
Abstract
Camptothecin (CPT) is an effective anticancer agent against various cancers but the clinical application is limited because of its poor water solubility, low bioavailability and severe toxic side effects. The aim of the present study was to evaluate the feasibility of using targeted NPs as a high-performance CPT delivery system that targets liver cancer cells through intravenous (i.v.) administration route. CPT was incorporated into biotin-F127-PLA or F127-PLA polymeric nanoparticles (NPs) by a dialysis method. The preparation of the targeting NPs was performed by conjugating biotin-F127-PLA NPs with anti-3A5 antibody. The antitumor effect of the CPT-loaded nanoparticles against H22 cells in vitro was determined using an MTT assay. Tissue distribution and tumor inhibition in vivo were also evaluated. Survivin mRNA expression was assessed by real-time polymerase chain reaction. Results showed that the targeted CPT NPs exhibited regular spherical shapes with a mean diameter of approximately 180 nm. In vitro release of the targeted CPT NPs exhibited an initial burst (40%) within 12 h, followed by a slow release. Cytotoxicity test against H22 cells indicated that the targeted CPT NPs exerted significant antitumor effects. Compared with free CPT and non-targeted CPT NPs, the targeted CPT NPs showed superior inhibition ratio against tumor in vivo, which may be associated with reduced survivin mRNA expression. The results suggested that the new targeted CPT NPs may be a promising injectable delivery system for cancer therapy.
Introduction
Camptothecin (CPT), a natural alkaloid, is present in the bark of Camptotheca acuminata. Numerous studies have shown that CPT has high antitumor activity against various cancers, such as cancers from liver, gastric, breast, ovarian and lung (Çirpanli et al., 2009). CPT and its analogs can inhibit the activity of DNA topoisomerase I, an enzyme required for DNA replication and transcription, through chemical binding between the enzyme and acyl position of the CPT lactone ring (Swaminathan et al., 2010). However, the clinical application of CPT against cancer is limited because of its poor water solubility, instability, low bioavailability and severe toxic side effects (Venditto et al., 2010). To overcome the poor solubility of the lactone form, CPT is commonly used as a sodium salt of the carboxylate form in clinical applications. However, the CPT half-life in this form is only 5 min, leading to a significant reduction in the drug efficacy as well as to additional adverse reactions. CPT has a chemically unstable E-ring (lactone function) that is essential for antitumor activity. However, the plasma pH favors conversion of the active lactone form to the inactive carboxylate form (Cırpanli et al., Citation2011). The latter form has minimal clinical value, lower solubility and higher toxicity (Kunii et al., Citation2007). Therefore, a suitable delivery system is important to the clinical application of CPT.
Several attempts have been made to develop various CPT delivery systems, including micelles (Fan et al., Citation2010; Guo et al. Citation2012), liposomes (Modi et al., Citation2012), polymer conjugations (Hong et al., Citation2010), and nanoparticles (NPs) (Bahadur et al., Citation2014). Among these drug delivery systems, polymeric NPs have become a hot research topic because of their unique characteristics. The majority of polymer drug carriers have no active but passive targeting characteristic. For example, CPT-loaded NPs with polyethylene glycol shells can stay in circulation for a long time (Bildstein et al., Citation2011; Luo & Jiang, Citation2012), thereby allowing the prolonged exposure of tumor cells to antitumor drugs. The drugs subsequently reach the target site during passive targeting through the enhanced permeability and retention effect. Passive targeting delivery system for CPT still presents numerous challenges due to low target-tissue concentrations and unpredictable toxicity (Paranjpe et al., Citation2004).
The ability to deliver highly effective dosages to specific sites in the human body has become a hotspot in drug delivery research. Polymeric NPs with targeting ligands are promising candidates for cancer therapy because they can preferentially localize in tumor tissue rather than in non-cancerous tissues in the body (Pardridge, Citation2007; Pulkkinen et al., Citation2008). One strategy that has been explored is the application of antibody-targeted NPs (Kocbek et al., Citation2007; Chen et al., Citation2008; Sun et al., Citation2008; Fay et al., Citation2011; Lei et al., Citation2011).
The attachment of antibody fragments onto NP surfaces has been used to enhance tumor targeting because of the high-binding specificity of the fragments. Antibody-conjugated NPs are mainly used to deliver specific drugs to the diseased site (Shukla et al., Citation2006; Cirstoiu-Hapca et al., Citation2010). For instance, the attachment of anti-Fas antibody onto CPT-loaded NPs can be used as a therapeutic strategy for the treatment of tumors that express death receptors (McCarron et al., Citation2008). Apart from some success in antibody targeting, another active targeting strategy focuses on the investigation of receptors, such as transferrin and folate, which are constitutively overexpressed in many tumor cell types because of their increased metabolic demand. To our knowledge, attempts to attach ligands onto particle surfaces have been achieved and further developed to target breast, lung and prostate cancers specifically through the use of tumor-specific ligands (Byrne et al., Citation2008). Active targeting NPs are regarded as promising carriers for the controlled delivery of anticancer drugs with high antitumor efficacies and low side effects. However, few examples have successfully entered clinical application. Some of the major bottlenecks are the high cost, and instability of new nano-delivery systems for cancer chemotherapy.
