitosan). The prepared formulations were subsequently spray-dried using a mini spray dryer equipped with a high-performance cyclone (Bilon-6000Y; Shanghai Bilon Instrument Co., Ltd, Shanghai, China) with a 0.9-mm two-fluid nozzle, using the following standard operating conditions: inlet temperature 200 °C; spray flow rate 1 L/h and evaporated water 1.5 L/h. These conditions resulted in an outlet temperature of 110 °C. The spray-dried microspheres were subsequently dispersed in a dehydrated alcohol solution containing 10% (v/v) of glutaraldehyde to cross-link for 2.5 h. Afterwards, the final product was obtained by centrifugation (4000 rpm, 5 min) and freeze-drying.
Surface morphology
The surface morphology of the microspheres was observed by scanning electron microscope (JCM-6000, Joel Ltd, Tokyo, Japan). The microspheres were placed on graphite surface and coated with gold using an ion sputter and were observed at 12 kV (Canan et al., Citation2003). The size and zeta potential of microspheres were measured by the light scattering method. The analyses were performed with a helium–neon laser (632.8 nm) at a scattering angle of 90° at 25 °C using a Nicomp™ 380 ZLS (Particle Sizing Systems, Port Richey, FL). The samples were diluted to the appropriate concentration using deionized water before measurement.
Drug content, entrapment efficiency and drug loading
The drug content analysis was determined from various batches of the microspheres. Briefly, the powdered microspheres were dissolved in adequate quantity of phosphate buffer pH 6.8 and then filtered. The drug content of LFX microspheres was detected by high-performance liquid chromatography (HPLC) (Sousa et al., Citation2013). The drug entrapment efficiency and drug loading was calculated by the following equations:
WLFX represents the amount of LFX loaded in the microspheres, Wmicrospheres represents the weight of the LFX microspheres.
In vitro release
Microsphere samples (200 mg) were placed into dialysis bags and suspended in 350 mL 0.1 N hydrochloric acid solution (pH 1.2) to simulate gastric fluid (Bose et al., Citation2014). The pH of the gastric fluid was later adjusted to 6.8 to simulate intestinal fluid through adding 150 mL of 0.2 M tribasic sodium phosphate solution and 6 mL pectinase enzyme solution (5300 unit/mL) which had previously equilibrated to 37 ± 0.1 °C and stirred at 100 rpm using the USP paddle method. At predetermined times of 0.25, 0.5, 1, 2, 3, 4, 6, 8, 10, 12, 16 and 20 h samples (2 mL) were withdrawn with a syringe filter (0.45 μm pore size) from the release media and replaced with an equal volume of the corresponding fresh media to maintain a constant volume. The test solution was analyzed by HPLC method as follows. Triplicates were conducted and the results averaged.
Stability
The stability protocol designed was based on the ICH guidelines. The microspheres were stored at 25 ± 2 °C and 60 ± 5% relative humidity (RH) for a period of 6 months and at 5 ± 3 °C for a period of 12 months, which is the accelerated storage temperature and long-term storage temperature, respectively for products which are intended to be refrigerated. The stored samples were tested for their drug content, particle size distribution and for any physical change. The drug content was determined by HPLC method. The testing was carried out at 0, 2, 4 and 6 months for accelerated storage condition and at 3-month interval for a period of 12 months for long-term storage condition as per ICH guideline.
Pharmacokinetics
Twelve rats were used to investigate the effect of microspheres formulation on the pharmacokinetics of LFX after oral administration. Rats were divided into two groups at random and given a single 200 mg/kg dose of the LFX-loaded microspheres and LFX tablets by intragastric administration. LFX tablets were crushed and mixed with 100 mL water before administration. Blood samples (0.5 mL) were collected into heparinized tubes from the caudal vein at 0, 1, 2, 4, 6, 8 and 12 h after intragastric administration. Blood was immediately processed for plasma by centrifugation at 4000 g for 10 min. Plasma samples were frozen and maintained at −70 °C until analysis. Pharmacokinetic parameters were calculated from the plasma concentration–time data. The elimination half-life (t1/2) was determined by linear regression of the terminal portion of the plasma concentration–time data. The area under the plasma concentration–time curve from zero to the last measurable plasma concentration point (AUC0–t) was calculated by the linear trapezoidal method. Extrapolation to time infinity (AUC0–∞) was calculated as follows: AUC0–∞ = AUC0–t + Ct/ke, where Ct is the last measurable plasma concentration and ke is the terminal elimination rate constant. In order to decrease the interference of creatinine clearance rate (CCR), we determined the CCR of each subject by the following formula: CCR = Ucr (mg/dL) × urine volume per min (mL/min)/Scr (mg/dL); Ucr, urine creatinine; Scr, serum creatinine.
