
Abstract
Background: Vorinostat (VRS), a histone deacetylases inhibitor, has significant cytotoxic potential in a large number of human cancer cell lines.
Objective: To clarify its promising anticancer potential and to improve its drawback related to physical properties and in vivo performance of VRS.
Methods: VRS was successfully incorporated into nanostructured lipid carriers (NLCs) by the hot microemulsion method using sonication following a homogenization technique.
Results: After the optimization process, VRS-loaded NLCs (VRS-NLCs) were obtained as ideal quality nanoparticles with a spherical shape, small size (∼150 nm), negative charge (∼−22 mV), and narrow size distribution. In addition, the high entrapment efficiency (∼99%) and sustained drug release profile were recorded. Cytotoxicity study in three different cell lines (A549, MCF-7, and SCC-7) demonstrated higher cytotoxicity of VRS-NLCs than free drug. Finally, the AUC of VRS (118.16 ± 17.35 µgh/mL) was enhanced ∼4.4 times compared with that of free drug (27.03 ± 3.25 µgh/mL).
Conclusion: These results suggest the potential of NLCs as an oral delivery system for enhancement of cellular uptake, in vitro cytotoxicity in cancer cell lines and the oral bioavailability of VRS.
Introduction
Chromatin structure and function were regulated by histone deacetylases (HDACs), which acted as catalyst for removal of the acetyl modification from lysine residues of histones (Marks et al., Citation2004). Treatment with HDAC inhibitors resulted in growth arrest, terminal differentiation, apoptosis, or autophagic cell death. Thus, development of HDAC inhibitors as therapeutic agents for cancer treatment has been attempted (Kelly et al., Citation2005; Bolden et al., Citation2006).
Vorinostat (VRS) (suberoylanilide hydroxamic acid or SAHA) is a potent candidate in the HDAC family. The anticancer activity of VRS is proposed to be due to drug-induced accumulation of acetylated proteins, including the core nucleosomal histones and other proteins (e.g. BCL6, p53 and Hsp90) (Richon, Citation2006; Marks, Citation2007). VRS was approved by the FDA for treatment of cutaneous T-cell lymphoma (Konsoula & Jung, Citation2008). ZOLINZA® capsule (100 mg) is a commercial product of VRS with a dose of 400 mg orally once daily (Mohamed et al., Citation2012). Such a high dose is due to low aqueous solubility and permeability leading to low bioavailability (Chandran et al., Citation2014; Tran et al., Citation2014). In addition, VRS exhibited a short half-life of 40 min following intravenous (IV) administration, compared with ∼2 h following oral administration and underwent extensive first-pass metabolism (Tran et al., Citation2014). In the effort, to overcome these problems, lipid nanoparticles can be an ideal system for enhancement of in vitro as well as in vivo drug performance, especially hydrophobic anticancer drug (Aznar et al., Citation2013; Minelli et al., Citation2013; Xu et al., Citation2013; Kumar et al., Citation2014).
Exploiting the advantages of nanostructured lipid carriers (NLCs) for high loading capacity, improvement of solubility, controlling release and then enhancing in vitro cytotoxicity, cellular uptake and bioavailability is the main objective of this study. NLCs were prepared using a hot emulsification method and characterized at various levels. Then, physical properties, including size, thermal dynamic state, morphology, and drug release were investigated in order to obtain the optimized formulation. In addition, cell study was performed in order to confirm efficiency of loaded drug in the system by MTT assay and confocal images. Eventually, the potential for the use of VRS-NLCs as a drug delivery system was proven via pharmacokinetics study through oral administration.
Materials and methods
Materials
Vorinostat was purchased from LC Laboratories (Woburn, MA). Precirol ATO 5 and Capryol 90 were procured from Gattefosse (Nanterre Cedex, France). Soybean lecithin was purchased from Junsei Co. Ltd (Tokyo, Japan). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) was obtained from Sigma (St. Louis, MO). NBD-PC was supplied by Avanti Polar Lipids, Inc. (Alabaster, AL) and LysoTracker Red was purchased from Thermo Fisher Scientific Inc (Waltham, MA). Human breast adenocarcinoma cells (MCF-7), human non-small cell lung cancer cells (A-549), and squamous cell carcinoma cells (SCC-7) were originally obtained from the Korean Cell Line Bank (Seoul, South Korea). All other chemicals and reagents used were at least of analytical grade.
