
Abstract
A water-insoluble anti-tumor agent, paclitaxel (PTX) was successfully incorporated into novel-targeted polymeric micelles based on tocopherol succinate-chitosan-polyethylene glycol-folic acid (PTX/TS-CS-PEG-FA). The aim of the present study was to evaluate the pharmacokinetics, tissue distribution and efficacy of PTX/TS-CS-PEG-FA in comparison to Anzatax® in tumor bearing mice. The micellar formulation showed higher in vitro cytotoxicity against mice breast cancer cell line, 4T1, due to the folate receptor-mediated endocytosis. The IC50 value of PTX, a concentration at which 50% cells are killed, was 1.17 and 0.93 µM for Anzatax® and PTX/TS-CS-PEG-FA micelles, respectively. The in vivo anti-tumor efficacy of PTX/TS-CS-PEG-FA, as measured by reduction in tumor volume of 4T1 mouse breast cancer injected in Balb/c mice was significantly greater than that of Anzatax®. Pharmacokinetic study in tumor bearing mice revealed that the micellar formulation prolonged the systemic circulation time of PTX and the AUC of PTX/TS-CS-PEG-FA was obtained 0.83-fold lower than Anzatax®. Compared with Anzatax®, the Vd, T1/2ß and MRT of PTX/TS-CS-PEG-FA was increased by 2.76, 2.05 and 1.68-fold, respectively. As demonstrated by tissue distribution, the PTX/TS-CS-PEG-FA micelles increased accumulation of PTX in tumor, therefore, resulted in anti-tumor effects enhancement and drug concentration in the normal tissues reduction. Taken together, our evaluations show that PTX/TS-CS-PEG-FA micelle is a potential drug delivery system of PTX for the effective treatment of the tumor and systematic toxicity reduction, thus, the micellar formulation can provide a useful alternative dosage form for intravenous administration of PTX.
Introduction
Paclitaxel (PTX), a microtubule stabilizing anti-proliferative agent, has been used in the treatment of a broad spectrum of human cancers, especially in carcinomas of the lungs, breast and ovary (Kim et al., Citation2001). Because of the low water solubility, PTX is commercially available in a solution composed of a 50:50 (v/v) mixture of Cremophor® EL (polyethoxylated castor oil) and dehydrated ethanol marketed as Taxol®. Unfortunately, Cremophor® EL induces some serious adverse reactions such as hypersensitivity, nephrotoxicity and neurotoxicity after intravenous (i.v.) administration (Kim et al., Citation2001). Application of Taxol® is also limited by its short-term physical stability since PTX tends to precipitate out of the aqueous media upon dilution (Emami et al., Citation2012). Hence, many attempts have been devoted to substitute the Cremophor® EL-based formulation with alternative carriers which enable delivering PTX selectively to the tumor tissues with significantly reduced adverse effects on normal cells. Numerous drug delivery systems including parenteral emulsions (He et al., Citation2003; Nornoo et al., Citation2008), liposomes (Fetterly et al., Citation2003; Soenpenberg et al., Citation2004), nanoparticles (Hawkins et al., Citation2008; Zhu et al., Citation2010) and polymeric micelles (Qu et al., Citation2009; Tao et al., Citation2012; Qu et al., 2013; Wang et al., Citation2013) have extensively been studied and evaluated for parenteral delivery of PTX into cancer cells. Few years ago, Abraxane® which is an albumin-bound PTX nanoparticle formulation, was approved by the food and drug administration (FDA) for metastatic breast cancer. In this formulation, PTX is incorporated within albumin particles to retain the pharmacotherapeutic effects of PTX and to eliminate the adverse effects associated with Cremophor® EL (Hawkins et al., Citation2008). However, pharmacokinetic studies of Abraxane® showed the rapid elimination of PTX from the blood circulation and most of the drug was distributed in the liver, lungs and spleen. Moreover, using human albumin in the preparation of Abraxane® makes the formulation very expensive which might not be affordable for every patient (Bernabeua et al., Citation2014). Recently, polymeric micelles fabricated from natural and synthetic polymers have received particular attention. Polymeric micelles are self-assemblies of amphiphilic block or grafted copolymers forming a core-shell structure in aqueous media and poorly soluble drugs can be solubilized within the hydrophobic inner core of the micelles (Qu et al., 2013). Although micellar systems have made a significant impact on the development of anticancer drug delivery systems, some improvements are still essential. An ideal polymeric micelle should ensure small particle sizes, high drug loading (DL) capacity, and in vivo stability to achieve high drug accumulation in tumor tissues. In most of earlier published reports, PTX has been physically entrapped in the core of micelles by weak hydrophobic interaction (Zhang et al., Citation1996, Kim et al., Citation2001, Liu et al., Citation2011). Thus, upon blood dilution following i.v. administration, micelles are easily disassembled resulting in the rapid loss of the loaded drug. On the other hand, polymeric micelles with particle sizes >200 nm are easily cleared from blood circulation by macrophages and mostly distributed in the liver, lungs and spleen (Hu et al., Citation2008). Accordingly, most of PTX-loaded polymeric micelles have been reported so far could not improve the pharmacokinetics of PTX (Taxol®). For instance, after i.v. injection of PAXCEED® and Genexol®-PM, two early developed micellar formulations of PTX, the area under the plasma PTX concentration–time curves (AUC) were dramatically lower than Taxol® (Zhang et al., Citation1996; Kim et al., Citation2001). Similarly, in vivo studies of PTX-loaded amphiphilic N-octyl chitosan derivatives showed 3.5-fold decrease in plasma AUC of PTX compared to Taxol® solution and the drug was mostly distributed in liver and spleen (Zhang et al., Citation2008; Liu et al., Citation2011). To prevent the uptake of micelles by macrophages and to prolong efficient plasma concentrations of loaded drugs, hydrophilic polymers such as polyethylene glycol (PEG) has been extensively used as shell forming polymer (Liu, Citation2007; Hu et al., Citation2008; Qu et al., Citation2009). In aqueous environment, PEG is highly hydrated and can form a dense, brush-like shell that stretches away from the core. This characteristic prevents the micelle interaction with adjacent micelles and protein adsorption (opsonization) to the surface of polymeric micelles which reduce the micelle aggregations and their uptake by the mononuclear phagocytic system (MPS) (Yang et al., Citation2012). To maximize site-specific