Evaluation of colon targeting
Sixty rats were used in the experiment to assess the effect of microspheres formulation on the intestines biodistribution of LFX after intragastric administration. The rats were divided into two groups at random and given a single 200 mg/kg dose of either the LFX microspheres or LFX tablets. At 2, 4, 6, 8 and 12 h after drug administration, each animal (n = 6 for each time-point) was euthanized, and colon and rectum samples were collected. Tissue samples were washed in ice-cold saline, blotted with paper towel to remove excess fluid, weighed and stored at −70 °C until assessed for drug concentration by HPLC.
Histopathologic studies
After 12 h of administration of the formulation, animals (including control group) were sacrificed by excess anesthesia and the colon and rectum were dissected and washed with cold saline. The organs were pressed between filter pads and weighed. Colon and rectum tissues were fixed in 10% neutral formalin using standard techniques and stained with hematoxylin and eosin for histopathologic examination. All tissue samples were examined and graded under light microscopy with 500× magnification (Nanji et al., Citation2002).
HPLC analysis
The amount of LFX in each sample was determined by HPLC (LC-10 A, Shimadzu Co. Ltd, Kyoto, Japan) equipped with a fluorescence detector (RF-20AXS). Chromatographic separation was achieved using a Dikma Diamonsil TM C18 column (Dikma Co Ltd, Beijing, China, 5 μm, 200 mm× 4.6 mm) and a precolumn (Nova-Pak, 10 μm, C18, Waters, Milford, MA, USA). About 0.1% aqueous formic acid (pH 3.0, triethylamine):acetonitrile:methanol = 80:5:15 (v/v) was used as mobile phase at a flow rate of 1.0 mL/min. The temperature of the column was maintained at ambient temperature (25 °C). The fluorescence detector was operated at an excitation wavelength of 260 nm and an emission wavelength of 455 nm.
Each 200 μL aliquot of plasma or tissue samples [100 mg tissue samples were homogenized in a mixed solution of acetonitrile and water (0.5 mL, 50:50, v/v)] was spiked with 10 μL of internal standard working solution (norfloxacin, NFX 1 mg/mL). Then, 600 μL of acetonitrile was added to the sample to precipitate plasma proteins. The mixture was vortex mixed for 30 s and centrifuged at 12 000 rpm for 5 min. The resulting supernatant (∼800 μL) was transferred to a glass tube and evaporated to dryness under a nitrogen stream at 45 °C. The residue was reconstituted with 100 μL of deionised water, vortex mixed for 1 min and placed in an ultrasonic bath at room temperature for 1 min. The reconstituted samples were transferred to 0.22-μm Spin-X centrifuge filters (Corning Inc., Corning, NY, USA) and centrifuged at 12 000 rpm for 1 min. The final filtered samples were placed in amber auto sampler vials for HPLC analysis.
Statistical analysis
The data was statistically analyzed by Student’s t-test. A p < 0.05 was considered statistically significant.
Results and discussion
Method validation
The analytical peaks of LFX and internal standard (NFX) were resolved with good symmetry and no endogenous sources of interference were observed at the retention time of the analyte. The LLOQ was 0.02 µg/mL (S/N = 10). Good linearity was observed over the concentration range of 0.02–10 µg/mL plasma (r = 0.9993). The intra-/inter-day precision and accuracy were determined. The RSD of LFX ranged from 5.26 to 6.98% for intra-day and 5.85 to 8.33% for inter-day, respectively. The relative error of LFX ranged from 1.3 to 2.6% for intra-day and −0.9 to 1.7% for inter-day, respectively. Freshly prepared solutions showed no evidence of degradation for either LFX or the internal standard. No significant degradation was observed for any analytes during the sample processing and extraction, including the drydown procedure. The stability of the sample solution in the auto sampler at 4 °C was also assessed. LFX in sample solution was found to be stable for ∼24 h since the found concentrations were within 98–101% of the initial concentrations. The results obtained after three freeze-thaw cycles demonstrated that 97.2 ± 4.1% of the initial content of LFX were recovered and that the analytes were stable under these conditions. Plasma samples collected from studies of LFX were evaluated before and after storage at −18 °C for stability and found to be stable for at least 3 months. The mean absolute recoveries for LFX were 88.2, 91.3 and 92.1% at the high, middle and low concentrations, respectively (n = 3).