Solubility study
Solubility studies of VRS were performed according to a published method (Poudel et al., Citation2012; Tran et al., Citation2013). An excess of VRS was vortex-mixed with 1 mL of each of the chosen carriers. The micro tubes (Axygen MCT-200, Sigma-Aldrich) were shaken at 37 °C for three days in a water bath at 100 strokes per min, and the obtained dispersions were then centrifuged at 12 000 rpm for 10 min and filtered through a 0.45 μm membrane filter. Drug solubility was evaluated using HPLC with the following conditions: flow rate of 1.0 mL/min, wavelength 241 nm, formic acid (0.1%)/acetonitrile (70/30) was used as a mobile phase (Cai et al., Citation2010). The solubility test was performed and results were expressed as the mean ± standard deviation of three determinations.
Preparation of VRS-loaded nanostructured lipid carriers
VRS-loaded NLCs (VRS-NLCs) were prepared using the method of emulsion at a high temperature under homogenization, followed by sonication. The obtained emulsion was solidified at a low temperature using ice (Tsai et al., Citation2012; Tran et al., Citation2014). 0.1% VRS (weight percentage of drug to the total volume), 550 mg of lipids consisting of Precirol and Capryol 90 and 50 mg of lecithin were weighed accurately, then melted and mixed at 75 °C. The aqueous phase including 1.5% (w/v) Tween 80 was heated to 75 °C, and then added to the lipid phase and mixed with mechanical agitation at 13 500 rpm in an Ultra Turrax® T-25 homogenizer (IKA®-Werke, Staufen, Germany) for 3 min. The mixture was homogenized continuously using a probe sonicator (Vibracell VCX130; Sonics, Newtown, CT) at 90% amplitude for 5 min. In order to remove free drug and other components, the obtained formulation was washed thrice by distilled water using centrifugal 10-kDa molecular weight cut-off devices (Amicon Ultra, Millipore, Billerica, MA).The final product was cooled at low temperature, which was maintained by ice.The obtained solutions were frozen at −80 °C and then lyophilized (FDA5518, IlShin, South Korea).
Physical characterizations
Particle size and zeta potential measurements
The mean particle size (z-average) and size distribution of NLCs were measured by the dynamic light scattering (DLS) technique using a Zetasizer Nano-Z (Malvern, Worcestershire, UK) at 25 °C and a 90° scattering angle. The zeta potential was determined according to the particle electrophoretic mobility in aqueous medium using the same instrument. All measurements were performed in triplicate with a 1:10 dilution using distilled water.
Determination of drug entrapment efficiency
The percentage of drug incorporated into NLC was determined indirectly after estimating free drug by ultracentrifugation. Briefly, the upper chamber was filled with 1 mL of NLC dispersion using centrifugal 10-kDa molecular weight cut-off devices (Amicon Ultra, Millipore, USA). The drug concentration in the filtrate collected in the lower chamber was analyzed using the HPLC method. Drug entrapment efficiency and drug-loading capacity were calculated using the following equations (Zhuang et al., Citation2010):
where EE is the entrapment efficiency; LC is the drug-loading capacity; W is the weight (mg).
Transmission electron microscope analysis
The morphology of NLCs was observed by transmission electron microscope (TEM) (H-7000, Hitachi, Shiga, Japan). A drop of NLC dispersion was diluted 10-fold with double-distilled water before negatively staining with 2% phosphotungstic acid for 30 s and spread on a copper grid. The grid was air-dried at room temperature and then images were taken by TEM.
Thermal analysis
Differential scanning calorimetry (DSC) characterization was performed using a DSC-200 differential scanning calorimeter (TA Instruments, New Castle, DE). The lyophilized samples were accurately weighed, then placed in aluminum pans and sealed with a lid. During the scanning process, an empty aluminum pan was used as the reference and the sample was heated at a rate of 10 °C/min at a temperature range between 40 and 180 °C with a nitrogen purge of 50 mL/min.
Powder X-ray diffraction analysis
Powder X-ray diffraction (PXRD) was used to determine the crystallite of VRS accommodated in the lipid matrix. PXRD studies were performed for pure VRS, Precirol, and freeze-dried VRS-NLCs using a powder X-ray diffractometer (X’Pert PRO MPD diffractometer, Almelo, The Netherlands) using Cu-Kα radiation. The samples were scanned over a 2θ range of 10–50°.
In vitro drug release
The release of VRS from the optimized NLC formulation was carried out using a modified dialysis membrane diffusion technique (Cirri et al., Citation2012). One milliliter of NLC dispersion was added to a dialysis bag with a molecular weight cut off (MWCO) of 3500. The dialysis bag was immersed in a Falcon tube including 40 mL media pH 1.2 and 6.8 for stimulation of gastric and intestinal conditions. The Falcon tube was kept in a shaking water bath (HST – 205 SW, Hanbaek ST Co., Gwangju, Korea) at 100 strokes/min and 37 ± 0.5 °C. At predetermined time intervals (0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h), for a total period of 24 h, 0.5 mL samples were taken and replaced with equal volumes of fresh medium. Finally, the drug concentration was evaluated using the HPLC method. The results were the mean values after experiments were performed in triplicate.