targeting and efficient anti-cancer effect, polymeric micelles could also be equipped with targeting ligands, which can recognize and adhere to the biomarkers on the surface of the corresponding cancer cells. Zhang et al. (Citation2011) prepared folic acid functionalized Pluronic P123/F127 mixed micelles for delivery of PTX. Although their formulation could prolong and maintain efficient plasma concentrations of PTX, too small particle sizes of the micelles (<50 nm) are found to be difficult to escape the vasculature system and interact with individual tumor cells. Moreover, DL percent was found to be <2%. Due to its high molecular weight, PTX could not be efficiently entrapped in the core of micelles, so in most formulations the DL has been reported to be <10% (Nam et al., 2013; Wang et al., Citation2013; Bernabeua et al., Citation2014). Low DL efficiency augments the usage of nanocarrier excipients which may display cytotoxicity as the concentration increased. On the whole, inferior in vivo stability and low DL are two main practical challenges in clinical application of polymeric micelles. In order to overcome these limitations, we have recently prepared novel non-targeted and targeted polymeric micelles through synthesizing tocopherol succinate-chitosan (TS-CS) and tocopherol succinate-chitosan-polyethylene glycol-folate (TS-CS-PEG-FA) for systemic administration of PTX. A prominent feature of TS-CS copolymer is the ability to self-assemble into polymeric micelles in aqueous solutions, which has a hydrophobic core formed by tocopherol chains and a hydrophilic corona formed by chitosan chains. α-Tocopherol is an excellent solvent for many poorly soluble drugs which provides sufficient capacity for hydrophobic drugs. Aromatic ring and bulk lipophilic portion of α-TS molecules lead to an increase in the hydrophobic interaction between PTX and the micelle core that promotes the formation of more stabilized and compact micellar core. Furthermore, the PEG units prevent uptake of the micelles by macrophages and folate molecules induce targeting ability. In this study, we attempted a systematic evaluation of the anti-tumor efficacy of our developed PTX-loaded micelles (PTX/TS-CS and PTX/TS-CS-PEG-FA) at the cellular and animal levels by performing intracellular accumulation, in vitro cytotoxicity, PTX/TS-CS-PEG-FA retention in blood circulation, tissue distribution, and in vivo study in tumor bearing Balb/c mice.
Methods
Materials
PTX, fluorescein isothiocyanate (FITC) and diazepam were purchased from Sigma Chemical Co. (St Louis, MO). High-performance liquid chromatography (HPLC) grade methanol and acetonitrile were procured from Caledon (Ontario, Canada). TS-CS and TS-CS-PEG-FA derivatives were synthesized in our laboratory. Anzatax® (30 mg/5 ml) was provided by Hospira Pty Ltd (Australia). Roswell Park Memorial Institute (RPMI)-1640 medium without folic acid, fetal bovine serum (FBS) and antibiotics for cell culture were provided by Sigma-Aldrich (St Louis, MO). Mouse breast cancer cell line (4-T1) was obtained from Pasteur Institute (Tehran, Iran). 3-(4,5–Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was supplied by Sigma-Aldrich.
Female Balb/c mice (5–6 weeks old, 25 ± 2 g) were obtained from Laboratory Animal Center of Faculty of Pharmacy and Pharmaceutical Science, Isfahan University of Medical science, Isfahan, Iran. All the animals were pathogen free and allowed to access food and water freely. All the animal experiments were carried out in accordance with Guide for the Care and Use of Laboratory Animals published by the National Institute of Health and approved by the Research Committee of Isfahan University of Medical Science.
Preparation and characterization of PTX-loaded TS-CS and PTX-loaded TS-CS-PEG-FA micelles
PTX was loaded into the core of micelles via membrane dialysis technique as described previously (Emami et al., Citation2014). Briefly, 10 mg TS-CS or TS-CS-PEG-FA was dissolved in 10 ml water and 5 mg PTX was dissolved in 1 ml ethanol. The PTX solution was then injected into the polymer solution, sonicated (JY 92-II Ultrasonic Processor, Shanghai, China) for 30 min and dialyzed against deionized water overnight at room temperature using dialysis membrane [molecular weight cutoff (MWCO): 10 kDa; Viskase Companies Inc., Darien, IL]. The micellar solution was centrifuged at 7000 rpm for 10 min and filtered through a 0.45-µm microfiltration membrane to collect the PTX-loaded micelles. The drug-loaded micelles were lyophilized (Freeze Dryer Model ALPHA 2-4 LD plus; Christ Company, Stuttgart, Germany) and kept 4 °C until further evaluation.
The size and zeta potential of the micelles were determined by dynamic light scattering using a Zetasizer (3000HS, Malvern Instruments Ltd., Worcestershire, UK). To measure the DL, 2 mg of the lyophilized sample was transferred into 50 ml acetonitrile and ultrasonicated for 10 min to dissolve PTX-loaded micelles. The resultant solution was filtered through 0.45-µm filter unit and PTX concentration was analyzed using HPLC method developed and validated in our laboratory.
The apparatus used was a Waters HPLC system model 746 (Milford, US), consisting of a model 515 intelligent solvent delivery pump, a 100-μl injection loop, a computerized system controller and a Waters 2487 UV detector. Chromatographic separation was performed using a reverse-phase C18-Bondapak (3.9 × 250 mm) column. The mobile phase consisted of potassium dihydrogen phosphate (0.01 M)/acetonitrile (35:65) with final pH adjusted to 3.5 ± 0.1 with orthophosphoric acid. The mobile phase eluted at the flow rate of 1.5 ml/min and the effluent was monitored at 227 nm. Column temperature was kept at 40 °C and 30 µl of the sample was injected into the HPLC column.
The calibration curve was constructed in the range of 0.5–20 µg/ml (r2 > 0.998), the inter- and intra-day precision and accuracy were <12.87%.
The drug entrapment efficiency (EE) and DL were calculated by the following equations:
Release studies of the drug-loaded micelles were performed in phosphate-buffered solution (PBS) (0.1 M, pH 7.4) containing 0.5% Tween-80 to provide sink condition with agitation of 200 rpm. The samples were taken from the release medium at the predetermined time intervals and replaced with fresh buffer. The content of PTX was determined by the above described HPLC method.
Cell culture
4T1 cells were maintained in RPMI-1640 (without folic acid) supplemented with 10% (v/v) FBS and 1% penicillin–streptomycin at 37 °C and 5% CO2. Cells were subcultured regularly using trypsin/EDTA.