Physical–chemical characteristics
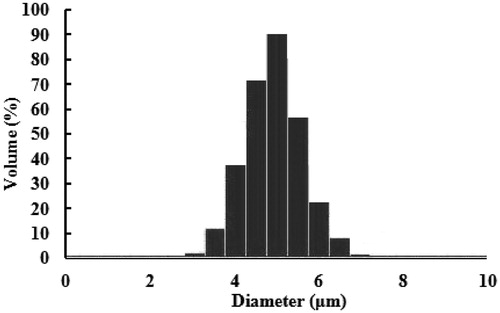
The freeze-dried microspheres were white powder with good appearance. Scanning electron microscopy was used to visualize the particle diameter, structural and surface morphology of the spray-dried microspheres (). As seen from , the surface morphology of the microspheres were smooth, discrete with a regular spherical to near-spherical shape, which may be due to cross-linking of coacervate to stabilize emulsion droplets and hence resulted in well-formed microspheres. Results of size and size distribution of LFX microspheres are shown in . Size of the microspheres after freeze-drying was 4.96 ± 0.76 μm and well-distributed. The zeta potential of microspheres was −29.3 ± 2.1 mV.
Figure 1. Scanning electron micrograph of LFX microspheres (5000×). Bar = 2.5 μm.
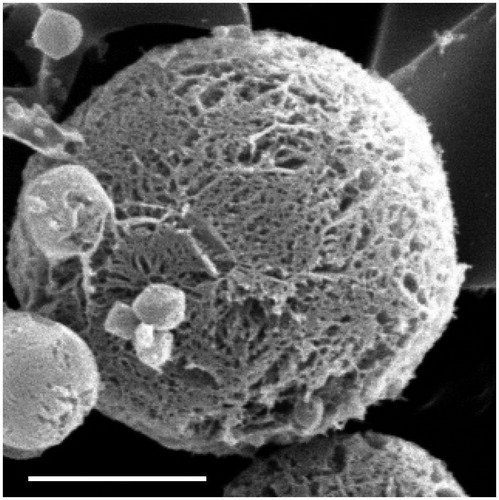
Figure 2. Mean size distribution of LFX microspheres.
The drug loading and encapsulation efficiency could be affected by several factors, such as the methods of preparation, the physicochemical characteristics of drug, the concentrations of drug and polymer, phase ratio, the concentrations of the stabilizer and stirring speed (Yen et al., Citation2001; Zhang et al., Citation2011). In this study, it was an efficient method to increase the drug loading and encapsulation efficiency by using the method of spray-dried. An average drug loading of 9.3 ± 0.4% and encapsulation efficiency of 81.1 ± 4.7% of LFX microspheres were obtained with the optimized preparation parameters, suggesting that the drug loading and encapsulation efficiency were high enough for further studies. At the same time, the method of spray-dried induced high yields (ranged from 62 to 70%).
In vitro release
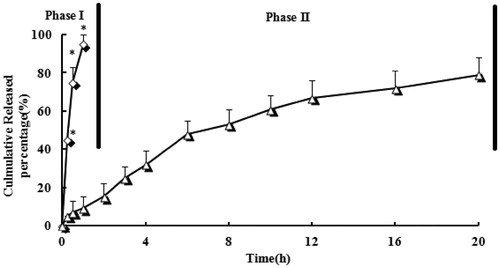
shows in vitro release for LFX microspheres and tablets, respectively. A very fast release behavior of LFX was observed in tablets, whereas the cumulative release rate of LFX microspheres was much slower followed by a sustained release. In tablets group, >60% of drug was released in the first hour of dissolution process in the simulated gastric medium. Almost 100% of LFX were released in the first 2 h. In contrast, only 15% of LFX were released from microspheres in the first 2 h (p < 0.05). LFX was graduated released from the microspheres as time lapsed (∼50% released in 6 h, ∼70% released in 12 h and ∼80% released in 20 h), suggesting that LFX was well-entrapped in chitosan microspheres.