Liver cancer is a common global disease and is the third highest cause of cancer-related deaths (Ferlay et al., Citation2010). Actively targeting drug carriers for liver cancer treatment has become a research hotspot. In our earlier work, polymeric NPs with targeting antibody that can increase the selectivity and efficiency of drug delivery to the target cells were developed (Xiong et al., Citation2012). In this study, CPT-loaded biotinylated pluronic F127/poly(lactic acid) (biotin-F127-PLA) NPs were prepared by a dialysis method. The NP particle size and drug loading were characterized by dynamic light scattering (DLS) and ultraviolet (UV) spectrophotometry, respectively. The targeted NPs were prepared by conjugating the biotin-F127-PLA NPs with anti-3A5 antibody, which can specifically bind to the surface of hepatoma H22 (Liang et al., Citation1999). In vitro release and cytotoxicity tests were also conducted. The tissue distribution, in vivo antitumor effect against H22 cells, and expression of survivin gene were investigated after intravenous (i.v.) administration of the CPT NPs. The purpose was to prove the feasibility of using targeted NPs as a high-performance CPT delivery system that targets liver cancer cells.
Materials and methods
Materials
CPT was kindly provided by Beijing Hui Tak Technology Limited Company (Beijing, China). Amphiphilic block copolymers Pluronic-PLA and biotin-Pluronic-PLA were synthesized by ring-opening polymerization (Li et al., Citation2010). Hepatocellular carcinoma cell line H22 was provided by Cancer Institute, Chinese Academy of Medical Sciences. Monoclonal anti-3A5 antibody was purchased from Abnova (Taiwan, China). RPMI 1640, MTT and dimethyl sulfoxide (DMSO) were supplied by Sigma Chemical Co. (St. Louis, MO). Fetal bovine serum (FBS) was purchased from Gibco BRL (Gaithersberg, MD). All other chemicals were of reagent grade.
Male ICR mice (20 ± 2 g) were obtained from Hunan SLAC Experiment Animal Limited Company (Changsha, China). All mice were allowed access to water and food ad libitum under the controlled conditions of temperature (22 ± 2 °C), humidity (55 ± 5%) and light (12 and 12 h of light and dark, respectively). They were housed in a plastic cage containing sterile paddy husk (procured locally) as bedding throughout the experiment. The animal study protocol was reviewed and approved by the Experimental Animal Committee of Jiangxi Institute of Occupational Disease Prevention.
Preparation and characterization of CPT NPs
Preparation of CPT NPs
A biotin-F127-PLA block copolymer (12 mg) and CPT (1 mg) were dissolved in tetrahydrofuran (THF). The mixture solution was then added dropwise to ultrapure water (15 g) under gentle stirring, and THF was removed under reduced pressure. The aggregated polymer solution was dialyzed against ultrapure water for 5 h using a cellulose membrane bag (molecular weight cut-off =12 000–14 000 Da) to remove non-encapsulated drug molecules. Water was replaced at 1 h intervals. Aqueous CPT-loaded F127-PLA NPs were also prepared using the same method with F127-PLA block copolymer.
Characterization of CPT NPs
The mean diameters, size distributions and zeta potential of the NPs were determined by the dynamic light scattering (DLS) method (Malvern Zetamaster ZEM 5002, Malvern, UK). Samples were diluted with super pure water for size measurement and solution containing sodium chloride to adjust the conductivity to 50 μS/cm for zeta potential measurement. All measurements were performed at a temperature of 25 °C.
To determine encapsulation efficiency, about 5 mL of the CPT NPs solution was lyophilized and then dissolved in THF (1 mL). The CPT content was determined by measuring the UV absorbance at 292 nm using Perkin Elmer Lambda 35 UV-Vis spectrophotometer (Perkin Elmer, Waltham, MA). A calibration curve was prepared using THF as the solvent. The entrapment efficiency and drug loading content were calculated using the following formulae:
Preparation of targeted CPT NPs
Targeted CPT NPs was prepared by conjugating biotinylated drug-loaded NPs with anti-3A5 antibody. Two hundred microliters of biotinylated drug-loaded NPs (15 mg/mL) were diluted with phosphate-buffered saline (PBS). The NPs were incubated at 37 °C for 30 min in the presence of excess streptavidin and were washed with PBS three times to remove unreacted streptavidin. The biotinylated anti-3A5 antibody was then added to the NPs, and the mixtures were incubated at 37 °C for 30 min. The resulting product was centrifuged at 10 000rpm for 8 min and subsequently washed with deionized water three times to remove unreacted biotinylated anti-3A5 antibody.
The unreacted biotinylated anti-3A5 antibody content was determined by measuring the UV absorbance at 280 nm using a UV-visible spectrophotometer. The antibody binding rate and coupling ratio of antibody to NPs were calculated using the following formulae:
where W(total), W(unreacted) and W(NPs) are the content of the total biotinylated anti-3A5 antibody, the unreacted biotinylated anti-3A5 antibody and the biotinylated drug-loaded NPs, respectively.