Preparation of FITC-labeled TS-CS FITC-labeled TS-CS-PEG-FA micelles
FITC-labeled micelles (FITC/TS-CS and FITC/TS-CS-PEG-FA) were prepared through the reaction between amino groups of chitosan and isothiocyanate of FITC. Briefly, 2.7 mg/ml of FITC solution in ethanol was prepared and 500 µl of this solution was dropped into the polymer solution. The molar ratio of the polymer to FITC was controlled at 1:2. The solution was stirred for 24 h at room temperature and in dark condition. The reaction product was dialyzed against deionized water using a dialysis membrane (MWCO 8 kDa) for 24 h to remove the unreacted FITC (Du et al., Citation2011). Then, the dialyzed products were lyophilized to obtain FITC/TS-CS or FITC/TS-CS-PEG-FA micelles.
Cellular uptake of FITC/TS-CS and FITC/TS-CS-PEG-FA micelles
In a 12-well culture plate (SPL Life Sciences Co. Ltd, Gyeonggi-Do, Korea), 4T1 cells were seeded at a density of 2 × 105 cells per well in 2 ml of growth medium and incubated for 24 h to attach. Cells were then incubated with FITC/TS-CS or FITC/TS-CS-PEG-FA micelles at concentration of 20 µg/ml of the polymer at different times. Cells were washed twice with PBS (pH: 7.4) and directly observed under a fluorescence microscope (CETI, 3100.5000 Triton ΙΙ, Oxon, UK).
To assay cellular uptake quantitatively, after washing the cells with PBS, 200 µl of trypsin PBS solution (2.5 mg/ml) was added to each well. After incubation for 5 min, the cells were harvested by addition of 1 ml PBS followed by sonication with probe-type ultrasonication for 2 min at room temperature to obtain the cell lysate. Finally, the cell lysate was centrifuged at 10 000 rpm for 10 min and the supernatant was supplied to fluorescence assay using spectrofluorometer (Jasco FP-750, Tokyo, Japan) (excitation: 490 nm, emission: 528 nm). The cellular uptake percentage of fluorescent micelles was calculated using following equation:
where, I is the fluorescence intensity at different time and I0 is the initial fluorescence intensity of the fluorescent micelles (Yuan et al., Citation2008).
In vitro cytotoxicity assay
The cytotoxicity of the free drug (PTX in dimethyl sulfoxide), Anzatax®, PTX/TS-CS micelles, PTX/TS-CS-PEG-FA micelles and blank micelles (TS-CS and TS-CS-PEG-FA micelles) were assessed against 4T1 cells by MTT assay. The cells were seeded at 1 × 104 cells/well in a 96-well culture plate (SPL Life Sciences). When the cell confluence reached to 75%, the cells were incubated with samples for 72 h at the equivalent PTX concentrations of 0.035–2.34 µM. Then, 20 µl MTT solution (5 mg/ml in 0.02 M PBS) was added to each well and the plate was incubated for another 4 h. After that, unreacted MTT and medium was removed and the formazan crystals in cells were dissolved in 150 µl dimethyl sulfoxide. The absorbance was measured at 570 nm using enzyme-linked immunosorbent assay reader (Stat Fax-2100; Awareness Technology Inc., Palm City, FL). Untreated cells were taken as the negative control with 100% viability and the blank culture medium was used as a blank control. Cell viability for each sample was calculated using following equation (Emami et al., Citation2012).
Folate competition assays
4T1 cells were exposed to a constant drug concentration of 1.17 µM, while being maintained in growth media supplemented with free folate molecules within the concentration rang of 0–500 mg/L. The viabilities of the cells were then quantified, as described above.
In vivo anti-tumor efficiency and histological studies
To evaluate the in vivo anti-tumor efficiency of the different formulations, 4T1 cells were injected subcutaneously (s.c.) at 2 × 106 cells in 0.2 ml of PBS into the right flank of female Balb/c mice (5–6 weeks old, 25 ± 2 g). Treatments were started when the tumor volumes reached to ∼50–80 mm3 and this day was designated Day 0. On Day 0, the mice were randomly divided into five groups (n = 6) and treated i.v. through the tail vein with an equivalent dose of 10 mg PTX per kg body weight of one of the following formulations: (i) saline as negative control, (ii) Anzatax®, (iii) PTX/TS-CS, (iv) PTX/TS-CS-PEG-FA and (v) blank TS-CS-PEG-FA micelles on Days 0, 3, 6 and 9. The tumor diameters were measured every other day with a vernier caliper in two dimensions. The tumor volume was calculated using the formula: V = (L × W2)/2, where, L is the largest and W is the smallest diameter. In addition, for safety evaluation of the different PTX formulations, the body weight of each mouse was determined every alternative day. After 25 days, the animals were sacrificed, and the tumor mass was harvested and weighed. The anti-tumor activities of each formulation were evaluated by tumor inhibition rate (TIR%) using the following formulation:
Finally the tumor tissue was fixed in 10% formalin and embedded into paraffin, sectioned at a thickness of 5 µm, stained with hematoxylin and eosin (H&E) and then observed under microscope.
Pharmacokinetics and tissue distribution of Anzatax® and PTX/TS-CS-PEG-FA micelles
Twenty one female Balb/c mice (5–6 weeks old, 25 ± 2 g) were inoculated s.c. into the right flank with mouse 4T1 breast cancer cells (2 × 106 cells). When the tumor volumes reached to ∼50–80 mm3, the mice were injected i.v. through the tail vein with Anzatax® or PTX/TS-CS-PEG-FA micelles at an equivalent dose of 10 mg/kg PTX versus the body weight. Three mice were sacrificed at 5 min, 30 min, 1 h, 2 h, 4 h, 8 h and 12 h after the drug administration. Samples of plasma, liver, kidneys, spleen, lungs, heart and tumor were harvested and stored at −70 °C until analyzed for PTX.
