
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Meloxicam (Mel) is a non-steroidal potent anti-inflammatory drug with effective analgesic effect for various situations; e.g. postoperative pain. The early systemic exposure to Mel and hence the rapid onset of pharmacological action is limited by its poor water solubility; a situation which may be more pronounced during acute pain episode because of reduced gastric motility that affects disintegration and dissolution of solid dosage forms. To overcome delayed absorption of Mel, improvement in the dissolution behavior of Mel is essential. Firstly, Mel spherical crystalline agglomerates (SCA) were prepared. Secondly, selected Mel SCA were integrated into intraoral fast disintegrating (OF) and edible (EF) films, they possess larger surface area that leads to rapid disintegration and release of the drug into the oral cavity within seconds and hence a rapid onset of action could be achieved. Stability study of formulations resulting in faster and higher extent of dissolution and suitable mechanical properties (G3 and G12) revealed their physical and chemical stability after three months of storage under different conditions. Both G3 and G12 successfully offered rapid absorption rate and accordingly an earlier systemic exposure to Mel compared to Mobic tablets as revealed by significantly earlier T max and higher AUC0–0.5h and AUC0–4h. T max following G3 fast disintegrating film administration was comparable to that reported following Mel parenteral administration but avoiding patient inconvenience. Both films may be suitable alternative to conventional oral and intramuscular Mel especially when earlier onset of action is required (in acute conditions).
Introduction
Meloxicam (Mel) is a cyclo-oxygenase-2 preferential non steroidal anti-inflammatory drug. It has been effectively utilized for its anti-inflammatory and analgesic effects (Davies & Skjodt, Citation1999; Hanft et al., Citation2001; Ahmed et al., Citation2005). It is also reported as an emerging promising drug for the treatment of cancer (Goldman et al., Citation1998; Naruse et al., Citation2006). Mel is a class II drug; i.e. exhibits low water solubility and high permeability. Early systemic exposure to Mel and rapid achievement of pharmacological response after oral administration is limited by poor water solubility and low dissolution rate of Mel. For situations requiring rapid onset of action (such as acute pain, acute flares of osteoarthritis, sciatica and exacerbations of rheumatism) parenteral forms are commercially available (Davies & Skjodt, Citation1999). However, parenteral route is not convenient for chronic use. Therefore, it would be appropriate to develop an oral formulation of meloxicam with faster onset of action. Independence of disintegration and dissolution of the oral developed formulation on the gastric motility and secretions that may be reduced during acute pain episode (Kulmatycki & Jamali, Citation2007) is of special importance as gastric motility plays a major role in the disintegration, dissolution and hence absorption rate of solid dosage forms.
Small oral daily dose of Mel (7.5–15 mg) makes the administration of Mel via intraoral route suitable.
Intraoral fast disintegrating and edible films (OF and EF, respectively) are non-bulky oral dosage forms that offer several advantages over conventional oral dosage forms including: ease of administration since they are easy to swallow with no need for water thus improving patient compliance particularly elderly and pediatric patients. Availability of larger surface area that leads to rapid disintegration and release of the drug into the oral cavity within seconds and hence a rapid onset of action could be achieved (Dixit & Puthli, Citation2009; Hoffmann et al., Citation2011). Owing to pregastric or oramucosal absorption, drugs can directly enter systemic circulation avoiding first-pass metabolism thus improving bioavailability allowing use of reduced doses and fewer side effects (Liang & Chen, Citation2001; Dixit & Puthli, Citation2009; Hoffmann et al., Citation2011).
To maximize the potential of Mel oramucosal absorption and accordingly faster onset of action, we attempted to improve dissolution rate of Mel before integration into film formulations via formation of Mel spherical agglomerated crystals. Spherical agglomeration is an efficient particle engineering technique that has been successfully employed to improve micrometric properties, solubility and dissolution rate of several drugs (Di Martino et al., Citation1999; Maghsoodi et al., Citation2008; Usha et al., Citation2008) during which crystallization and agglomeration can be carried out in one step. Spherical agglomeration is expected to improve dissolution rate of Mel avoiding somewhat sophisticated techniques (e.g. nanosizing) and techniques that may add to the bulkiness of the dosage form (e.g. solid dispersion) which are not suitable especially when designing an intraoral dosage form.
The aim of the current study was to improve the dissolution properties of the poorly water-soluble drug, Mel, by spherical agglomeration followed by incorporation in different film formulations seeking for reducing its delayed absorption and accordingly an earlier pharmacological response particularly required for acute pain episodes and exacerbated rheumatic situations. Since there appears to be a link between plasma concentration and pharmacological response (Laska et al., 1986), comparison of selected promising film formulation(s) pharmacokinetic parameters to those of marketed solid dosage form to investigate the possibility of increasing pharmacological effects was performed.