Intracellular uptake and cell cytotoxicity
MCF-7, A-549, and SCC-7 cells were cultured in RPMI-1640 medium supplemented with 10% fetal bovine serum, penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C and 5% CO2. Intracellular uptake of VRS-NLCs was performed by confocal microscopy technique (Hong et al., Citation2014). MCF-7 and SCC-7 cells were attached to coverslips and placed in a 12-well plate (2 × 105 cells/well) and grown for 24 h. NBD-PC was used as a lipophilic fluorescence agent stand for lipid particles. The cells were treated with NBD-PC-NLC at a concentration of 1 µg/mL and further incubated for 0.5 h. After 20 min, 100 nM LysoTracker Red was added for 10 min for staining of endosome/lysosome. The cells were then washed three times with cold PBS and fixed in 4% paraformaldehyde. The coverslips were removed and mounted on microscope slides and images were observed on a confocal laser scanning microscope (Olympus FV1000-IX81, Tokyo, Japan).
Assessment of cell viability was performed for evaluation of VRS in cancer cell lines (Ramasamy et al., Citation2013; Tran et al., Citation2014). Three different cells (A549, MCF-7, and SCC-7) were cultured in 96-well plates prior to treatment (1 × 104 cells/well). Samples (blank NLC, free VRS, and VRS-NLC) were applied to cells in a range of concentrations (0.1–50 µg/mL). The free VRS was prepared by dissolving VRS in 0.2% (v/v) DMSO and then diluted accordingly (Bondi et al., Citation2007). Following incubation for 24 h in a humidified incubator (95% air and 5% CO2) at 37 °C, the cells were washed once with phosphate buffered saline (PBS). 100 µL of MTT reagent (1.25 mg/mL in medium) was added to each well. After incubation at 37 °C for 4 h, cells were exposed to 100 μL of DMSO. Absorbance of each sample was measured at 570 nm using a spectrophotometric plate reader (Multiskan EX, Thermo Scientific, Waltham, MA). Cell viability was calculated using the following formula:
Pharmacokinetics study
Male Sprague-Dawley rats weighing 300 ± 10 g were fed in an animal house maintained at 25 ± 2 °C and 50–60% RH. The procedures for the animal studies were approved by the Institutional Animal Ethical Committee, Yeungnam University, South Korea. Two groups, each group containing four rats, were fasted for 12 h prior to the experiments. Samples (drug suspension in 1% methylcellulose and VRS-NLCs) were administrated orally to the rats at a dose of 30 mg/kg (Tran et al., Citation2014). In case of VRS-NLCs, the lyophilized powder was dissolved in distilled water at VRS concentration of 3 mg/mL, accordingly. Blood samples (300 µL) were collected from the right femoral artery in heparin-containing tubes (100 IU/mL) at pre-determined times (0.25, 0.5, 1, 2, 3, 4, 6, 8, 12, and 24 h) after the administration of these formulations and centrifuged (Eppendorf, Hauppauge, NY) at 14 000 rpm for 10 min. The plasma supernatant was collected and stored at −20 °C until further analysis.
VRS was extracted from plasma by the addition of acetonitrile for 15 min. After vortexing, the protein was precipitated and discarded using centrifugation at 13 000 rpm for 10 min. The quantities of drug from the supernatant layer were evaluated using the HPLC method.
Win-NonLin pharmacokinetic software (v4.0, Pharsight Software, Mountain View, CA) was used for the calculation of pharmacokinetics data, including areas under the curve and half-life time, based on the non-compartmental method. The maximum drug concentration (Cmax) and the time to Cmax (Tmax) were obtained directly from experimental profiles.
Statistical analysis
All experiments were performed at least three times. Mean data are presented as the mean ± SD. Statistical comparisons were determined by the analysis of variance (ANOVA) among at least three groups or Student’s t-test between two groups. p < 0.05 and p < 0.01 were considered statistically significant.