Figure 3. Drug release profiles of LFX microspheres (triangle) and tablets (diamond) in 0.1 N hydrochloric acid solution to simulate gastric fluid (pH 1.2) for 2 h (phase I) and followed by adding tribasic sodium phosphate and pectinase enzyme solution to simulate intestinal fluid (pH 6.8) for remaining time (phase II). n = 3, *p < 0.05 microspheres versus tablets.
Due to its excellent biocompatibility and biodegradability, chitosan can be completely absorbed by the body. The derivatives of chitosan like semi-synthesis of succinic acid, chitosan, phthalic acid–chitosan, etc. have alkali dependency and expanse into gel in acidic medium. This prevents drug delivery and delays drug release rate. It can thus tolerate the gastric acid of UGIT and different digestive enzymes. After the chitosan arrives in colon, it will be decomposed by the enzymes, which are produced by the colon flora. The drugs carried are then released. At the same time, biologic adhesive of chitosan may keep adhere to the colonic mucosa. This results the prolonging of duration of drug action and therefore achieving a more effective cure. Therefore, researches of application in colon targeted drug delivery are seeing in a rising number (Zhang et al., Citation2002; Shimono et al., Citation2003).
Several models describing drug release from immediate and modified release dosage forms were adopted. The model that fitted best for LFX released from the microspheres was Higuchi kinetic model with correlation coefficient r = 0.9992, revealing that LFX could be controlled released from the microspheres.
Stability
The drug content in long-term storage conditions did not vary to a large extent in the microsphere formulations; the maximum variation of 1.5% from the initial concentration was seen in microspheres, 12 months from the date of manufacture. The formulations were stable for 6 months under accelerated storage conditions of 25 ± 2 °C and 60 ± 5% RH. A maximum decrease of 4.2% from the initial concentration was observed. The average particle size did not vary appreciably. And the physical–chemical characteristics changes were found to be negligible and had no impact on the quality of the formulations.
In vivo studies
The HPLC method for analysis has been validated. Linearity in the standard curves was demonstrated over the concentration range studied, and endogenous components had no interference in the chromatograms. Plasma concentration–time profiles of LFX after intragastric administration of microspheres and tablets was shown in . The pharmacokinetics parameters were shown in . As shown in , drug plasma concentration attained at a higher level (>7 μg/mL) in the initial 2 h after intragastric administration of LFX tablets. This was correspondent with drug initial burst release from tablets. And then drug concentration decreased markedly after 2 h and kept at a lower level of 0.4–4.7 μg/mL. Drug plasma was below to detect limitation after the time of 10 h. In contrast, microspheres altered the distribution of LFX in vivo and the half-life after intragastric administration was prolonged remarkably than those of tablets (4.3 versus 1.2). The result indicated that the microspheres had sustained release efficacy. The AUC0–∞ of the microspheres formulation were 1.77-fold higher than that of tablets, suggesting that the encapsulated LFX had almost been absorbed in rats over the period of 2 days.
Figure 4. Mean plasma concentration–time profiles of LFX after intragastric administration of microspheres and tablets. (Each group represents the mean ± standard deviation of six rats). *p < 0.05 microspheres versus tablets.
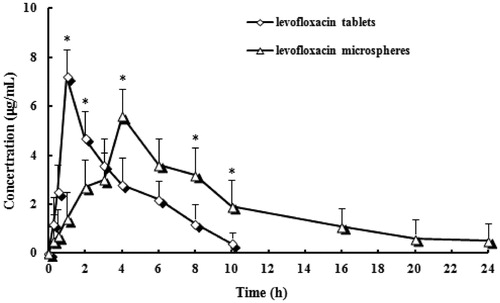
Table 1. Pharmacokinetic parameters of LFX after intragastric administration of microspheres and tablets (n = 6).