Materials and methods
Materials
Meloxicam and aspartame (kind gifts from Amoun Pharmaceutical Co, Cairo, Egypt). Piroxicam (kindly supplied by South Egypt Drug Industries Co, Giza, Egypt). Hydroxyethyl cellulose (HEC, gift from El-Nile Pharmaceutical Co, Cairo, Egypt). Hydroxypropyl methylcellulose (HPMC E5 and HPMC E15, Dow Chemicals Co, Michigan, USA). Hydroxypropyl-β-cyclodextrin (HPβCD, Sigma-Aldrich Chemie GmbH, Steinheim, Germany). Polyvinyl alcohol (PVA, Fluka Chemie GmbH, Buchs, Switzerland). Glycerol, propylene glycol (PG), hydrochloric acid, citric acid, chloroform and sodium hydroxide (El-Nasr Pharmaceutical Chemicals Co, Cairo, Egypt). Acetonitrile, methanol (HPLC grade, Merk Co, Whitehouse Station, NJ, USA). Mobic® tablet (15 mg meloxicam, Boehringer Ingelheim International GmbH, Ingelheim am Rhein, Germany). All other reagents were of analytical grade.
Methods
Preparation of Mel spherical crystalline agglomerates
Mel spherical crystalline agglomerates (SCA) were prepared using neutralization technique. Mel powder was dissolved in 0.1 N sodium hydroxide (good solvent) with the aid of heat until a clear solution was obtained. The drug solution was poured into 0.1 N hydrochloric acid (poor solvent) containing HPβCD at a concentration of 0.0125% w/v under continuous stirring at 600 rpm (Dixit et al., Citation2011) with a propeller at room temperature. After the crystallization of Mel, chloroform (bridging solvent) was added, in two portions and the system was agitated for 30 min then the prepared agglomerates were collected by filtration through Whatman® filter paper. The spherical crystals were washed with distilled water and left for drying at 45 °C for 12 h and then the spherical agglomerates were kept in a desiccator until further experiments.
Solid state characterization of spherical crystalline agglomerates
Differential scanning calorimetric studies
Samples of Mel or its SCA (5 mg) were heated in hermetically sealed aluminum pans at a heating rate of 10 °C/min over temperature range of 20–400 °C (Shimadzu-DSC 50, Kyoto, Japan).
Powder X-ray diffraction
Philips X-Ray diffractometer (model PW/1710 with monochromator, Pila, Poland) was used at 40 kV, 35 mA and a scanning rate of 0.02°/s at room temperature over a range of 2–60 2θ, using Cu-Kα1 radiation of wavelength 1.542 Å.
Preparation of different Mel films
Preparation of Mel intraoral fast disintegrating films
The film forming material was dissolved in cold (HPMC, HEC) or hot distilled water (PVA). The active ingredient (raw Mel or SCA) was added in the required quantity. Then remaining ingredients were added in the proportions given in . The formed suspension was stirred for 0.5 h to ensure homogenous dispersion of the ingredients and then degassed twice to remove air bubbles after that it was casted in a Teflon plate and then kept for drying at 75 °C for the first 30 min and then the temperature was decreased to 45 °C for next 24 h (El-Setouhy & Abd El Malak, Citation2010; Prabhakara et al., Citation2011).
Table 1. Composition of different Mel intraoral fast disintegrating films.
Preparation of Mel edible films
Water solution (4 g) containing 0.5 g of glycerin was heated to 80 °C. The polymer (10% w/w HPMC E5 or E15) was then added and stirred until well dispersed and the temperature was decreased to room temperature to dissolve the polymer completely. The components were mixed until a clear solution was obtained. The polymeric solutions were left until the removal of air bubbles. The polymer solution (5 g) was then added to 5 g of either Mel powder or SCA dispersed in water, mixed for 0.5 h and then degassed to remove air bubbles. The final suspension was then casted on a glass Petri dish. The film was then dried overnight in an oven at 42 °C (Lucas et al., Citation2012). The composition of edible films is shown in .
Table 2. Composition of different Mel edible films.
The resultant films were cut into the dimension of 2 × 2 cm2 in size.
Evaluation of the prepared Mel films
The surface pH of fast disintegrating films was determined, the oral film was made slightly wet and the pH was measured by bringing electrode in contact with surface of the oral film (Hanna instrumental 9417, Póvoa de Varzim, Portugal.
Film thickness
The thickness of the film was measured using Vernier caliper micrometer (Shanghai, China). The thickness was measured at five different locations (four corners and one at center) and the average was taken.
Determination of drug content
The content uniformity of dosage units of the oral film preparation was determined and the results were expressed as mean of five. Films of 2 × 2 cm2 were cut at five different places of the casted films. Each film was dissolved in phosphate buffer pH 6.8 in 100 ml volumetric flask. An aliquot of 1 ml was taken and diluted with the same buffer up to 10 ml. The absorbance of the solution was measured spectrophotometrically at 362 nm using UV using phosphate buffer as blank.