The targeted CPT NPs surface morphology was observed by transmission electron microscopy (TEM, Tecnai G2 20, FEI Co., Eindhoven, The Netherlands). Samples for TEM observation were redispersed in super pure water, then dropped on copper grids stained with phosphotungstic acid (PTA) solution (2%, w/v) and dried in air at room temperature before examined under a microscope (Yang et al., Citation2007).
In vitro release studies
The CPT-loaded NP suspensions (5 mL) were placed in the dialysis membrane, which was immersed in 30 mL PBS (pH 7.4). The solution was then stirred (100 rpm) with a magnetic stirrer at 37 °C in a water bath for the drug release study. At selected time intervals, 2 mL of the solution outside the dialysis membrane were collected, and fresh PBS solution (2 mL) was added to the incubation medium to maintain a constant volume. The sample solution was acidified with glacial acetic acid and then stored at −20 °C prior to HPLC analysis. The total amount of released CPT was measured using an Agilent 1100 HPLC (Waldbronn, Germany) equipped with an analytical C18 column and a UV-Vis detector set at 292 nm. A mixture of acetonitrile and aqueous PBS solution (50/50, v/v) was used as the mobile phase. The flow rate was 1.0 mL/min. The retention time of CPT was around 4.8 min.
In vitro antitumor effect of CPT NPs
H22 hepatocellular carcinoma cell lines were routinely cultured in RPMI 1640 containing 10% fetal calf serum (FCS) and then incubated at 37 °C in a 5% CO2 humidified incubator. Briefly, 190 μL of the cells were seeded on a 96-well plate at a density of approximately 1 × 104 cells per well. The cells were subsequently incubated at 37 °C in a 5% CO2 humid incubator for 24 h. Free CPT was dissolved in an RPMI 1640 solution containing 10% (v/v) dimethyl sulfoxide (DMSO), and the CPT NPs were suspended in RPMI 1640. The incubated cells were exposed to 10 μL of free CPT, non-targeted NPs, and targeted NPs at CPT concentration ranges of 5 μg/mL to 80 μg/mL, respectively, and further incubated continuously for 72 h. Cells in the control group were treated with blank NPs for comparison. Cell viability was determined by the MTT colorimetric assay. The medium was replaced by 200 μL of MTT solution (0.5 mg/mL in PBS) and stained for 4 h. The MTT dye solution was then removed, and the cells were treated with 100 μL of DMSO. The absorbance intensity at 490 nm was measured using Bio-Rad Model 550 microplate spectrophotometer (Bio-Rad, Hercules, CA). The in vitro cell viability was calculated using the following formula:
where A(test) and A(untreated) are the absorbance of NP-treated live cells and untreated cells, respectively.
In vivo antitumor effects of CPT NPs
Ascites were collected after mice inoculation of H22 sarcoma cells for 7 days. The H22 cells were adjusted to a concentration of 1 × 105 cells/mL in normal saline. For tumor implantation, the dilute H22 suspensions were subcutaneously inoculated into imprinting control region mice under the armpit of the left forelimb. After 1 week, the tumors formed, and the tumor-bearing mice were randomly divided into four groups of 10 mice each: one group receiving PBS, treatment group with free CPT (control), non-targeted CPT NPs and targeted CPT NPs. All groups were administered daily by tail vein injection for 14 days. At the end of the experiment, the mice were sacrificed and the tumors were separated. Tumor weight was obtained and tumor inhibition rate was calculated according to the formula:
Tissue distribution study
H22 tumor cells were inoculated subcutaneously to ICR mice under the armpit at left forelimb. The H22 bearing mice were randomly divided into eight groups (five per group) seven days after the inoculation. Four groups were administered non-targeted CPT NPs and the other four groups were given targeted CPT NPs at the same dose level through tail vein. At time 1.0, 4.0, 8.0 and 12.0 h after drug administration, blood was drawn from the carotid artery, placed into heparinized test tubes and centrifuged at 1000 rpm for 10 min to obtain plasma samples. Afterwards, the mice were dissected and each test organ was exorcised. Heart, lung, liver, spleen, kidney and brain tissues were collected, weighed and homogenized with saline and acidified to pH 3.0 with glacial acetic acid. Then tissue homogenates and plasma samples were treated with double volume cold mixture of acetonitrile/methanol (1/1 v/v) to precipitate proteins and extract CPT. Concentrations of CPT in samples were determined using the HPLC method described in the section “In vitro release studies”.
Survivin mRNA expression
Total RNA was extracted from tumor tissue by disruption and homogenization using TRIzol reagent (Gibco BRL, Gaithersburg, MD). RNA (2 mL, 1 mg/mL) was used to synthesize cDNA according to the manufacturer’s protocol. The reaction mixture was initially incubated for 5 min at 25 °C, followed by 60 min at 50 °C, and then by enzyme inactivation for 5 min at 85 °C. The cDNA samples were stored at −80 °C until use.