Results and discussion
Solubility study
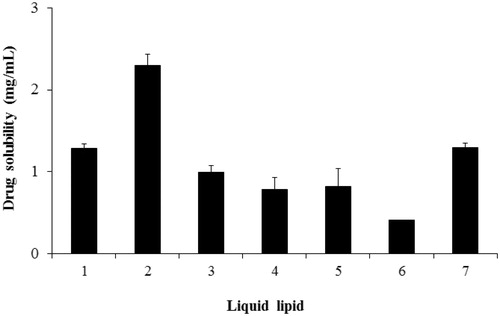
NLCs are the new generation of lipid nanovehicles, consisting of solid lipid and liquid lipid. Precirol ATO 5 has been reported as a solid lipid with favorable properties in many studies (Beloqui et al., Citation2013; Brugè et al., Citation2013), therefore we selected it for this work. Liquid lipid, which avoids crystals and expulsion phenomenon during storage and impacts on drug loading capacity, was carefully selected through solubility testing. Among various carriers tested, Capryol 90 was selected as a liquid lipid due to its highest solubility (2.30 ± 0.14 mg/mL) ().
Figure 1. Drug solubility in various vehicles. (1) Capryol™ PGMC, (2) Capryol™ 90, (3) Lauroglycol-FCC, (4) castor oil, (5) Labrafil M1944CS, (6) Labrafil M2125 CS, and (7) Peceol. Data represent the mean ± standard deviation (n = 3).
Optimization of NLCs
VRS-NLCs were prepared using the hot emulsification method. Capryol 90 and Preciol were used as the liquid and solid lipid, respectively. In order to obtain proper formulation, we investigated several factors, having an impact on physical properties, including particle size, PDI, zeta potential, or drug loading capacity, as shown in . First, out of 5.5% m/v lipid, the particle size was augmented when liquid lipid concentration increased (). It was supposed that most of the liquid lipid proportion was located inside solid lipid, resulting in this increase. Due to favorable PDI, 1% of liquid lipid was selected for further experiments. The next step was to evaluate the effect of organic phase and aqueous phase. The particle size increased with increasing lipid concentration as well as decreasing surfactant concentration (). This is reasonable because higher viscosity level of the organic phase was obtained at higher lipid concentrations, which could lead to improved particle size with wide size distributions (Chen et al., Citation2010). Meanwhile, the augmentation of surfactant concentration in aqueous phase facilitates stronger surface-active properties so that the particle size is reduced (Bunjes et al., Citation1996). Except drug, the main components determined were described as formulation 11 ().
Figure 2. Effect of compositions on formulation parameters: particle size, polydispersity index (PDI), and zeta potential (ZP). (A) Liquid lipid concentration, (B) lipid concentration, (C) Tween 80 concentration, (D) drug concentration. Data are expressed as the mean ± standard deviation (n = 3).
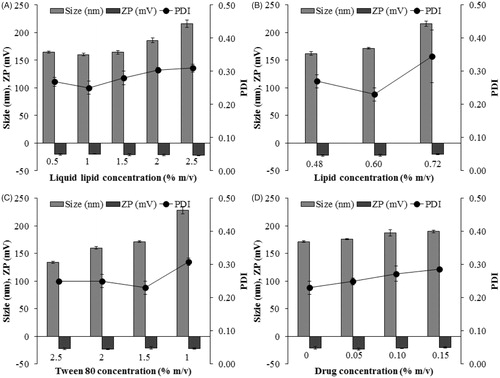
Table 1. Compositions of VRS-NLCs.
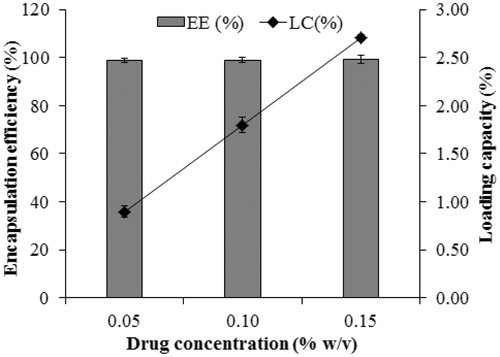
In addition, loading capacity is an important factor for carrying hydrophobic drug. As shown in , the particle size increased slightly with the increase of drug concentration. In addition, loading capacity showed improvement, although the drug entrapment efficiency remained almost constant at high level (∼99%, ), which might be attributed in part to the presence of liquid lipid. After long-term storage for three months, F14 with 0.1 % m/v was more stable (data not shown).Therefore, it was selected as the optimized formulation for further studies.
Figure 3. Drug entrapment efficiency and loading capacity.Data are expressed as the mean ± standard deviation (n = 3).
shows TEM photomicrographs of formulations demonstrating the spherical shape of VRS-NLCs. Homogenous particles without any crystal of free drug, indicating good drug encapsulation are shown. The observed particle diameters were found to be consistent with the DLS data.
Figure 4. TEM image of VRS-NLCs.