In vitro disintegration time of intraoral films
Disintegration time is taken as the time when a film starts breaking when brought in contact with water or saliva. United State Pharmacopoeia (USP) disintegration apparatus was used to study the disintegration time (Dahiya et al., Citation2009). The test was done in triplicate.
Mechanical characteristics of the prepared Mel films
This is done by measuring the tensile strength, percent elongation and modulus of elasticity.
Chacillon force measurement apparatus (Greenspora, NC 27409, Mumbai, India) was used. The apparatus has two clamps, the upper one is fixed and the lower is movable. The film sample (1 × 3 cm) was clamped between the two clamps. The force at tearing and elongation were determined. The percent elongation (%E) was calculated using the following equation:
(1)
(1)
where, Lo is the original length and Ls is the length of the film after elongation.
The modulus of elasticity of films was calculated using the following equation:
(2)
(2)
where, F = breaking load (N), A = cross-sectional area of the film, and EM is the modulus of elasticity.
In vitro dissolution study
In vitro dissolution of Mel films was studied in USP dissolution test apparatus I (Pharma Test, PTW, Hainburg, Germany). Simulated saliva fluid (SSF pH 6.8) was used as dissolution medium for fast disintegrating films, while dissolution testing of edible films was done in both simulated gastric fluid (SGF pH 1.2) and SSF. The volume of dissolution medium was 900 ml stirred at 50 rpm (Patel et al., Citation2009) and the temperature was maintained at 37 ± 0.5 °C throughout the experiment. Samples of 5 ml were withdrawn by means of syringe fitted with pre-filter at predetermined time intervals and analyzed by measuring the absorbance spectrophotometrically at λmax 362 nm using SSF as blank or at λmax 345 nm using SGF as blank. The volume withdrawn at each time interval was replaced with fresh quantity of dissolution medium. Cumulative percent of meloxicam dissoluted was calculated and plotted against time. The study was performed in triplicate.
The dissolution testing of Mel SCA was carried out adopting USP dissolution method type II rotated at 100 rpm (Kürti et al., Citation2011) in both SGF pH 1.2 and SSF pH 6.8.
Accelerated stability studies
The best formula obtained from fast disintegrating films (G3) and that of edible films (G12) were chosen for the study. Firstly, the film was wrapped in a butter paper then above it an aluminum foil was wrapped tightly. The storage conditions were 30 °C/60% relative humidity (RH) and 40 °C/75% RH (Shimoda et al., Citation2009; Tomar et al., Citation2012). The samples were evaluated for changes in appearance, thickness, drug content, disintegration time and surface pH after 30, 60 and finally 90 d.
Pharmacokinetic study in healthy human volunteers
Study design
The pharmacokinetics of the developed film formulations (G3 and G12) in comparison with marketed Mobic® tablets was carried out in four healthy human volunteers (age: 25–40 years; weight: 60 ± 5 kg). The study protocol was approved by research ethics committee, Faculty of Pharmacy, Cairo University (Approval No. PI 336). The study was performed on three phases: phase I; all volunteers received Mobic® 15 mg tablets, phase II; all volunteers received G3 fast dissolving film, phase III; all volunteers received G12 edible film. Both film formulae contain an amount of Mel SCA equivalent to 15 mg Mel. Blood samples were obtained at 0, 0.5, 1, 1.5, 2, 3, 4, 6, 6 and 24 h post-dose. All samples were immediately centrifuged at 3000 rpm for 10 min and the plasma was frozen and stored at −20 °C until analysis.
Analytical procedure for determination of Mel in plasma
The HPLC method of Abd El-bary et al. (Citation2001) for the determination of Mel in plasma was adopted with slight modification. The HPLC system (Shimadzu Corp, Kyoto, Japan) consists of isocratic pump (LC-10 AS), UV/Vis Detector (SPD 10 A) and C18 reversed-phase column (Waters Corporation, Milford, Massachusetts, USA, 3.9 × 3 M mm). The mobile phase consisted of methanol: phosphate buffer (60:40) v/v, pH 3.2 adjusted with phosphoric acid. The flow rate was 2 ml/min; the detection wavelength was 362 nm. All assays were performed at ambient conditions.
Standard solution and sample preparation
Mel stock standard solution (100 µg/ml) was prepared by dissolving 10 mg of drug in 100 ml methanol. Standard calibration curve for in-vivo study was prepared by making serial dilutions of stock solution and spiking them into 1 ml drug-free plasma to give concentrations of 0.02–2 µg/ml. Piroxicam was adopted as an internal standard (IS) at a fixed concentration of 10 µg/ml.