Analysis of PTX
Plasma and tissue concentrations of PTX were determined by reverse-phase HPLC with UV detection developed and validated in our laboratory. Briefly, 50 μl of sodium acetate buffer (pH 5), 25 μl of internal standard (25 µg/ml diazepam in methanol) and 6 ml of diethyl ether were added into 250 μl of plasma samples. Sample tubes were vortexed for 2 min and centrifuged at 5000 rpm for 10 min. The upper organic layer was transferred to the clean tubes and evaporated to dryness under nitrogen gas. The residue was reconstituted with 100 μl of mobile phase and 80 μl aliquot was injected into the HPLC column. Chromatographic separation was achieved using a reverse-phase C18-Bondapak column (3.9 × 250 mm) at 58 °C. The mobile phase consisted of acetate buffer (0.01 M)/acetonitrile (58/42, pH 5 ± 0.1) was eluted at a flow rate of 1.9 ml/min, and effluent was monitored at 227 nm using a UV detector. Quantitation was achieved by measurement of the peak area ratios of the drug to the internal standard. The pharmacokinetic parameters including the area under the plasma concentration–time curve from zero to infinity (AUC0–∞), the apparent volume of distribution (Vd), systemic plasma clearance (CL), distribution half-life (T1/2α), elimination half-life (t 1/2ß) and mean residence time (MRT) were calculated for each formulation by standard methods. To determine the amount of PTX in different organs, tissue samples were homogenized (Heidolph, SilentCrusher S, Schwabach, Germany) with 2-fold weight of normal saline. About 250 µl of each organ homogenate was transferred into a glass tube and extracted in the same manner as described for plasma specimen.
Statistical analysis
Data were expressed as means of three separate experiments and compared by independent sample t-test for two groups, and one-way analysis of variance for multiple groups. A p value <0.05 was considered statistically significant in all experiments.
Results and discussion
Preparation and characterization of PTX-loaded TS-CS and PTX-loaded TS-CS-PEG-FA micelles
The physicochemical characterization and drug-loading parameters of PTX-loaded micelles are summarized in . The DL and EE of PTX/TS-CS-PEG-FA were 28.2 ± 2.1 and 82.5 ± 7.3, respectively which were significantly higher than those of the PTX/TS-CS micelles (p < 0.05). The higher DL of the targeted micelles could be attributed to the shield effect of PEG that prevents the aggregation of the micelles in dialysis tube and consequently the drug loss during centrifugation. The size of PTX-loaded targeted micelles was larger than non-targeted micelles resulted from the stretch of PEG chains on the micelle surface toward water phase. The zeta potentials of TS-CS-PEG-FA were a little lower than that of CS-TS micelles which might be due to the barrier shield effect of PEG chain on the micelles and the reduced amino groups of chitosan after PEGylation. Hu et al. (Citation2008) prepared a series of PEGylated chitosan-stearic acid micelles for delivery of mitomycin. Similarly, the particle size of the micelles increased with increasing the modification ratio of PEG, whereas the zeta potentials decreased (Hu et al., Citation2008).
Table 1. The physicochemical characterization of PTX-loaded polymeric micelles (n = 3).
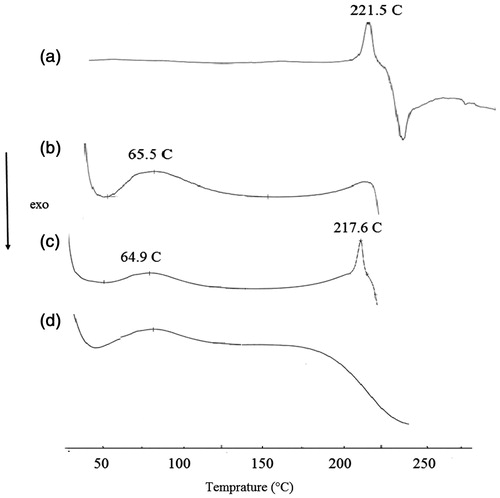
X-ray powder diffraction (XRD) and differential scanning calorimetry (DSC) were used to further characterize the drug-loaded micelles. As shown in , diffraction characteristic peaks of PTX were visible in the patterns obtained for physical mixture of PTX and TS-CS-PEG-FA derivative, while disappeared in the pattern of PTX-loaded micelles. It demonstrates that PTX is either in molecular state or in an amorphous state dispersed in the polymer after lyophilization. DSC thermograms () revealed PTX endothermic peak at 221 °C and one exothermic peak at 240 °C, which are attributed to the melting and decomposition temperature of PTX, respectively. Blank micelles presented the endothermic peak ∼60 °C, which could be attributed to water loss of chitosan derivative (Liu et al., Citation2011). When the two samples were physically mixed, all characteristic peaks of PTX were present only with minor shifts. The PTX-loaded micelles showed a curve similar to blank micelles suggesting that PTX existed in an amorphous state and there was no free drug on the surface of micelles. In vitro release studies indicated no significant differences between the release rate of PTX from TS-CS and TS-CS-PEG-FA micelles. Approximately 20–30% of PTX was released within 20 h at 37 °C, which was followed by a sustained manner and almost completed during 160 h. The sustaining release behavior from the micelles might be due to the relatively high lipophilicity of PTX and also strong hydrophobic interaction between PTX and the core of micelles.
Figure 1. XRD spectra of (a) PTX, (b) TS-CS-PEG-FA, (c) physical mixture of PTX and TS-CS-PEG-FA and (d) PTX-loaded TS-CS-PEG-FA.
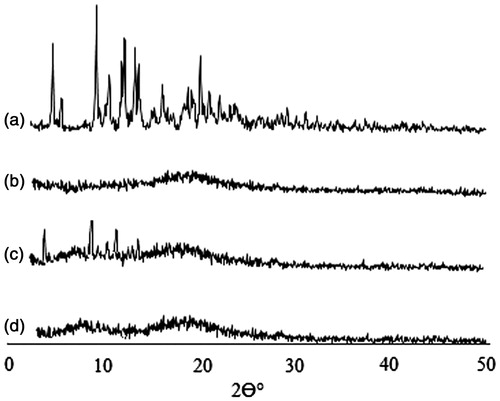
Figure 2. DSC spectra of (a) PTX, (b) TS-CS-PEG-FA, (c) physical mixture of PTX and TS-CS-PEG-FA and (d) PTX-loaded TS-CS-PEG-FA.