A real-time fluorescent quantitative polymerase chain reaction (PCR) system was used to analyze the survivin (target) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH, endogenous control) gene expression. cDNA was then added to a final volume of 25 mL, with 0.5 mL of each primer. PCR was performed using a total reaction mixture volume of 20 μL. The reaction mixture contained 1 × TaqMan universal master mix with AmpErase uracil N-glycosylase, 600 nmol/L of each primer, 200 nmol/L TaqMan probe, and 1 μL of an unknown cDNA or 2 μL of a standard template. The cycling program was as follows: 25 °C for 5 min, 42 °C for 60 min, and 70 °C for 5 min. The corresponding primers to mice survivin were as follows: forward, 5′-AAC TAC CGC ATC GCC ACC TT-3′; reverse, 5′-ATC GGG TTG TCA TCG GGT TC-3′. The predicted product was 176 bp. GAPDH was used as an internal standard, and its mRNA was amplified with the following primers: forward, 5′-GGT GCT GAG TAT GTC GTG GAG T-3′; reverse, 5′-TCT TCT GGG TGG CAG TGA TG-3′. The amplification was performed using the previously described method, except for the annealing temperature, which was set at 60 °C. The predicted size of PCR product was 289 bp. Fluorescence signals were detected at the end of each cycle.
A three-step PCR consisted of an initial denaturation at 95 °C for 3 min followed by 40 cycles at 95 °C for 15 s; denaturation at 50 °C to 58 °C for 30 s and at 72 °C for 30 s; and a final extension step at 72 °C for 5 min. A two-step PCR protocol consisted of an initial denaturation at 95 °C for 3 min followed by 40 cycles at 95 °C for 15 s, then at 60–68 °C for 60 s of annealing/extension, and a final extension step at 72 °C for 5 min. In both cases, a melt cycle starting from 55 °C to 95 °C was included, with a 0.5 °C increase cycle of 10 s for 80 cycles. GAPDH mRNA transcript levels were used as internal controls. Gene expression levels were expressed as fold changes relative to the controls.
Statistical analysis
The experiments were repeated three times, and the results were expressed as mean ± S.D. Statistical analysis was done by using SPSS 10.0 (Chicago, IL). Comparisons among all groups were performed with the one-way analysis of variance (ANOVA) test. In all cases, p < 0.05 was considered to be statistically significant.
Results and discussion
Characterization of NPs
The clinical efficacy of CPT is significantly restricted by its insolubility and instability. Free CPT is water insoluble. CPT exists in two forms depending on pH, with an active lactone form existing at pH < 5 and an inactive carboxylate form existing at physiological or basic pH. When CPT was successfully loaded into the NPs, the solubility and stability of CPT in the aqueous solution increased. As shown in , the entrapment efficiencies of both biotin-F127-PLA NPs and F127-PLA NPs were 29.3 ± 1.05% and 33.0 ± 2.06%, corresponding to drug loadings of 8.8 ± 0.56% and 9.9 ± 0.98%, respectively. The particle sizes of the biotin-F127-PLA NPs and F127-PLA NPs were 165.7 ± 19.2 and 158.4 ± 16.7 nm, respectively. Two kinds of NPs had a net negative surface charge, and the presence of biotin appeared to reduce the zeta potential (−9.2 ± 0.3 mV) of NPs compared to F127-PLA NPs (−14.5 ± 0.7 mV), which may be shielded by the biotin molecules binding on the surfaces of NPs. The value of zeta potential can be used as an index of electrostatic interaction between NPs, and the electric repulsion plays an important role in the stability of NPs dispersions.
Table 1. Characterization of CPT NPs.
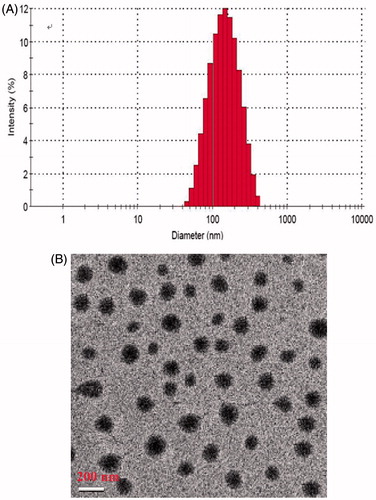
shows size distribution of the CPT-loaded biotin-F127-PLA NPs with a polydispersity index of 0.183, as determined by dynamic light scattering (DLS). The mean particle size was 165 nm, and a single-peak size distribution was observed.
Figure 1. (A) Size distribution of CPT-loaded biotin-F127-PLA NPs and (B) transmission electron micrograph of the targeted CPT NPs.
Targeted CPT NPs was prepared by conjugating biotinylated drug-loaded NPs with anti-3A5 antibody in presence of streptavidin. To assess the influence of anti-3A5 antibody content on the coupling ratio of antibody to NPs, biotinylated CPT NPs were incubated with different concentration of anti-3A5 antibody solution. As seen in , the higher antibody content induced the lower antibody binding rate, but coupling ratio of antibody to NPs increased from 0.18 ± 0.02 to 0.81 ± 0.08% with increasing biotinylated antibody concentration from 10 to 60 μg/mL. The results illustrated that the higher the antibody content is taken, the more the antibody molecules are likely to be coupled with NPs. When the concentration of the antibody was above 60 μg/mL, it was found that the binding of antibody saturated and no increase in coupling ratio occurred.