One milliliter acetonitrile used as extracting solvent was added and the mixture was vortexed for 20 s and centrifuged for 10 min at 3000 rpm. The upper layer was transferred to another tube, filtered through 0.45 µm Millipore filter, and 20 µl were injected to the column for analysis. A standard plasma calibration curve was constructed by plotting the peak area ratio of Mel to that of IS against Mel concentrations in plasma.
Pharmacokinetic parameters and statistical calculations
Peak concentrations (Cmax) and peak times (Tmax) were derived directly from the experimental points. The area under the plasma concentration–time curve (AUC) was calculated using the linear trapezoidal method. The terminal elimination rate constant (Kel) was estimated from the slope of the terminal phase of the plasma concentration curve using the log-transformed concentrations, and then the terminal elimination half-life (t1/2) was calculated as 0.693/Kel.
The pharmacokinetic parameters of the two tested formulae were compared by the one-way analysis of variance using the software SPSS (SPSS Inc., Chicago, IL). The significance of the difference was determined at α = 0.05.
Results
Particle engineering of Mel into crystalline spherical agglomerates positively affected dissolution of the Mel compared to raw drug both in pH 1.2 and 6.8.
In pH 1.2 after 60 min, Mel SCA (% dissolved = 15.87% ± 0.76) showed 13.03 times and 4.94 times improvement of dissolution compared to raw Mel (1.218% ± 0.45) and physical mixture (3.21% ± 0.396), respectively.
In pH 6.8, Mel SCA (100% ± 0.83) showed 38.46 times and 25.64 times improvement of dissolution compared to raw Mel (2.6% ± 1.14) and physical mixture (3.9% ± 0.59), respectively, after 5 min. Composition of physical mixture is the same as that of SCA formula.
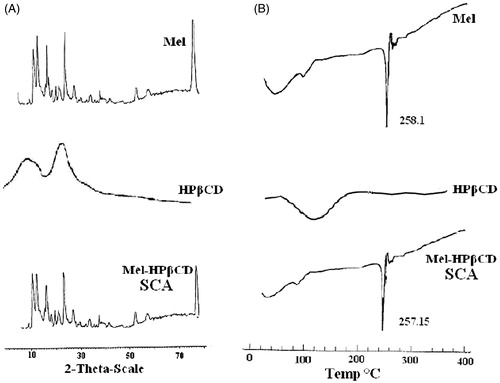
Powder X-ray diffraction (PXRD; ) studies revealed the retention of characteristic diffraction peaks of Mel in SCA but the intensity of the peaks has been reduced suggesting the decreased crystallinity and/or decreased primary particle size (Usha et al., Citation2008). DSC examination of Mel SCA showed a non-appreciable change in peak melting endotherm of Mel in SCA compared to raw Mel () indicating that no interaction has occurred between Mel and the polymer during crystallization of particles. However, an enthalpy change from ΔH = −338.1 J/g to −86.75 J/g, for raw Mel and Mel SCA was observed, respectively.
Figure 1. X-Ray diffractograms (A) and DSC patterns (B) of Mel spherical crystalline agglomerate (SCA).
Physicochemical evaluation of prepared Mel films
All films except G1 had smooth surface and they were elegant enough to see. G1 formula did not make a film may be due to the low concentration of the low viscosity HPMC E5 polymer. The results of the physicochemical evaluation of the different prepared fast disintegrating and edible films are demonstrated in .
Table 3. Physicochemical and mechanical characteristics of the prepared Mel films.
The surface pH was found to be in the range of 6.25–6.89, which is close to the salivary pH, indicating that the films have less potential to irritate the mucosal lining of the oral cavity and hence, more acceptable by the patients (Bhanja et al., Citation2010).
The thickness varied in the range of 0.12 ± 0.019 to 0.275 ± 0.022 mm. It is essential to ascertain uniformity in the thickness of film as this is directly related to accuracy of dose distribution in the film.
The drug content values were found to be between 96.76% ± 0.55 and 99.04% ± 1.53 for the different formulations suggesting that drug was uniformly dispersed throughout all films. The drug content was within the required compendial specifications, i.e. within 92–107.5% (British Pharmacopoeia III, Citation2003).
Mechanical characteristics of the prepared Mel films
The mechanical characteristics of the prepared Mel films are shown in . Films should possess moderate tensile strength, high % elongation (%E), and low elastic modulus (EM) (Mashru et al., Citation2005). The results revealed that all the films showed moderate tensile strength values except those prepared by PVA alone and as the concentration of PVA increased, the tensile strength increased and the film became more rigid. Hydroxypropyl methylcellulose-based films showed the best mechanical properties in comparison with those containing HEC or PVA.
Among the tested OF, G3 showed significantly highest %E and lowest EM compared with other formulae. While amongst the tested EF, G12 showed the highest %E, lowest EM compared to other edible formulae.