Physical properties analysis
Differential scanning calorimetry (DSC) was performed for the analysis of thermal characteristics of compounds. DSC thermograms of free VRS, Precirol, and the lyophilized VRS-NLCs are shown in . Precirol and free VRS showed a melting endothermic peak at 55 °C and 163 °C, respectively, which was related to their natural crystallites. In contrast, endothermic peak of VRS-NLCs totally disappeared near range melting point of the drug (163 °C) and a low-intensity peak still existed at ∼53 °C, indicating molecular dispersion of VRS within the lipid matrix or crystal form transformed to amorphous state (Jia et al., Citation2010).
Figure 5. (A) Differential scanning calorimetric (DSC) thermograms and (B) X-ray diffraction (XRD) patterns of solid lipid, free VRS, and VRS-NLCs.
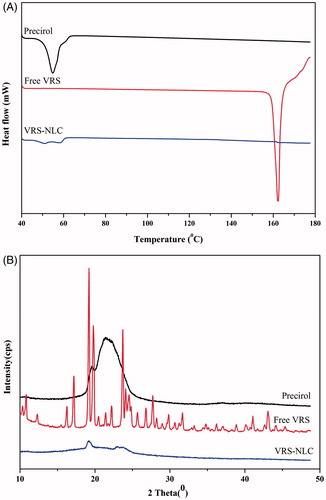
X-ray diffraction analysis was performed for further investigation of physicochemical properties of VRS formulations. The crystalline VRS is indicated as corresponding to several sharp peaks on the XRD pattern of free drug (). This may be attributed in part to high crystallinity of VRS in natural state. On the other hand, VRS-NLCs were found to have an XRD pattern with a shape similar to that of Precirol and all characteristic peaks of free VRS disappeared. The XRD data are consistent with DSC analysis, in that VRS was well entrapped in the lipid core and remained as molecular dispersion or transformed to amorphous form. Meanwhile, the intensity reduction of Precirol enabled the drug to have more space for accommodation, resulting in high loading capacity as well as low-drug expulsion (Lin et al., Citation2007).
In vitro drug release
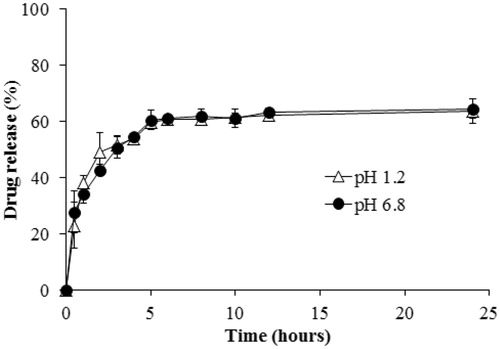
shows the in vitro release of VRS from NLCs in medium pH 1.2 and 6.8. Drug release of VRS from NLCs was similar under both pH conditions and showed a biphasic pattern. In detail, the initial burst release occurred within 5 h and dissolution rate reached approximately 60%, then the second stage was sustained without significant augmentation of drug release. The first rapid stage might be due to the rapid diffusion of amorphous drug onto the surface of NLCs, whereas the following sustained release could indicate strong interaction between hydrophobic drug and lipid core (Jia et al., Citation2012). The drug release data were greatly supported by physiochemical results and also provide potential for conduct of further study.
Figure 6. In vitro drug release from VRS-NLCs under different conditions: pH 1.2 (Δ) and pH 6.8 (•).
Cellular uptake and in vitro cytotoxicity
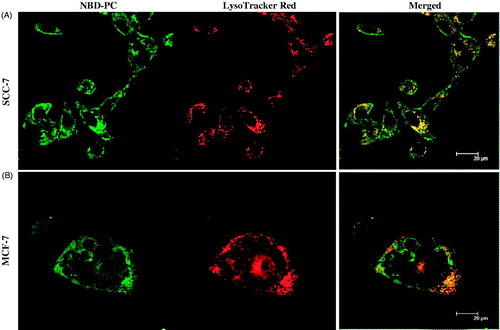
Confocal microscopy was used for observation of the intracellular distribution of the internalized nanoparticles in SCC-7 and MCF-7 cell lines. As shown in , green color of NBD-PC NLCs was localized in cytoplasm, whereas endosomes were stained and visualized as red fluorescence from LysoTracker Red. Overlap of red and green fluorescence was observed as yellow color in cells incubated with NBD-PC-NLCs for 30 min, which showed that the nanoparticles had penetrated rapidly and were localized in the endosomes after internalization following postulated endocytosis mechanisms (Delgado et al., Citation2011).