Table 2. Evaluation of antibody binding rate and coupling ratio of antibody to NPs.
After conjugation of the biotinylated CPT NPs with the anti-3A5 antibody, the morphology of the targeted CPT NPs was determined by TEM, and the result is shown in . The NPs exhibited regular spherical shapes, with a mean diameter of approximately 180 nm.
In vitro release profile of CPT from the NPs
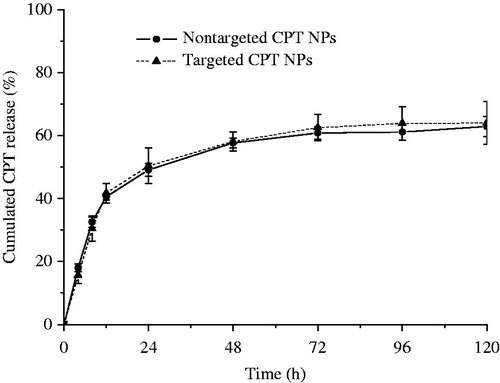
In vitro release behaviors of CPT from the non-targeted and targeted NPs in PBS solutions at 37 °C were investigated using dialysis, and the results are shown in . The cumulative release curves of the non-targeted CPT NPs showed an initial burst release (40%) within 12 h, followed by a slow release. Almost 3 days (72 h) later, the drug release profile reached a plateau (approximately 57%). The release rate of CPT from the targeted CPT NPs was nearly the same as that from non-targeted CPT NPs. The CPT–polymer interaction as well as the hydrophilicities of the polymers may play an important role in the release rate of CPT from the NPs.
Figure 2. In vitro release profile of CPT from NPs in phosphate buffer solutions.
In vitro antitumor effects on the H22 cell lines
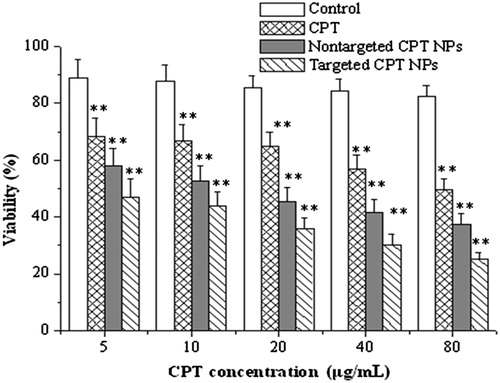
In vitro antitumor effect of free CPT, non-targeted CPT NPs, and targeted CPT NPs against H22 cells was evaluated by a cell viability test. shows that both the non-targeted and targeted CPT NPs exhibited higher antitumor effects than the free CPT. This result explained the necessity for CPT to enter the interior of the tumor cells and exert its antitumor efficacy, as well as the potency of the lactone form of CPT. Free CPT exists mainly in its inactive carboxylate form at pH 7.4. The anionic nature of the CPT carboxylate prevented its easy entrance into the cell interior. By contrast, the encapsulated CPT was the active lactone form. The CPT NPs can be internalized by endocytosis because of their nanometer-scale size. If the lactone-formed CPT was also released inside the cells, a prominent antitumor effect can be expected. The incubation time for the samples with H22 cells was sufficient for the release of the drug from the polymer matrix. Compared with CPT in the non-targeted NPs, CPT in the targeted NPs exerted stronger anticancer effects against H22 cells. This result can be attributed to the specific interaction between B-F127-PLA/avidin/biotinylated anti-3A5 antibody and the related antigen on the H22 cell surfaces. The cytotoxicities of the CPT NPs and the free CPT against H22 cells obviously increased with increased CPT concentration. The cytotoxicities of the blank NPs formulations were also evaluated to confirm that the difference in the effects was not due to the cytotoxicity of the vehicle but due to that of the drug. This result indicated that the PLA homopolymer and Pluronic F127 were biocompatible polymers. Earlier results indicated that the biotin-F127-PLA block polymer also had high biocompatibility (Li et al., Citation2010).
Figure 3. In vitro antitumor effects of CPT NPs on H22 cell lines. The concentrations of the blank NPs (control group) were set at the same level as those in the drug-loaded NPs group. **p < 0.01 compared with the control group.
Some studies have shown that most CPT forms (80%) in PBS (pH 7.4, 37 °C) are converted into the inactive carboxylate form only after 4 h (Çirpanli et al., 2009). However, CPT in the present study was encapsulated in the NPs and remained in its active lactone form during the in vitro release. The CPT proportions in the lactone ring and in the carboxylate forms were critical in predicting the tumor response to CPT because the lactone form exerted a significantly higher antitumor efficacy than the carboxylate form. Once entrapped within a PLA-based NP, CPT was exposed to an acidic environment because of PLA degradation, which stabilized the active lactone structure of CPT. This behavior was similar to that observed for PLGA-based NPs (Shenderova et al., Citation1999).