In vitro dissolution study
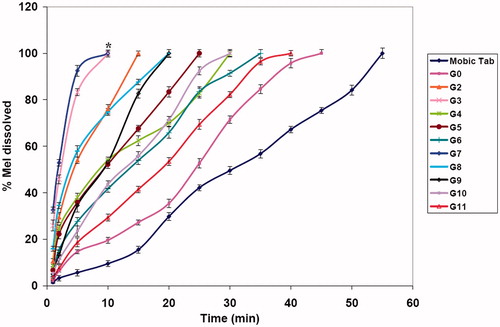
Dissolution data for Mobic® 15 mg tablet, fast disintegrating and edible films are graphically illustrated in and .
Figure 2. Dissolution profiles of Mel edible films in SGF pH 1.2 (A) and SSF pH 6.8 (B).Statistically significant differences are indicated as *p < 0.05.
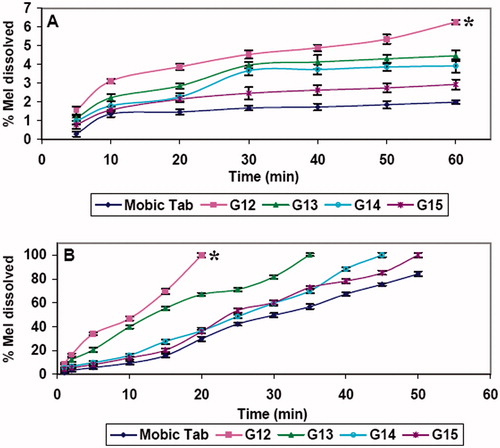
Figure 3. Dissolution profiles of Mel fast disintegrating films in SSF pH 6.8. Statistically significant differences are indicated as *p < 0.05.
In SGF, G12 and G13 edible films () gave higher initial dissolution; 5 min {(1.54% ± 0.21 and 1.12% ± 0.16, respectively) and at 60 min (6.25% ± 0.119 and 4.45% ± 0.3 correspondingly)} than Mobic tablet (0.28% ± 0.14 and 1.98% ± 0.11 after 5 and 60 min, respectively).
Matching pH-dependant solubility of Mel, the in-vitro dissolution of Mel from Mobic tablets and edible films in SSF was higher than that in SGF (). Those edible films (G12 and G13) containing Mel SCA showed greater dissolution rate than films containing raw Mel, which is consistent with higher dissolution of Mel SCA compared with raw Mel.
G13 showed significantly lower dissolution rate at 20 min (66.96% ± 1.33, p < 0.05) compared to G12 (100% ± 1.78), which is related to higher viscosity of the film forming polymer (HPMC E15) of G13 than that of G12 (HPMC E5).
Concerning Mel fast disintegrating films (), all of the prepared films showed higher initial dissolution and at 10 min compared to Mobic tablet. Both G3 and G7 produced highest initial dissolution at 1 min (25.86% ± 2.36 for G3 and 32.56% ± 1.23 for G7 compared to 1.46% ± 0.53 of Mobic tablet) and at 10 min (100% ± 1.05 for G3 and 100% ± 1.45 for G7 compared to 9.46% ± 1.34 of Mobic tablet).
The drug dissoluted from all formulae decreased as the amount of the polymer increased, this was in agreement with Rebecca et al. (Citation2013).
G12 edible film and G3 fast disintegrating film showing highest dissolution rate and suitable mechanical properties were chosen for further studies.
Accelerated stability studies
The chosen best fast disintegrating and edible film formulae (G3 and G12, respectively) were stored at 30 °C ± 2 (60% ± 5 RH) and 40 °C ± 2 (75% ± 5 RH) for 3 months in a stability chamber, the samples were withdrawn at three time intervals after 30 d, 60 d and finally 90 d.
Fast disintegrating and edible films were found to be physically and chemically stable at the selected temperature and humidity with no significant change in terms of physical characteristics and drug content. Under storage conditions 30 °C ± 2 (60% ± 5 RH); G3 showed % drug content of 98.91 ± 1.08, 97.95 ± 0.13 and 97.32 ± 0.24 after 30 d, 60 d and 90 d, respectively, compared to 99.04 ± 1.53 at zero time. While, G12 showed % drug content of 98.19 ± 1.32, 97.96 ± 1.45 and 96.98 ± 1.53 compared to 98.2 ± 1.54. Under storage conditions 40 °C ± 2 (75% ± 5 RH); G3 showed % drug content of 98.81 ± 1.18, 97.95 ± 0.15 and 97.12 ± 0.21 after 30 days, 60 days and 90 days, respectively. While, G12 showed % drug content of 98.15 ± 1.13, 97.99 ± 1.64 and 96.78 ± 1.56, respectively.