Figure 7. Intracellular uptake of NLCs in (A) SCC-7 cell, and (B) MCF-7 cells. NLCs contain NBD-PC (green) as a fluorescent probe. The LysoTracker Red stained for lysosome (red).
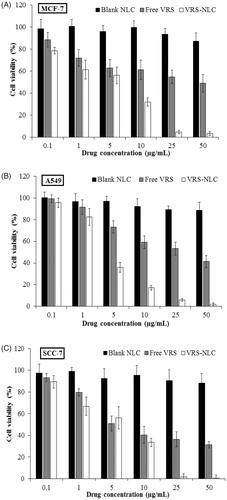
The rapid internalization of NLCs into cells is potential for efficient therapy. To confirm actual efficacy of the formulations against cancer cells (A549, SCC-7, and MCF-7), in vitro cytotoxicity was assessed at various concentrations of VRS (0.1, 1, 5, 10, 25, and 50 µg/mL). The results clearly demonstrated that after incubation for 24 h, free VRS and VRS-NLCs exhibited a high cytotoxicity and dose-dependence (). In contrast, blank carriers were safe at a range of concentrations with cell viability of more than 80%. Notably, in all cell lines, VRS-NLCs showed better performance than free VRS at most concentrations. The lipophilicity of NLCs might increase the interaction between particles with the membranes of cells resulting in high internalization into cytosol (Mussi et al., Citation2013). In addition, slow release profile and rapid penetration provided safety in blood circulation as well as efficacy in killing the cancer cell.
Figure 8. Cell viability following exposure of MCF-7, A549, and SCC-7 cells to blank NLCs, free VRS, and VRS-NLCs for 24 h. Data are expressed as the mean ± standard deviation (n = 8).
Pharmacokinetics studies
The plasma concentration–time profile of free VRS and VRS-NLCs is shown in . The pharmacokinetics parameters were subjected to non-compartmental analysis and a summary is shown in . The drug concentration of free VRS was under limited detection after 12 hour sampling, however, VRS-NLCs still showed good concentration up to 24 h. The Cmax of VRS-NLCs was 9.76 ± 0.91 µg/mL, which was significantly higher than that obtained with free VRS suspension (8.95 ± 0.12 µg/mL) (p < 0.05). Although it takes a longer period of time (4 h compared to 1 h) to reach the highest concentration, the half-life and mean residence time of VRS-NLCs are also longer (11.99 ± 0.55, 15.28 ± 3.36, respectively). It is not only improving 4.37-fold AUC0–∞ (118.16 ± 17.35 µgh/mL), but also maintaining sufficient drug concentration to reach the tumor site. Eventually, the drug is useful for effective therapy and the frequency of administration is reduced. As with many hydrophobic drugs, VRS is associated with a number of problems, including low solubility, short life-time, and first-pass metabolism. NLCs seemed to provide several solutions that have shown advantages in this study. First, the sustained release and addition of hydrophilic surfactant (Tween 80) on the surface prolonged drug in blood circulation (Tiwari & Pathak, Citation2011; Tsai et al., Citation2011). In addition, drug was shielded by a lipid layer in order to protect it from harsh gastric conditions and to overcome the first-pass by lymphatic uptake (Bhandari & Kaur, Citation2013). On the other hand, the surfactants might contribute to enhanced permeability of the drug through the intestinal membrane (Dwivedi et al., Citation2014). Increasing adhesion into intestinal membrane of the colloidal system and higher solubility facilitated faster and more continuous saturated drug concentration between the intestinal membrane and blood vessel, resulting in enhanced absorption (Yuan et al., Citation2013). Finally, the particle size of VRS-NLCs was ∼150 nm, which can provide a large surface area, consequently serving a high concentration of VRS for absorption and thus can enhance its oral performance (Tiwari & Pathak, Citation2011; Dudhipala & Veerabrahma, Citation2014). Therefore, according to this result, NLCs would have potential for enhancing bioavailability of a poorly soluble drug such as VRS.
Figure 9. Plasma concentration-time profile of VRS after oral administration at a dose of 30 mg/kg of free VRS (□) or VRS-NLCs (▪). Data are expressed as the mean ± standard deviation (n = 4).
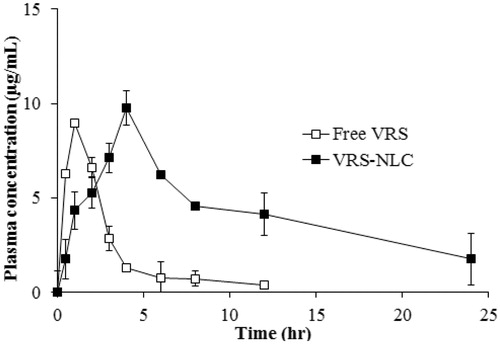
Table 2. Pharmacokinetic parameters of VRS in rats after oral administration of free VRS and VRS-NLCs at a dose of 30 mg/kg.