Tissue distribution
Tissue distribution of CPT after intravenous administration of non-targeted CPT NPs and targeted CPT NPs in mice at 1.0, 4.0, 8.0 and 12.0 h were shown in . In the targeted CPT NPs group, the highest CPT concentration was found in the liver, followed by spleen, lung and kidney. At 8.0 and 12.0 h after dosing, drug levels of CPT-loaded targeted NPs in tumor tissue remained higher compared with the other tissues (p < 0.05), which suggested that targeted NPs could increase the accumulation of CPT within tumor and achieve great targeting ability. It could be explained by the antibody-targeted effect in that anti-3A5 antibody-specific bind to the liver tumor tissues.
Figure 4. Tissue distribution of CPT in tumor-bearing mice after intravenous administration of non-targeted NPs (A) and targeted NPs (B). *p < 0.05 compared with the plasma.
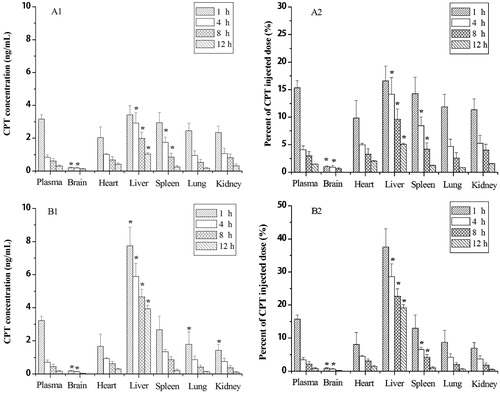
In the non-targeted CPT NPs group, CPT concentrations in liver and spleen tended to be greater than that in other tissue. Tissue distributions of intravenously injected NPs are mainly influenced by their size, surface features and opsonization. Opsonins adsorbed on the NPs surface can promote particle recognition by the reticuloendothelial system. Therefore, conventional NPs are rapidly taken up by the reticuloendothelial system in the liver and spleen after intravenous administration (Jia et al., Citation2010). However, showed that the concentration of CPT in liver tumor tissue for targeted CPT NPs was significantly higher than that for non-targeted CPT NPs. As also shown in , the percent of CPT injected dose in each organ were consistent with the CPT concentration in each organ, where the percent of CPT in liver tumor tissue was the highest among all organ, and the trend for targeted CPT NPs was significantly greater than that for non-targeted CPT NPs, which provides important evidence for supporting reduced side effect from targeted CPT NPs.
In vivo antitumor effect in H22-bearing mice
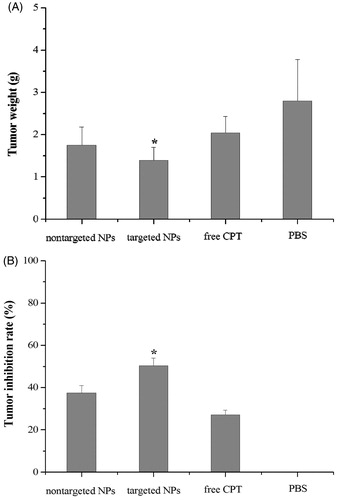
The in vivo antitumor effects of three different CPT formulations at the same dose were evaluated after i.v. injection of free CPT, non-targeted CPT NPs, and targeted CPT NPs in tumor-bearing nude mice. There were no significant changes in body weight of the mice during the treatment period (data not shown). lists the tumor weight and the tumor inhibition rates of all the test groups. All groups, except for the PBS group treated with normal saline, yielded significant tumor inhibition (inhibition rates above 25%). Within 14 days, where tumors from the untreated mice (PBS group) were the heaviest, and the groups injected with free CPT exhibited 2.04 g tumor weight and 27.14% tumor inhibition rate. Tumor tissues had high interstitial fluid pressure that prevented conventional chemotherapeutic agents to enter the tumors. Only 5 to 10% of drugs that accumulated in normal viscera entered the tumor tissues (Heldin et al., Citation2004). By contrast, the tumors from mice treated with targeted CPT NPs were the smallest (1.39 g) and statistically significant tumor inhibition rate (50.36%) compared to free CPT group (p < 0.05), whereas the group treated with non-targeted CPT NPs exhibited 1.75 g tumor weight and 37.50% tumor inhibition rate. These results indicated that treatment with the targeted CPT NPs had a higher antitumor efficacy than treatment with non-targeted CPT NPs, which may be associated with the higher tumor uptake and retention of the targeted CPT NPs as a result of the tumor-bound biotinylated anti-3A5 antibody/avidin complex.
Figure 5. In vivo antitumor effect of CPT in either targeted or non-targeted CPT NPs in H22-bearing mice. Tumor weight (A) and tumor inhibition rate (B). *p < 0.05 compared to the free CPT group.