In vivo biodistribution behavior of LFX after intragastric administration of the microspheres to rats was investigated with tablets as a control. The amounts of drug distributed in unit mass of colon and rectum at various times were measured. present the mean concentration–time profiles of LFX in unit mass of each organ in rats. The total amount of drug accumulated in each organ within 24 h (AUC0–t) was calculated, and the results are shown in . The drug concentration was significantly difference in colon and rectum between the microspheres and tablets (p < 0.05). The LFX AUC0–t of the microspheres was 6-fold and 6.7-fold higher compared to tablets in colon and rectum, respectively. Hence, LFX chitosan microspheres are thought to have the potential to maintain LFX concentration within target ranges for a long time, decreasing side effects caused by concentration fluctuation, ensuring the efficiency of treatment and improving patient compliance by reducing dosing frequency.
Figure 5. Distribution in tissue in rats after following intragastric administration of a single 200 mg/kg dose of LFX tablets and microspheres (A: colon tissue; B: rectum tissue). (Each point represents the mean ± standard deviation of six rats). *p < 0.05 microspheres versus tablets.
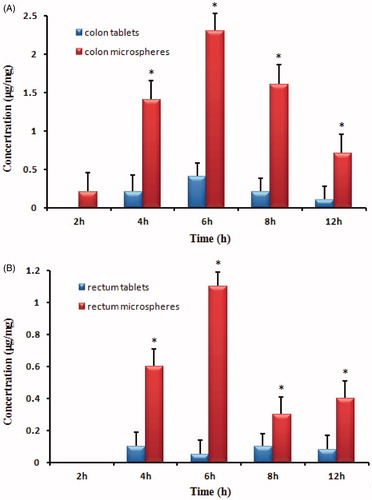
Table 2. The AUC0–24 h of LFX in colon and rectum tissues after intragastric administration of tablets and microspheres to rats (n = 6).
Histopathologic studies
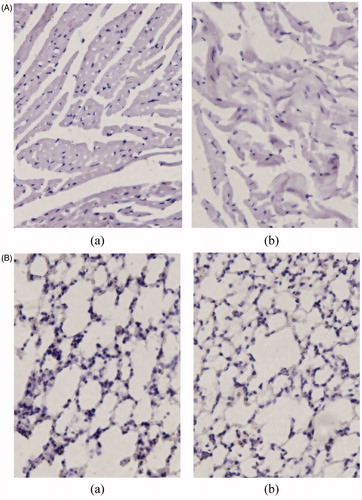
The histopathologic examination of colon and rectum was carried out for detection of any damage to the tissue. The microphotographs were taken of colon and rectum following incubation with microsphere formulations for >12 h (). Saline was the control. Nothing of the severe signs like appearance of epithelial necrosis and sloughing of epithelial cells was detected.
Figure 6. Histopathologic studies of colon (A) and rectum (B). (a) Saline and (b) LFX microspheres.
Conclusion
The results of our present study clearly represent promising capabilities of LFX load–chitosan microspheres for orally drug delivery as well as it could be viewed as substitute to conventional dosage form. The microspheres showed the particle size of ∼4.96 μm which could be prepared by spray-dried method. The microspheres had not only good sphericity but also uniform distribution of particle size. In vitro release study, only a little drug were released from microspheres in the initial time and then was graduated released from the microspheres as time lapsed, suggesting that LFX was well-entrapped in chitosan microspheres. In vivo studies showed that chitosan microspheres are thought to have the potential to maintain LFX concentration within target ranges for a long time, decreasing side effects caused by concentration fluctuation, ensuring the efficiency of treatment and improving patient compliance by reducing dosing frequency. It also does not cause any harmful or toxic effect in colon and rectum as evaluated by histopathologic studies.
Declaration of interest
The authors report no conflicts of interest.