Conclusions
VRS-NLCs were successfully developed by sonication following a homogenization technique. The optimized VRS-NLCs showed a nano-sized spherical shape with narrow distribution. The high encapsulation of drug in the lipid matrix was confirmed by loading capacity and thermal characterization. In vitro release study showed that VRS-NLCs exhibited sustained release after an initial burst release and were pH-independent.Of particular importance, VRS-NLCs promoted significant enhancement of the in vitro antitumor activity and intracellular uptake, compared to the free VRS. Finally, the plasma concentration profile and pharmacokinetic parameters of VRS were significantly improved upon oral administration. These findings suggest that NLCs are a promise delivery system for VRS for chemotherapy.
Declaration of interest
This research was supported by a National Research Foundation of Korea (NRF) grant funded by the Ministry of Education, Science and Technology (No. 2012R1A2A2A02044997 and No. 2012R1A1A1039059).
References
- Aznar MÁ, Lasa-Saracíbar B, Estella-Hermoso De Mendoza A, et al. (2013). Efficacy of edelfosine lipid nanoparticles in breast cancer cells. Int J Pharm 454:720–6
- Beloqui A, Solinís MÁ, Gascón AR, et al. (2013). Mechanism of transport of saquinavir-loaded nanostructured lipid carriers across the intestinal barrier. J Controll Release 166:115–23
- Bhandari R, Kaur IP. (2013). Pharmacokinetics, tissue distribution and relative bioavailability of isoniazid-solid lipid nanoparticles. Int J Pharm 441:202–12
- Bolden JE, Peart MJ, Johnstone RW. (2006). Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov 5:769–84
- Bondì ML, Craparo EF, Giammona G, et al. (2007). Nanostructured lipid carriers-containing anticancer compounds: preparation, characterization, and cytotoxicity studies. Drug Deliv 14:61–7
- Brugè F, Damiani E, Puglia C, et al. (2013). Nanostructured lipid carriers loaded with CoQ10: effect on human dermal fibroblasts under normal and UVA-mediated oxidative conditions. Int J Pharm 455:348–56
- Bunjes H, Westesen K, Koch MH. (1996). Crystallization tendency and polymorphic transitions in triglyceride nanoparticles. Int J Pharm 129:159–73
- Cai Y, Yap C, Wang Z, et al. (2010). Solubilization of vorinostat by cyclodextrins. J Clin Pharm Ther 35:521–6
- Chandran P, Kavalakatt A, Malarvizhi GL, et al. (2014). Epigenetics targeted protein-vorinostat nanomedicine inducing apoptosis in heterogeneous population of primary acute myeloid leukemia cells including refractory and relapsed cases. Nanomed-Nanotechnol 10:721–32
- Chen C-C, Tsai T-H, Huang Z-R, et al. (2010). Effects of lipophilic emulsifiers on the oral administration of lovastatin from nanostructured lipid carriers: physicochemical characterization and pharmacokinetics. Eur J Pharm Biopharm 74:474–82
- Cirri M, Bragagni M, Mennini N, et al. (2012). Development of a new delivery system consisting in “drug – in cyclodextrin – in nanostructured lipid carriers” for ketoprofen topical delivery. Eur J Pharm Biopharm 80:46–53
- Delgado D, Del Pozo-Rodríguez A, Solinís MÁ, et al. (2011). Understanding the mechanism of protamine in solid lipid nanoparticle-based lipofection: the importance of the entry pathway. Eur J Pharm Biopharm 79:495–502
- Dudhipala N, Veerabrahma K. (2014). Candesartan cilexetil loaded solid lipid nanoparticles for oral delivery: characterization, pharmacokinetic and pharmacodynamic evaluation. Drug Delivery Early online:1–10. doi:10.3109/10717544.2014.914986
- Dwivedi P, Khatik R, Khandelwal K, et al. (2014). Pharmacokinetics study of arteether loaded solid lipid nanoparticles: an improved oral bioavailability in rats. Int J Pharm 466:321–7
- Hong W, Chen D, Jia L, et al. (2014). Thermo- and pH-responsive copolymers based on PLGA-PEG-PLGA and poly(l-histidine): synthesis and in vitro characterization of copolymer micelles. Acta Biomater 10:1259–71