Pharmacokinetic study
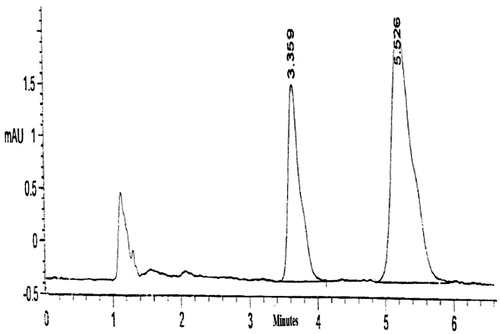
The standard plasma calibration curve of Mel showed a linear response across the concentration range used from 0.02 to 2 μg/ml with a determination coefficient R2 = 0.997. The curve was used to estimate the concentrations of Mel in the unknown plasma samples at different time intervals. No endogenous plasma components were observed at the retention times corresponding to both the drug and internal standard which proves the specificity of the method, .
Figure 4. Chromatogram of Mel (RT = 5.526) and piroxicam internal standard (RT = 3.359) in plasma.
and illustrated the mean pharmacokinetic parameters and the mean plasma concentration of Mel in (µg/ml) versus time in h, respectively, following oral administration of Mobic® tablet 15 mg, G3 and G12 to four human volunteers.
Figure 5. Mel plasma concentration-time profiles after administration of Mobic® tablets, G3 fast disintegrating film and G12 edible film.
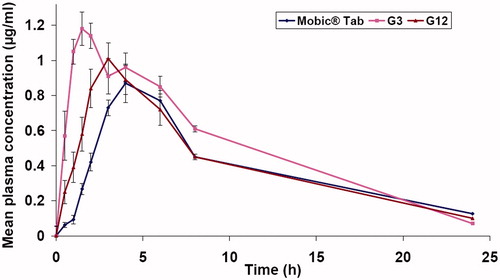
Table 4. Mean pharmacokinetic parameters of Mel following administration of different formulae.
Orally administrated Mobic® 15 mg tablets exhibited slower absorption rate than both G3 and G12 with Cmax of 0.89 ± 0.02 µg/ml and Tmax of 4.67 ± 1.15 h. Administration of G3 and G12 films resulted in faster and higher Mel plasma levels up to Cmax of 1.23 ± 0.026 µg/ml (1.38-fold higher) with Tmax of 1.67 ± 0.29 h and Cmax of 1.03 ± 0.076 µg/ml (1.15-fold higher) with Tmax of 2.67 ± 0.58 h for G3 and G12, respectively. A significantly higher early exposure (AUC0–0.5h was 0.1425 ± 0.013 and 0.0625 ± 0.013 µg h/ml for G3 and G12, respectively, compared to 0.016 ± 0.0012 of Mobic tablets) and higher AUC0–4h (average Tmax of Mobic tablet is ∼4 h) followed administration of G3 and G12 films indicating faster Mel absorption.
The overall exposure of Mel, AUC0–24 was improved in the order of G3 administration (1.36-fold higher) > G12 administration (1.09-fold higher) > Mobic administration. However, the AUC0–24 obtained following administration of G12 edible films was not significantly different from that of AUC0–24 obtained following Mobic tablet administration.
Discussion
Choice of neutralization technique employed for the preparation of Mel spherical crystalline agglomerates and the type and the concentration of the polymer (HPβCD, 0.0125% w/v) used was based on a study performed by our work team (Farid et al., Citation2014). The chosen Mel SCA for the current study is amongst other formulae showing highest dissolution rate at pH 1.2 and 6.8 yet it is the one with the lowest polydispersity index.
Seeking for possible effect of change in Mel crystalline state during crystallization process on such dissolution enhancement; DSC and XRD studies were carried out.
Solid state characterization of Mel SCA illustrated lower heat of solution, referring to that spherical agglomerate is more energetic and hence more soluble than the drug (Najib et al., Citation1986); decreased crystallinity and/or decreased primary particle size of processed Mel relative to raw one, which may be attributable to the improved dissolution rate of the prepared Mel SCA. Also, surface modification due to deposition of polymer on to the agglomerate surface, and higher porosity of agglomerates evaluated through bulk and tapped density determination and scanning electron microscopy examination (Farid et al., Citation2014) may account for improved dissolution of Mel.
In the dissolution study of different prepared films, particle engineering of Mel followed by impregnation into different films positively affected dissolution rate of Mel.
G12 and G13 edible films presented the highest dissolution rate at 5 min and 60 min compared to Mobic® tablets which can be attributed to the solubility enhancing effect of Mel SCA. In addition, availability of larger surface area that leads to rapid disintegration and release of the drug unlike Mobic® tablet.