In vitro cellular uptake of targeted and non-targeted micelles
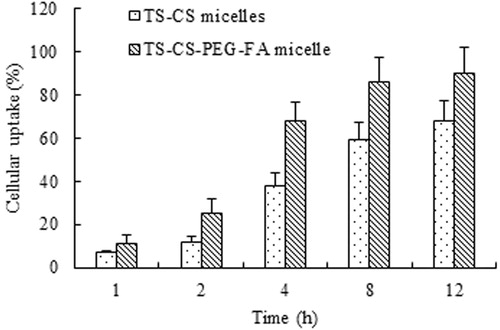
The cellular uptake of TS-CS and TS-CS-PEG-FA micelles were qualitatively and quantitatively evaluated by using FITC incorporated micelles and employing fluorescence microscopy and also the measurement of fluorescence intensity. shows the fluorescence image when the cells were incubated with FITC/TS-CS and FITC/TS-CS-PEG-FA micelles for 1, 2, 4, 8 and 12 h, respectively. There was no significant fluorescence inside 4T1 cells after incubation with FITC/TS-CS in 2 h. After 2 h, the obvious fluorescence images were observed and the fluorescence intensity was enhanced with the incubation time indicating that the cellular uptakes of TS-CS micelles are time-dependent process. Nanoparticles are generally taken up into cells via receptor mediated endocytosis, fluid phase endocytosis and phagocytosis (Yuan et al., Citation2008). To date, numerous efforts have been devoted to improve the specific internalization of drug carriers into cancer cells. An efficient strategy is to modify the nanoparticle surfaces with ligands that can recognize and adhere to the biomarkers on the surface of the corresponding cancer cells. Folic acid has been frequently attached to drugs, proteins, liposomes and polymeric micelles as targeting ligand for enhanced uptake by cancerous cells (Liu et al. Citation2007; Hong et al. Citation2008; Wang et al. Citation2012). The obvious fluorescence images were found after the cells were incubated with FITC/TS-CS-PEG-FA micelles in 2 h. From , the rapid cell internalization ability of TS-CS-EG-FA was clearly visible compared to TS-CS micelles. This result was consistent with previously published reports (Yuan et al., Citation2008; Hami, Citation2014). Furthermore to evaluate the cellular uptake of the micelles quantitatively, the fluorescence intensity was measured after the cells were incubated with FITC/CS-TS and FITC/TS-CS-PEG-FA for different time. shows the cellular uptake percentage of the fluorescent micelles against incubation time. The quantitative results also indicated that the introduction of PEG-FA into the micelles could significantly (p < 0.05) enhance the cellular uptake of the micelles.
Figure 3. Fluorescence image after the cells were incubated with fluorescent (a) TS-CS micelles and (b) TSCS.
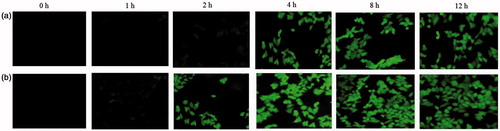
Figure 4. The cellular uptake percentage of FITC/CS-TS and FITC/TS-CS-PEG-FA micelles in 4T1 cells (the polymer concentrations were 20 μg/ml).
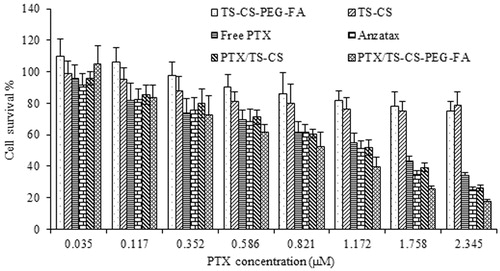
In vitro cytotoxicity
In vitro cell cytotoxicity of the free drug, Anzatax®, blank micelles, and PTX incorporated in targeted and non-targeted micelles was studied. The IC50 values of PTX, a concentration at which 50% cells are killed, were 1.63, 1.17, 1.27 and 0.93 µM for free PTX, Anzatax®, PTX/TS-CS and PTX/TS-CS-PEG-FA micelles, respectively. As shown in , Anzatax® and free PTX at the concentration ranges from 0.035 to 0.82 µM showed similar cytotoxicity against 4T1 cells after 72 h incubation. However, in concentrations > 1.172 µM, the cell cytotoxicity of Anzatax® was significantly greater than free PTX (p < 0.05) which might be attributed to the cytotoxicity of Cremophor® EL and low solubility of free PTX at higher concentrations. The viability of the cells treated with PTX/TS-CS-PEG-FA did not demonstrate a significant difference compared with Anzatax® at low drug concentrations (<0.35 µM, p > 0.05), but the inhibiting activity of PTX/TS-CS-PEG-FA increased with increase in PTX concentration from 0.35 to 2.3 µM, with a significant difference at 0.82 µM compared with the activity of Anzatax® (p < 0.05). The increased cytotoxicity of PTX/TS-CS-PEG-FA micelles could have attributed to the faster cellular uptake of the targeted micelles mediated by folate receptors overexpressed on the surface of 4T1 cells. In addition earlier published reports demonstrated higher intracellular accumulation of PEGylated particles due to the high solubility of PEG in cell membrane (Hu et al., Citation2008; Yuan et al., Citation2008). On the contrary, PTX/TS-CS displayed a slightly higher viability compared with Anzatax® in all of concentration ranges (p > 0.05) which might be due to the slow release of PTX from the micelles and lower cellular uptake of the micelles compared with TS-CS-PEG-FA because of the absence of targeting moiety on the surface of the micelles. In addition, blank TS-CS and TS-CS-PEG-FA without PTX showed negligible cytotoxicity.
Figure 5. In vitro cytotoxicity of various PTX formulations and blank micelles against 4T1 cell line after 72 h incubation. Data were plotted as the mean ± SD of three measurements.
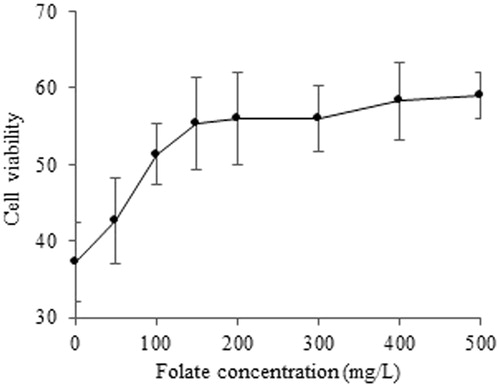
In order to further evaluate the role of folate in the cellular uptake of PTX/TS-CS-PEG-FA micelles, the cells were exposed to a constant drug concentration of 1.17 µM, while being maintained in growth media supplemented with increasing concentrations of free folate. It is found that cell viability increased with increasing folate concentration (). For instance, the cell viability was 37% in the absence of folate but increased to 58% in the presence of 500 mg/L free folate. These results suggest that free folate molecules hamper the cellular uptake of the micelles by competitive binding to the folate receptors on the cell surfaces.