Survivin mRNA expression in H22-bearing mice
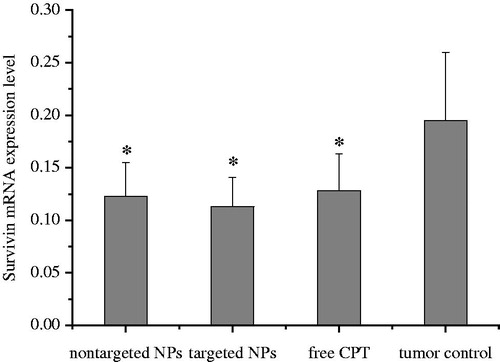
Survivin, an inhibitor of apoptosis protein, is not expressed in normal tissues but is highly expressed in cancer cells (Stauber et al., Citation2007). Nearly all common cancer tissues exhibit an overexpression of the survivin gene. Survivin has been proposed as an attractive target for new anticancer interventions (Okamura et al., Citation2009). Anti-apoptotic genes may also contribute to the development and progression of cancer. shows that targeted and non-targeted CPT NPs can significantly reduce gene expression level of survivin in mice bearing H22 tumor tissue compared with the tumor control group without CPT (p < 0.05). A significant difference with the tumor group was also observed. Survivin gene expression in the targeted NP group exhibited the lowest level. CPT exerts significant antitumor activity against various cancers by inhibiting the activity of DNA topoisomerase I, which is required for DNA replication and transcription (Topcu, Citation2001; Garcia-Carbonero & Supko, Citation2002). In this study, the targeted nanoparticles could stabilize the active lactone structure of CPT and delivery CPT to tumor tissue at the most high level. CPT increased the tumor inhibition rate in vivo, which may be associated with reduced survivin mRNA expression.
Figure 6. Survivin mRNA expression of mice bearing H22 tumor tissue. *p < 0.05 compared with the control group.
Conclusions
CPT was incorporated into biotin-F127-PLA or F127-PLA polymeric NPs by a dialysis method. The targeting NPs were prepared by conjugating biotin-F127-PLA NPs with anti-3A5 antibody. The in vitro release of the targeted CPT NPs exhibited an initial burst (40%) within 12 h, followed by a slow release. The cytotoxicity test against H22 cells showed that the targeted CPT NPs exerted stronger in vitro antitumor effects than the non-targeted CPT NPs or the free drug. Compared with free CPT and non-targeted CPT NPs, the targeted CPT NPs exhibited superior in vivo antitumor effects which may be associated with reduced survivin mRNA levels during the treatment period. The results indicated that the new targeted CPT NPs may be a promising injectable delivery system for cancer therapy.
Acknowledgements
We thank Zachary Y. Huang for improving the manuscript.
Declaration of interest
This work was supported by Research Foundation for Natural Science Foundation of Jiangxi Province (Project No. 20122BAB204001), Science and Technology Support Program of Jiangxi Province (Project No. 20111BBF60025, Project No. 2008160) and Young Scientist Training Program of Jiangxi Province (20122BCB23006).
References
- Bahadur RKC, Chandrashekaran V, Cheng B, et al. (2014). Redox potential ultrasensitive nanoparticle for the targeted delivery of camptothecin to HER2-positive cancer cells. Mol Pharmaceut 11:1897–905
- Bildstein L, Dubernet C, Couvreur P. (2011). Prodrug-based intracellular delivery of anticancer agents. Adv Drug Deliv Rev 63:3–23
- Byrne JD, Betancourt T, Brannon-Peppas L. (2008). Active targeting schemes for nanoparticle systems in cancer therapeutics. Adv Drug Deliv Rev 60:1615–26
- Chen H, Gao J, Lu Y, et al. (2008). Preparation and characterization of PE38KDEL-loaded anti-HER2 nanoparticles for targeted cancer therapy. J Control Release 128:209–16
- Cırpanlı Y, Allard E, Passirani C, et al. (2011). Antitumoral activity of camptothecin-loaded nanoparticles in 9L rat glioma model. Int J Pharm 403:201–6
- Çirpanli Y, Bilensoy E, Dogan AL, Çalis S. (2009). Comparative evaluation of polymeric and amphiphilic cyclodextrin nanoparticles for effective camptothecin delivery. Eur J Pharm Biopharm 73:82–9
- Cirstoiu-Hapca A, Buchegger F, Lange N, et al. (2010). Benefit of anti-HER2-coated paclitaxel-loaded immuno-nanoparticles in the treatment of disseminated ovarian cancer: therapeutic efficacy and biodistribution in mice. J Control Release 144:324–31
- Fan HL, Huang J, Li YP, et al. (2010). Fabrication of reduction degradable micelle based on disulfide-linked graft copolymer-camptothecin conjugate for enhancing solubility and stability of camptothecin. Polymer 515:107–14
- Fay F, McLaughlin KM, Small DM, et al. (2011). Conatumumab (AMG 655) coated nanoparticles for targeted pro-apoptotic drug delivery. Biomaterials 32: 8645–53
- Ferlay J, Shin HR, Bray F, et al. (2010). Estimates of worldwide burden of cancer in 2008: GLOBOCAN 2008. Int J Cancer 127:2893–917