References
- Bellows CF, Mills KT, Kelly TN, et al. (2011). Combination of oral non-absorbable and intravenous antibiotics versus intravenous antibiotics alone in the prevention of surgical site infections after colorectal surgery: a meta-analysis of randomized controlled trials. Tech Coloproctol 15:385–95
- Bose A, Elyagoby A, Wong TW. (2014). Oral 5-fluorouracil colon-specific delivery through in vivo pellet coating for colon cancer and aberrant crypt foci treatment. Int J Pharm 468:178–86
- Cai J, Zhang Y, Du W, Nan K. (2011). Preparation and in vitro release of spray-dried chitosan microspheres for levofloxacin delivery. J Control Release 152:e70–1
- Canan H, Nursin G, Nevin E. (2003). Mucoadhesive microspheres containing gentamicin sulphate for nasal administration: preparation and in vitro characterization. Farmaco 58:11–6
- Cannon JA, Altom LK, Deierhoi RJ, et al. (2012). Preoperative oral antibiotics reduce surgical site infection following elective colorectal resections. Dis Colon Rectum 55:1160–6
- Delgado A, Sorianoil SE, Oliva ME. (2000). Radiolabelled biodegradable microspheres for lung imaging. Eur J Pharm Biopharm 50:227–36
- Dhanaraj SA, Gowthamarajan K, Shanthi K, et al. (2001). Albumin microsphere containing methotreate: a lung specific delivery system. Ind J Pharm Sci 63:196–9
- Dimick JB, Chen SL, Taheri PA, et al. (2004). Hospital costs associated with surgical complications: a report from the private-sector National Surgical Quality Improvement Program. J Am Coll Surg 199:531–7
- Dodane V, Amin Khan M, Merwin JR. (1999). Effect of chitosan on epithelial permeability and structure. Int J Pharm 182:21–32
- Giordano P, Weber K, Gesin G, Kubert J. (2007). Skin and skin structure infections: treatment with newer generation fluoroquinolones. Ther Clin Risk Manag 3:309–17
- Kelly M, Sharp L, Dwane F, et al. (2012). Factors predicting hospital length-of-stay and readmission after colorectal resection: a population-based study of elective and emergency admissions. BMC Health Serv Res 12:77
- Lewis RT. (2002). Oral versus systemic antibiotic prophylaxis in elective colon surgery: a randomized study and meta-analysis send a message from the 1990s. Can J Surg 45:173–80
- McGregor JC, Allen GP, Bearden DT. (2008). Levofloxacin in the treatment of complicated urinary tract infections and acute pyelonephritis. Ther Clin Risk Manag 4:843–53
- Nanji AA, Jakelainen K, Fotouhinia M, et al. (2002). Increased severity of alcoholic liver injury in female rats: role of oxidative stress, endotoxin and chemokines. Am J Physiol Gastrointest Liver Physiol 281:348–56
- Nelson RL, Glenny AM, Song F. (2009). Antimicrobial prophylaxis for colorectal surgery. Cochrane Database Syst Rev 1:CD001181
- Ravi Kumar MNV. (2000). A review of chitin and chitosan applications. React Funct Polym 46:1–27
- Shimono N, Takatori T, Ueda M, et al. (2003). Multiparticulate chitosan-dispersed system for drug delivery. Chem Pharm Bull 51:620–4
- Shixuan Z, Xiujuan G, Kaihua S, et al. (2009). Evaluation of poly (d,l-lactide-co-glycolide) microspheres for the lung-targeting of yuanhuacine, a novel DNA topoisomerase I inhibitor. J Drug Target 17:286–293, 1029–2330
- Sogias IA, Williams AC, Khutoryanskiy VV. (2008). Why is chitosan mucoadhesive? Biomacromolecules 9:1837–42
- Sousa J, Alves G, Campos G, et al. (2013). First liquid chromatography method for the simultaneous determination of levofloxacin, pazufloxacin, gatifloxacin, moxifloxacin and trovafloxacin in human plasma. J Chromatogr B Analyt Technol Biomed Life Sci 930:104–11
- Townsend CM, Beauchamp RD, Evers BM, et al. (2012). Sabiston textbook of surgery: the biological basis of modern surgical practice. 19th ed. : Elsevier Saunders
- Yen SY, Sung KC, Wang JJ, et al. (2001). Controlled release of nalbuphine propionate from biodegradable microspheres: in vitro and in vivo studies. Int J Pharm 220:91–9
- Zhang H, Alsarra IA, Neau SH. (2002). An in vitro evaluation of a chitosan-containing multiparticulate system for macromolecule delivery to the colon. Int J Pharm 239:197–205
- Zhang Z, Bi X, Li H, et al. (2011). Enhanced targeting efficiency of PLGA microspheres loaded with Lornoxicam for intra-articular administration. Drug Deliv 18:536–44
- Zhou M, Peng Z, Liao S, et al. (2014). Design of microencapsulated carbon nanotube-based microspheres and its application in colon targeted drug delivery. Drug Deliv 21:101–9