Figure 6. Effect of free folic acid on viability of 4T1 cells incubated with PTX/TS-CS-PEG-FA micelles at PTX concentration of 1.17 μM.
In vivo anti-tumor effect and histological studies
In vivo anti-tumor activity of Anzatax®, PTX/TS-CS and PTX/TS-CS-PEG-FA micelles, and respective empty micelles was studied using 4T1 mouse breast cancer cells bearing Balb/c mice. Approximately 10 days after s.c implantation of cancer cells, the tumors were well-developed (mean volume: 50–80 mm3) and the treatments were started. Anzatax® and micellar solutions were injected through the tail vein on Day 0, 3, 6 and 9. The effectiveness and safety of different formulations were compared by measuring tumor volume and body weight of Balb/c mice after tumor implantation. As shown in , the tumor volume of the saline and the blank micelle groups were excessively enlarged (1400 mm3, after 25 days), while all PTX containing formulations decreased tumor growth rate. PTX/TS-CS-PEG-FA demonstrated the greatest inhibitory effect of tumor growth, showing the least tumor volume between other treatment groups (p < 0.05). The data presented in clarified that the weight of tumor treated with PTX/ TS-CS-PEG-FA was 2.83- and 3.71-fold lower than those of PTX/TS-CS and Anzatax® at the end of 25 days, respectively. In Anzatax®-receiving group, initial treatment decreased tumor growth rate but the tumor volume became the largest in the later time among other treated groups. In addition, the change in body weight of animals was monitored during 25 days as an index for systemic toxicity of formulations. In negative control group receiving saline, and in the group injected by blank micelles, the body weight was significantly increased up to ∼20% during 25 days after the treatment. Meanwhile, in PTX-loaded micelles there was a slight decrease in body weight at initial stage of the treatment which was gradually recovered after cessation of the drug treatment. The significant body loss (15%) observed in Anzatax® group could reflect the toxicity of the formulation. Moreover, in some cases, immediate ataxia and decreased activity were observed after i.v. administration of Anzatax® and two mice died during treatment, whereas the PTX-loaded micelles were well-tolerated. As shown in , the significant reduction of tumor mass was observed after treatment with PTX/TS-CS-PEG-FA compared to Anzatax®. The TRF value of PTX/TS-CS-PEG-FA was 85.6%, which was the highest among other groups followed by 55.2% for PTX/TS-CS, and 45.6% for Anzatax® (). In comparison with Anzatax®, the slight enhanced anti-tumor activity of PTX/TS-CS could be explained by the increased local accumulation of the micelles to the tumor sites due to enhanced permeability and retention (EPR) effect. PTX/TS-CS-PEG-FA micelles were the most effective formulation in terms of tumor growth suppression which was attributed to the rapid uptake of micelles by cancer cells. To evaluate the necrosis level in tumors from different groups treated by Anzatax®, PTX/TS-CS, PTX/TS-CS-PEG-FA and saline, the tumors were excised from the mice 25 days after the treatments, and tumor slices stained by H&E were analyzed via optical microscopy. Qualitatively, necrosis area in the PTX/TS-CS-PEG-FA-treated tumor was explicitly the largest among all of the tested groups. In contrast, the Anzatax-treated tumor displayed a much lower necrotic level with a large amount of living tumor cells existing (). These findings reinforce the conclusion that PTX/TS-CS-PEG-FA exhibited a greater therapeutic efficacy than either Anzatax® or PTX/TS-CS.
Figure 7. Tumor volume change in mice with PTX/TS-CS, PTX/TS-CS-PEG-FA, Anzatax®, blank micelles or saline (n = 6) (a) images of whole mice were taken at 25 days after tumor inoculation (b).
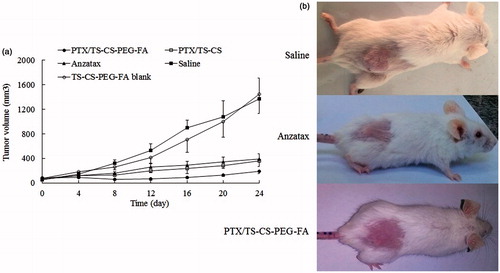
Figure 8. H&E-stained tumor slices from mice on the 25nd day after treatment with (a) saline, (b) Anzatax®, (c) PTX/TS-CS and (d) PTX/TS-CS-PEG-FA. Images were obtained under Olympus IX71 optical microscope.
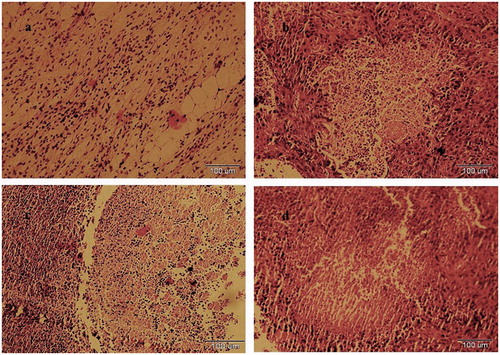
Table 2. In vivo anti-tumor effect of PTX formulations against 4T1 tumor in Balb/c mice (n = 6).