The significantly higher improvement of Mel dissolution observed in pH 6.8 compared to pH 1.2 is attributed to that, Mel is a zwitterionic drug with two pKa values, 1.09 and 4.18 (Luger et al., Citation1996), accordingly it shows a pH-dependent solubility; decreasing pH leads to an increase in the ratio of non-ionized to ionized drug combined with a decrease in solubility. On the contrary, in aqueous solution at pH values >4.18 Mel is ionized and the dominant species is the anion form.
As the results showed, fast disintegrating films; G3 and G7 produced highest initial dissolution at 1 min and 10 min compared to Mobic® tablet. Such results could be explained as follows:
- Shorter disintegration time of fast disintegrating films compared to longer time required for conventional Mobic tablet (higher initial dissolution rate).
- Solubility enhancing properties of Mel SCA formula upon which different prepared OF are based.
The role of PVA films in enhancing dissolution of Mel may be due to swelling of PVA forming a gel layer on the exposed film surfaces. The loosely bound PVA molecules were easily eroded, allowing the release of the drug (Fawzia et al., Citation2010).
As is shown in the results of in-vivo study, the earlier Tmax and higher Cmax levels correlate with the higher initial and overall dissolution rate of Mel SCA and Mel SCA films compared to raw Mel and Mobic tablets, respectively. Mel being a BCS class II drug, its bioavailability is rate-limited by its dissolution, and a small increase in dissolution profile may result in a large increase in bioavailability (Lobenberg & Amidon, Citation2000). Also, pregastric absorption may be attributable for such results.
The characteristic rapid disintegration and dissolution of different film formulations may also contribute to the improved bioavailability of Mel; independence of film disintegration and dissolution on gastric motility and secretions is of particular importance in the management of acute pain conditions, where the gastric motility facilitating disintegration of solid dosage forms may be impaired and subsequently the dissolution and the bioavailability of oral conventional tablets and capsules of Mel are expected to be delayed.
The Tmax achieved following G3 administration was comparable to the reported Tmax (1–1.5 h) following Mel intramuscular administration (Davies & Skjodt, Citation1999; Euller-Ziegler et al., Citation2001) yet circumventing patient non-compliance associated with parenteral therapy.
Mel edible film offered a rapid absorption of Mel without affecting the prolonged exposure to Mel. This is illustrated by comparable overall systemic exposure of Mel (insignificantly different AUC0-24) following administration of G12 and Mobic tablets.
Both of intraoral fast disintegrating and edible films were able to achieve earlier higher systemic exposure to Mel compared to Mel conventional tablets, revealed by shortened Tmax and higher drug exposure (higher AUC0–0.5h and AUC0–4h) implying faster absorption of Mel hence a rapid onset of action required for efficacious treatment of acute pain conditions and acute attack of rheumatism.
Conclusion
There were marked improvements in both dissolution and pharmacokinetic behaviors of Mel via spherical crystallization and different film formulations.
Spherical crystalline Mel-based films showed improved early and overall systemic exposure to Mel providing rapid pharmacological effect required for acute pain and rheumatic episodes. Intraoral fast disintegrating film showed an earlier Tmax than conventional tablets, which is comparable to that of intramuscular route but expectedly avoiding invasiveness, patient inconvenience and non-compliance associated with parenteral therapy.
Developed film formulations may be promising alternatives to Mel conventional oral dosage forms and intramuscular Mel to achieve earlier onset of action particularly in severe pain and acute rheumatic conditions.
Declaration of interest
There is no declaration of interest.
References
- Abd El-bary A, Foda N, Elkhateeb M. (2001). Reversed phase liquid chromatographic determination of meloxicam in human plasma and its pharmacokinetic application. Anal Lett 34:1175–87
- Ahmed M, Khanna D, Frust DE. (2005). Meloxicam in rheumatoid arthritis. Expert Opin Drug Metab Toxicol 1:739–51
- Bhanja SB, Ellaiah P, Martha SK, et al. (2010). Buccoadhesive drug delivery system of captopril: formulation and in vitro evaluation. J Pharm Res 3:335–40
- British Pharmacopoeia III. (2003). London: Her Majesty's Stationery Office Ltd, 2598–9
- Dahiya M, Saha S, Sahiwala A. (2009). A review on mouth dissolving films. Curr Drug Deliv 6:469–76