- Garcia-Carbonero R, Supko JG. (2002). Current perspectives on the clinical experience, pharmacology, and continued development of the camptothecins. Clin Cancer Res 8:641–61
- Guo Q, Luo P, Luo Y, et al. (2012). Fabrication of biodegradable micelles with sheddable poly(ethylene glycol) shells as the carrier of 7-ethyl-10-hydroxy-camptothecin. Colloid Surface B 100:138–45
- Heldin CH, Rubin K, Pietras K, Ostman A. (2004). High interstitial fluid pressure – an obstacle in cancer therapy. Nat Rev Cancer 4:806–13
- Hong M, Zhu S, Jiang Y, et al. (2010). Novel anti-tumor strategy: PEG-hydroxycamptothecin conjugate loaded transferring-PEG-nanoparticles. J Control Release 141:22–9
- Jia LJ, Zhang DR, Li ZY, et al. (2010). Nanostructured lipid carriers for parenteral delivery of silybin: biodistribution and pharmacokinetic studies. Colloids Surf B 80:213–18
- Kocbek P, Obermajer N, Cegnar M, et al. (2007). Targeting cancer cells using PLGA nanoparticles surface modified with monoclonal antibody. J Control Release 120:18–26
- Kunii R, Onishi H, Machida Y. (2007). Preparation and antitumor characteristics of PLA/(PEG-PPG-PEG) nanoparticles loaded with camptothecin. Eur J Pharm Sci 67:9–17
- Lei TJ, Srinivasan S, Tang Y, et al. (2011). Comparing cellular uptake and cytotoxicity of targeted drug carriers in cancer cell lines with different drug resistance mechanisms. Nanomed Nanotechnol 7:324–32
- Li ZL, Xiong XY, Li YP, et al. (2010). Synthesis and self-assembling behaviors of biotinylated pluronic/poly(lactic acid) biocompatible block copolymers in aqueous solutions. J Appl Polym Sci 115:1573–80
- Liang FY, Zhang SH, Pan Y, et al. (1999). Use of monoclonal antibody norcantharidin conjugate in experimental targeting therapy of tumor. Chin J Zoology 34:18–22
- Luo ZL, Jiang JW. (2012). pH-sensitive drug loading/releasing in amphiphilic copolymer PAE-PEG: Integrating molecular dynamics and dissipative particle dynamics simulations. J Control Release 162:185–93
- McCarron PA, Marouf WM, Quinn DJ, et al. (2008). Antibody targeting of camptothecin-loaded PLGA nanoparticles to tumor cells. Bioconjugate Chem 19:1561–9
- Modi S, Xiang TX, Anderson BD. (2012). Enhanced active liposomal loading of a poorly soluble ionizable drug using supersaturated drug solutions. J Control Release 162:330–9
- Okamura K, Koike H, Sekine Y, et al. (2009). Survivin and its spliced isoform gene expression is associated with proliferation of renal cancer cells and clinical stage of renal cancer. Cancer Epidemiol 33:137–41
- Paranjpe PV, Chen Y, Kholodovych V, et al. (2004). Tumor-targeted bioconjugate based delivery of camptothecin: design, synthesis and in vitro evaluation. J Control Release 100:275–92
- Pardridge WM. (2007). Drug targeting to the brain. Pharm Res 24:1733–44
- Pulkkinen M, Pikkarainen J, Wirth T, et al. (2008). Three-step tumor targeting of paclitaxel using biotinylated PLA-PEG nanoparticles and avidin-biotin technology: formulation development and in vitro anticancer activity. Eur J Pharm Biopharm 70:66–74
- Shenderova A, Burke TG, Schwendeman SP. (1999). The acidic microclimate in poly(lactide-co-glycolide) microspheres stabilizes camptothecins. Pharm Res 16:241–8
- Shukla R, Thomas TP, Peters JL, et al. (2006). HER2 specific tumor targeting with dendrimer conjugated anti-HER2 mAb. Bioconjugate Chem 17:1109–15
- Stauber RH, Mann W, Knauer SK. (2007). Nuclear and cytoplasmic Survivin: molecular mechanism, prognostic, and the therapeutic potential. Cancer Res 67:5999–6002
- Sun B, Ranganathan B, Feng SS. (2008). Multifunctional poly(D, L-lactide-co-glycolide)/montmorillonite (PLGA/MMT) nanoparticles decorated by Trastuzumab for targeted chemotherapy of breast cancer. Biomaterials 29:475–86
- Swaminathan S, Pastero L, Serpe L, et al. (2010). Cyclodextrin-based nanosponges encapsulating camptothecin: physicochemical characterization, stability and cytotoxicity. Eur J Pharm Biopharm 74:193–201
- Topcu Z. (2001). DNA topoisomerases as targets for anticancer drugs. J Clin Pharm Therap 26:405–16
- Venditto VJ, Simanek EE. (2010). Cancer therapies utilizing the camptothecins: a review of the in vivo literature. Mol Pharm 7:307–49
- Xiong XY, Guo L, Gong YC, et al. (2012). In vitro & in vivo targeting behaviors of biotinylated Pluronic F127/poly(lactic acid) nanoparticles through biotin-avidin interaction. Eur J Pharm Sci 46:537–44
- Yang AS, Yang L, Liu W, et al. (2007). Tumor necrosis factor alpha blocking peptide loaded PEG-PLGA nanoparticles: preparation and in vitro evaluation. Int J Pharm 331:123–32