- Jia L, Shen J, Zhang D, et al. (2012). In vitro and in vivo evaluation of oridonin-loaded long circulating nanostructured lipid carriers. Int J Biol Macromol 50:523–9
- Jia L-J, Zhang D-R, Li Z-Y, et al. (2010). Preparation and characterization of silybin-loaded nanostructured lipid carriers. Drug Deliv 17:11–18
- Kelly WK, O’connor OA, Krug LM, et al. (2005). Phase I study of an oral histone deacetylase inhibitor, suberoylanilide hydroxamic acid, in patients with advanced cancer. J Clin Oncol 23:3923–31
- Konsoula R & Jung M. (2008). In vitro plasma stability, permeability and solubility of mercaptoacetamide histone deacetylase inhibitors. Int J Pharm 361:19–25
- Kumar A, Ahuja A, Ali J, et al. (2014). Curcumin-loaded lipid nanocarrier for improving bioavailability, stability and cytotoxicity against malignant glioma cells. Drug Delivery Early online:1–16. doi:10.3109/10717544.2014.909906
- Lin X, Li X, Zheng L, et al. (2007). Preparation and characterization of monocaprate nanostructured lipid carriers. Colloid Surface A 311: 106–11
- Marks P. (2007). Discovery and development of SAHA as an anticancer agent. Oncogene 26:1351–6
- Marks PA, Richon VM, Miller T, et al. (2004). Histone deacetylase inhibitors. Adv Cancer Res 91:137–68
- Minelli R, Occhipinti S, Gigliotti C, et al. (2013). Solid lipid nanoparticles of cholesteryl butyrate inhibit the proliferation of cancer cells in vitro and in vivo models. Brit J Pharm 170:233–44
- Mohamed EA, Zhao Y, Meshali MM, et al. (2012). Vorinostat with sustained exposure and high solubility in poly (ethylene glycol)-b-poly (dl-lactic acid) micelle nanocarriers: characterization and effects on pharmacokinetics in rat serum and urine. J Pharm Sci 101:3787–98
- Mussi SV, Silva RC, Oliveira MCD, et al. (2013). New approach to improve encapsulation and antitumor activity of doxorubicin loaded in solid lipid nanoparticles. Eur J Pharm Sci 48:282–90
- Poudel BK, Marasini N, Tran TH, et al. (2012). Formulation, characterization and optimization of valsartan self-microemulsifying drug delivery system using statistical design of experiment. Chem Pharm Bull 60:1409–18
- Ramasamy T, Tran TH, Cho HJ, et al. (2013). Chitosan-based polyelectrolyte complexes as potential nanoparticulate carriers: physicochemical and biological characterization. Pharm Res 31:1302–14
- Richon V. (2006). Cancer biology: mechanism of antitumour action of vorinostat (suberoylanilide hydroxamic acid), a novel histone deacetylase inhibitor. Brit J Cancer 95:S2–6
- Tiwari R, Pathak K. (2011). Nanostructured lipid carrier versus solid lipid nanoparticles of simvastatin: comparative analysis of characteristics, pharmacokinetics and tissue uptake. Int J Pharm 415:232–43
- Tran TH, Poudel BK, Marasini N, et al. (2013). Preparation and evaluation of raloxifene-loaded solid dispersion nanoparticle by spray-drying technique without an organic solvent. Inter J Pharma 443:50–7
- Tran TH, Ramasamy T, Truong DH, et al. (2014). Development of vorinostat-loaded solid lipid nanoparticles to enhance pharmacokinetics and efficacy against multidrug-resistant cancer cells. Pharm Res 31:1978–88
- Tsai M-J, Huang Y-B, Wu P-C, et al. (2011). Oral apomorphine delivery from solid lipid nanoparticleswith different monostearate emulsifiers: pharmacokinetic and behavioral evaluations. J Pharm Sci 100:547–57
- Tsai M-J, Wu P-C, Huang Y-B, et al. (2012). Baicalein loaded in tocol nanostructured lipid carriers (tocol NLCs) for enhanced stability and brain targeting. Int J Pharm 423:461–70
- Xu P, Yin Q, Shen J, et al. (2013). Synergistic inhibition of breast cancer metastasis by silibinin-loaded lipid nanoparticles containing TPGS. Int J Pharm 454:21–30
- Yuan H, Chen C-Y, Chai G-H, et al. (2013). Improved transport and absorption through gastrointestinal tract by PEGylated solid lipid nanoparticles. Mol Pharm 10:1865–73
- Zhuang C-Y, Li N, Wang M, et al. (2010). Preparation and characterization of vinpocetine loaded nanostructured lipid carriers (NLC) for improved oral bioavailability. Int J Pharm 394:179–85