Pharmacokinetic and tissue distribution of Anzatax® and PTX/TS-CS-PEG-FA micelles
The mean plasma concentration time profiles of PTX in blood after i.v. administration of PTX/TS-CS-PEG-FA and Anzatax® are shown in . The related pharmacokinetic parameters are listed in and analyzed for statistical significance by unpaired Student's t-test. The decline in plasma concentrations following i.v. administration of Anzatax® and the micellar formulation described by two-compartment open model. The plasma concentrations of PTX/TS-CS-PEG-FA micelles were slightly lower than Anzatax® injection at all sampling time. A rapid decline in distribution phase was observed after dosing with two preparations. In contrast, the elimination half-life of PTX/TS-CS-PEG-FA (4.641 ± 0.384 h) was significantly (p < 0.01) longer than that of Anzatax® (2.486 ± 0.23 h). Moreover, the MRT and Vd (apparent volume of distribution) of PTX/TS-CS-PEG-FA micelles were significantly greater than those of Anzatax® injection. No significant difference was observed between plasma AUC and total body clearance (CL) of targeted micelles and Anzatax® (p > 0.05). The pharmacokinetic results demonstrated the long circulation properties and high stability of PTX/TS-CS-PEG-FA micelles to achieve efficient accumulation of the micelles in tumor tissue and high intracellular concentration of the drug. In our previous work, it was demonstrated that PTX/TS-CS-PEG-FA micelles show higher plasma AUC, much longer systemic circulation time and much slower plasma elimination rate than those of PTX/TS-CS micelles after i.v. injection of these two formulations into healthy Balb/c mice. This long blood circulation could be attributed to the stealth behavior of PTX/TS-CS-PEG-FA micelles induced by the hydrophilic shell created by PEG. In aqueous environment, PEG is highly hydrated and forms a dense, brush-like shell that stretches away from the core. This characteristic prevents the plasma protein adsorption (opsonization) to the surface of micelles and reduces their uptake by MPS (Hu et al., Citation2008). Here, the pharmacokinetic behavior and anti-tumor efficiency in cancerous mice illustrated the potential utility of PTX/TS-CS-PEG-FA micelles as long circulating carriers for targeted delivery of hydrophobic anticancer drugs, which were consistent with the report by Wang et al. (Citation2012) and Xin et al. ( Citation2010). In order to quantify and identify the critical factors influencing bio-distribution of various drug carriers, the tissue distribution profiles of PTX/TS-CS-PEG-FA and Anzatax® were compared in tumor bearing mice. The concentration of PTX in different organs after i.v. administration of 10 mg equivalent PTX per kg body weight for the micelle and Anzatax® are presented in . The maximum concentration in different organs and the time for maximum concentration values were obtained by visual inspection of the data. As anticipated, the drug concentration in tumor following injection of PTX/TS-CS-PEG-FA micelles was significantly higher than that of Anzatax® injection over a 12 h period after dosing. For both formulations, the distribution half-life was short (0.23–0.34 h) and the drug concentrations declined quickly in this phase. The PTX concentration in spleen and liver following injection of the micelles was lower than PTX concentration following injection of Anzatax®. While the difference in lung between the micelles and PTX injection was not significant. This observation is consistent with other studies on PEGylated drug delivery systems (Qu et al., Citation2009; Xin et al., Citation2010). summarizes the AUC and MRT values of PTX in various tissues following i.v. administration of Anzatax® and PTX/TS-CS-PEG-FA micelles. In contrast to Anzatax®, the micelle formulation showed a significant higher AUC in tumor, indicating that PTX/TS-CS-PEG-FA micelles are more preferable for targeting to tumor tissue. This phenomenon might be attributed to the fact that tumor tissue is characterized by hypervascular permeability and impaired lymphatic drainage to facilitate significant accumulation of nanoparticles in the tumor tissue. In addition, folic acid on the surface of micelles mediates active targeting and increases the intracellular concentration of PTX. Furthermore, the coupling of PEG to the composition of the micellar systems could greatly decrease plasma protein adsorption and elimination of the micelles by MPS which results in prolonged residence time in blood and further accumulation in tumor tissue enhancing endocytosis via folate receptors (Hong et al., Citation2008). In order to evaluate the targeting efficiency of PTX/TS-CS-PEG-FA micelles used in this study, a parameter targeting value [Re, defined as Re = AUC (PTX/TS-CS-PEG-FA)/AUC (Anzatax®)] was introduced (Liu et al., Citation2011) which demonstrates the relative targeting efficiency of the two delivery systems toward one tissue. As shown in , PTX/TS-CS-PEG-FA micelle showed a tumor targeting value of 1.81, indicating that the micellar formulation was more efficient in delivering PTX to the tumor tissue compared with Anzatax®. A higher amount of PTX accumulated in the kidneys (Re = 1.34) suggesting the increased kidney elimination of the micelles. Meanwhile, lower AUC values for the micelles in the heart (Re = 0.69) might decrease the toxic effect of PTX in this organ.
Figure 9. Mean concentration–time profile of PTX in Balb/c mice plasma after i.v. administration of Anzatax® and PTX/TS-CS-PEG-FA. Each point represents the mean ± SD (n = 3).
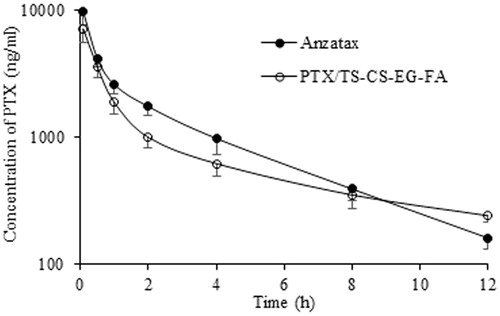
Figure 10. The PTX concentration in mice receiving Anzatax® or PTX/TS-CS-PEG-FA in tissues at different time points (0.083, 0.5, 1, 2, 4, 8 and 12 h) (mean ± SD, n = 5).
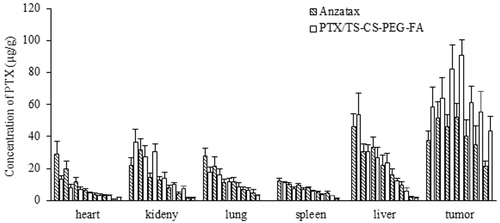
Table 3. Pharmacokinetic parameters of PTX in the two formulations after i.v. administration.
Table 4. AUC and MRT of PTX in tissue after i.v. administration of 10 mg/kg dose of Anzatax® and PTX/TS-CS-PEG-FA micelles.