- Davies NM, Skjodt NM. (1999). Clinical pharmacokinetics of meloxicam. A cyclo-oxygenase-2 preferential nonsteroidal anti-inflammatory drug. Clin Pharmacokinet 36:115–26
- Di Martino P, Barthélémy C, Piva F, et al. (1999). Improved dissolution behavior of fenbrufen by spherical crystallization. Drug Dev Ind Pharm 25:1073–81
- Dixit M, Kulkarni PK, Selvam RP, et al. (2011). Preparation and characterization of spherical agglomerates of piroxicam by neutralization method. Am J Drug Discov Devel 1:188–99
- Dixit RP, Puthli SP. (2009). Oral strip technology: overview and future potential. J Control Release 139:94–107
- El-Setouhy DA, Abd El-Malak NS. (2010). Formulation of a novel tianeptine sodium orodispersible films. AAPS Pharm Sci Tech 11:1018–25
- Euller-Ziegler L, Vélicitat P, Bluhmki E, et al. (2001). Meloxicam: a review of its pharmacokinetics, efficacy and tolerability following intramuscular administration. Inflamm Res 50:S5–9
- Farid M, El-Setouhy DA, El-Nabarawi MA, El-Bayomi T. (2014). Recrystallized agglomerated meloxicam: evaluation of anti-nociceptive effect. J Drug Deliv Sci Tec, accepted for publication
- Fawzia H, Abdel Azeem M, Gihan F, Mohamed S. (2010). Mucoadhesive buccal patches of lornoxicam: i-development and in-vitro characterization. Bull Pharm Sci, Assiut University 33:59–68
- Goldman AP, Williams CS, Sheng H, et al. (1998). Meloxicam inhibits the growth of colorectal cancer cells. Carcinogenesis 19:2195–9
- Hanft G, Türck D, Scheuerer S, Sigmund R. (2001). Meloxicam oral suspension: a treatment alternative to solid meloxicam formulations. Inflamm Res 50:535–7
- Hoffmann EM, Breitenbach A, Breitkreutz J. (2011). Advances in orodispersible films for drug delivery. Expert Opin Drug Deliv 8:299–316
- Kulmatycki KM, Jamali F. (2007). Drug disease interactions: role of inflammatory mediators in pain and variability in analgesic drug response. Int J Pharm Pharm Sci 10:554–66
- Kürti L, Kukovecz Á, Kozma G, et al. (2011). Study of the parameters influencing the co-grinding process for the production of meloxicam nanoparticles. Powder Technol 212:210–17
- Laska EM, Sunshine A, Marrero I, et al. (1986). The correlation between blood levels of ibuprofen and clinical analgesic response. Clin Pharmacol Ther 40:1–7
- Liang AC, Chen LH. (2001). Fast-dissolving intraoral drug delivery systems. Expert Opin Ther Pat 11:981–6
- Lobenberg R, Amidon GL. (2000). Modern bioavailability, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur J Pharm Biopharm 50:3–12
- Lucas SF, Anagha B, Jackeline I, et al. (2012). Preparation and characterization of hdroxypropyl methyl cellulose films containing stable BCS Class II drug nanoparticles for pharmaceutical applications. Int J Pharm 423:496–508
- Luger P, Daneck K, Engel W, et al. (1996). Structure and physicochemical properties of meloxicam, a new NSAID. Eur J Pharm Sci 4:175–87
- Maghsoodi M, Taghizadeh O, Martin GP, Nokhodchi A. (2008). Particle design of naproxen-disintegrant agglomerates for direct compression by a crystallo-co-agglomeration technique. Int J Pharm 2351:45–54
- Mashru RC, Sutariya VB, Sankalia MG, Parikh PP. (2005). Development and evaluation of fast-dissolving film of salbutamol sulphate. Drug Dev Ind Pharm 31:25–34
- Najib NM, Suleiman M, Malak A. (1986). Characteristics of the in-vitro release of ibuprofen from polyvinylpyrrolidone solid dispersion. Int J Pharm 32:229–36
- Naruse T, Nishida Y, Hosono K, Ishiguro N. (2006). Meloxicam inhibits osteosarcoma growth, invasiveness and metastasis by COX-2 dependent and independent routes. Carcinogenesis 27:584–92
- Patel R, Naik S, Baria A. (2009). Formulation development and evaluation of mouth melting film of ondansetron. Arch Pharm Sci Res 1:121–7
- Prabhakara P, Ravi M, Marina K, et al. (2011). Formulation and evaluation of fast dissolving films of levocitirizine dihydrochloride. Int J Pharm Investig 1:99–104
- Rebecca A, Kinam P, Alyssa P. (2013). Water soluble polymer films for intravascular drug delivery of antithrombotic biomolecules. Eur J Pharm Biopharm 84:125–31
- Shimoda H, Taniguchi K, Nishimura M, et al. (2009). Preparation of a fast dissolving oral thin film containing dexamethasone: a possible application to antiemesis during cancer chemotherapy. Eur J Pharm Biopharm 73:361–5
- Tomar A, Sharma K, Chauhan NS, et al. (2012). Formulation and evaluation of fast dissolving oral film of dicyclomine as potential route of buccal delivery. Int J Drug Dev Res 4:408–17
- Usha AN, Mutalik S, Reddy MS, et al. (2008). Preparation and, in vitro, preclinical and clinical studies of aceclofenac spherical agglomerates. Eur J Pharm Biopharm 70:674–83