Conclusion
In this study, a micelle delivery system based on TS-chitosan-PEG-folic acid was compared with Anzatax® as delivery system of PTX. In vitro cell viability studies using 4T1cell line revealed that PTX-loaded micelles could produce higher cytotoxicity than PTX injection due to the folate receptor-mediated endocytosis. As demonstrated by pharmacokinetic assay, tissue distribution and anti-tumor study in Balb/c tumor-bearing mice, the PTX/TS-CS-PEG-FA micelles displayed prolonged residence time in circulation, and increased accumulation in tumor, thus, enhanced anti-tumor effects and reduced toxicity. Taken together, our in vitro and in vivo evaluation showed that TS-CS-PEG-FA micelles could be a potential drug delivery system of PTX for the effective treatment of the tumor that overcome the disadvantages of conventional formulation containing Cremophor® EL and provide a desirable therapeutic efficacy.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Bernabeua E, Helguera G, Legaspia M, Gonzalez L. (2014). Paclitaxel-loaded PCL–TPGS nanoparticles: in vitro and in vivo performance compared with Abraxane®. Colloids Surf B Biointerfaces 113:43–5
- Du Y-Z, Wang L, Yuan H, Hu F-Q. (2011). Linoleic acid-grafted chitosan oligosaccharide micelles for intracellular drug delivery and reverse drug resistance of tumor cells. Int J Biol Macromol 48:215–22
- Emami J, Rezazadeh M, Rostami M, et al. (2014). Co-delivery of paclitaxel and α-tocopherol succinate by novel chitosan-based polymeric micelles for improving micellar stability and efficacious combination therapy. Drug Dev Ind Pharm 1–11. Early online, DOI: 10.3109/03639045.2014.935390
- Emami J, Rezazadeh M, Varshosaz J, et al. (2012). Formulation of LDL targeted nanostructured lipid carriers loaded with paclitaxel: a detailed study of preparation, freeze drying condition, and in vitro cytotoxicity. J Nanomaterials. doi:10.1155/2012/358782
- Fetterly GJ, Straubinger RM. (2003). Pharmacokinetics of paclitaxel-containing liposomes in rats. AAPS Pharm Sci 5:123–9
- Hami Z, Aminib M, Ghazi-Khansaric M, et al. (2014). Synthesis and in vitro evaluation of a pH-sensitive PLA–PEG–folatebased polymeric micelle for controlled delivery of docetaxel. Colloids Surf B Biointerfaces 116:309–17
- Hawkins MJ, Soon-Shiong P, Desai N. Protein nanoparticles as drug carriers in clinical medicine. Adv Drug Deliv Rev 2008:60:876–85
- He L, Wang G, Zhang Q. (2003). An alternative paclitaxel microemulsion formulatiom: hypersensitivity evaluation and pharmacokinetic profiles. Int J Pharm 250:45–50
- Hong G, Yuan R, Liang B, et al. (2008). Folate-functionalized polymeric micelle as hepatic carcinoma-targeted, MRI-ultrasensitive delivery system of antitumor drugs. Biomed Microdevices 5:693–700
- Hu FQ, Meng P, Dai YQ, et al. (2008). PEGylated chitosan-based polymer micelle as an intracellular delivery carrier for anti-tumor targeting therapy. Eur J Pharm Biopharm 70:749–57
- Kim SC, Kim DW, Shim YH, et al. (2001). In vivo evaluation of polymeric micellar paclitaxel formulation: toxicity and efficacy. J Control Release 72:191–202
- Liu J, Li H, Chen D, et al. (2011). In vivo evaluation of novel chitosan graft polymeric micelles for delivery of paclitaxel. Drug Deliv 18:181–9
- Liu J, Zeng F, Allen C. (2007). In vivo fate of unimers and micelles of a poly(ethylene glycol)-block-poly (caprolactone) copolymer in mice following intravenous administration. Eur J Pharm Biopharm 65:309–19
- Nam J-P, Park S-C, Kim T-H, Jang J-Y, Choi Ch, Jang M-K, Nah J-W. (2013). Encapsulation of paclitaxelinto lauric acid-o-carboxymethyl chitosan-transferin micelles for hydrophobic drug delivery and site specific targeted delivery. Int. J. Pharmaceutic 457:124–35
- Nornoo AO, Osborne DW, Chow DS. (2008). Cremophor-free intervenous microemulsions for paclitaxel: formulation, cytotoxicity and hemolysis. Int J Pharm 349:108–16
- Qu G, Yao Z, Zhang C, et al. (2009). PEG conjugated N-octyl-O-sulfate chitosan micelles for delivery of paclitaxel: in vitro characterization and in vivo evaluation. Eur J Pharmaceutic Sci 37:98–105
- Qu D, Lin H, Zhang N, et al. (2013). In vitro evaluation on novel modified chitosan for targeted antitumor drug delivery. Carbohydr Polym 92:545–54
- Soenpenberg O, Sparreboon A, Dejongy MJA, et al. (2004). Real-time pharmacokinetics guiding clinical decisions:phase Ι study of a weekly scheduleof liposome encapsulated paclitaxel in patients with solid tumours. Eur J Cancer 40:681–8
- Tao Y, Xu J, Chen M, et al. (2012). Core cross-linked hyaluronan-styrylpyridinium micelles as a novel carrier for paclitaxel. Carbohyd Polym 88:118–24
- Wang F, Chen Y, Zhang D, et al. (2012). Folate-mediated targeted and intracellular delivery of paclitaxel using a novel deoxycholic acid-O-carboxymethylated chitosan-folic acid micelles. Int J Nanomed 7:325–37
- Wang F, Shen Y, Xu X, et al. (2013). Selective tissue distribution and long circulation endowed by paclitaxel loaded PEGylated poly(ε-caprolactone-co-l-lactide) micelles leading to improved anti-tumor effects and low systematic toxicity. Int J Pharm 456:101–12
- Xin HL, Chen LC, Gu JJ, et al. (2010). Enhanced anti-glioblastoma efficacy by PTX-loaded PEGylated poly(ε-caprolactone) nanoparticles: in vitro and in vivo evaluation. Int J Pharm 402:238–7
- Yang C, Attia A, Tan PK, et al. (2012). The role of non-covalent interactions in anticancer drug loading and kinetic stability of polymeric micelles. Biomaterials 33:2971–9
- Yuan H, Miao J, Du YZ, et al. (2008). Cellular uptake of solid lipid nanoparticles and cytotoxicity of encapsulated paclitaxel in A549 cancer cells. Int J Pharm 348:137–45
- Zhang X, Jackson JK, Burt HM. (1996). Development of amphiphilic diblock copolymers as micellar carriers of taxol. Int J Pharm 132:195–206
- Zhang C, Qu G, Sun Y, et al. (2008). Pharmacokinetics, biodistribution, efficacy and safety of N-octyl-o-sulfate chitosan micelles loaded with paclitaxel. Biomaterials 29:1233–41
- Zhang W, Shi Y, Chen Y, et al. (2011). Multifunctional Pluronic P123/F127 mixed polymeric micelles loaded with paclitaxel for the treatment of multidrug resistant tumors. Biomaterials 32:2894–906
- Zhu ZS, Li Y, Li XL, et al. (2010). Paclitaxel-loaded poly(N-vinylpyrrolidone)-b-poly(ε-caprolactone) nanoparticles: preparation and antitumor activity in vivo. J Control Release 142:438–46