
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Context: Development of carvedilol-loaded transfersomes for intranasal administration to overcome poor nasal permeability and hepatic first pass effect so as to enhance its bioavailability.
Objective: The purpose of this study was to develop carvedilol-loaded transfersomes containing different edge activators (EAs) then evaluating the in vivo behavior of the optimized formula in rabbits.
Methods: The vesicles were prepared by incorporating different EAs including Span 20, Span 60, Tween 20, Tween 80, and sodium deoxycholate (SDC) in the lipid bilayer and each EA was used in three different ratios with respect to phosphatidylcholine (PC) including 95:5%, 85:15%, and 75:25% w/w (PC:EA). Evaluation of transfersomes was carried out in terms of shape, size, entrapment efficiency (EE), in vitro release, ex vivo permeation, confocal laser scanning microscopy (CLSM), and stability studies. The pharmacokinetic study of the optimized formula was conducted in rabbits.
Results: The mean diameter of the vesicles was in the range of 295–443 nm. Transfersomes prepared with 95:5% (w/w) (PC:EA) ratio showed highest EE% where Span 60 gave the highest values. Whereas those prepared using 85:15% w/w ratio showed highest percentages of drug release where SDC was superior to other EAs. The developed transfersomes exhibited significantly higher amounts of carvedilol permeated through nasal mucosa. CLSM of formula T14 containing SDC with 85:15% (w/w) (PC:EA) ratio revealed high permeation across the nasal mucosa.
Conclusion: The nanotransfersomal vesicles were significantly more efficient in nasal delivery of carvedilol with absolute bioavailability of 63.4%.
Introduction
Great attention has been paid to the nasal route as a non-invasive drug application route that offers many advantages for the delivery of drugs into systemic circulation. Its major advantage is avoiding the hepatic first pass effect. Additionally, it is characterized by quick onset of administered drugs as a result of their rapid absorption (Ozsoy et al., Citation2009).
Different types of nasal dosage forms have been reported in the literature including microspheres (Rajanikanth, Citation2003; Mao et al., Citation2004; Nema et al., Citation2013), liposomes (Vyas et al., Citation1995; Muramatsu et al., Citation1999), sprays (Quadir et al., Citation2000; Al-Ghananeem et al., Citation2008), gels (Varshosaz et al., Citation2006; Mahajan & Gattani, Citation2010), and solutions (Chavanpatil & Vavia, Citation2004; Harris et al., Citation2006).
Ultradeformable vesicles known as transfersomes which composed of phospholipids and an edge activator (EA) are a new approach in vesicular drug delivery. The idea of transfersomes was first introduced in 1992 by Cevc and co-workers. Transfersomes are characterized by their enhanced elasticity compared with traditional liposomes. The characteristic deformity of transfersomes is due to the presence of an EA, which is generally a single chain surfactant that is able to weaken the lipid bilayers of the vesicles (Cevc & Blume, Citation2003).
Several advantages of vesicular systems have been reported including: (1) their effectiveness as pharmaceutical carriers (Aburahma, Citation2014); (2) their ability to attain sustained release of drugs; (3) targeting organs for drug delivery; (4) lack of toxicity, and (5) capacity of increasing bioavailability of drugs (Dubey et al., Citation2008; Gupta et al., Citation2012; Kumar et al., Citation2012).
Vesicular systems have shown promising results in nasal delivery of many drugs such as levonorgestrel (Ding et al., Citation2007), acyclovir (Alsarra et al., Citation2008), and rivastigmine (Arumugam et al., Citation2008). Efficient brain delivery of olanzapine by the nasal route was carried out through formulation of transfersomes (Salama et al., Citation2012a,Citationb).
Carvedilol is a non-cardioselective beta blocker used in the treatment of hypertension and angina pectoris (Packer et al., Citation2002). It is well absorbed from the gastrointestinal tract but is subject to considerable first-pass hepatic metabolism; its oral absolute bioavailability is about 25% (Thummel & Shen, Citation2001).
The aim of present work was to develop nanotransfersomal vesicular systems for nasal delivery of carvedilol for enhancement of its bioavailability. An anionic as well as non-ionic surfactants (EAs) were utilized in the preparation of the carvedilol vesicles. The effect of different EAs with different ratios with respect to phosphatidylcholine was investigated. Moreover, the in vivo behavior of the optimized vesicular system was tested in rabbits.
Materials and methods
Materials
Carvedilol was obtained as a gift sample from Global Napi Pharmaceutical Company (Cairo, Egypt), l-α phosphatidylcholine (soyabean lecithin), sorbitan monolaurate (Span 20), sorbitan monostearate (Span 60), sodium deoxycholate, acetonitrile, formic acid, Poloxamer 407, Poloxamer 188, polyacrylic acid (carbopol 971P), polyoxyethylene sorbitan monolaurate (Tween 20), polyoxyethylene sorbitan monooleate (Tween 80), propylene glycol, and dialysis bags with a molecular weight cut off of 12 000 kDa were purchased from Sigma-Aldrich (St. Louis, MO), Triton X-100 and Rhodamine B were purchased from Loba Chemie (Mumbai, India). All other ingredients used were of analytical grade.
Preparation of carvedilol-loaded transfersomal vesicles
Different carvedilol transfersomal vesicles were prepared according to the thin film hydration technique (Bangham et al., Citation1965). The mixed lipid consisted of phosphatidylcholine and EA (100 mg) was added to a constant weight of drug (10 mg) and dissolved in chloroform:methanol, 2:1 v/v in a round bottom flask. The organic solvents were removed under vacuum in a rotary evaporator (Stuart rotary evaporator, RE300, Wolf Laboratories, North Yorkshire, UK with Stuart vacuum pump, RE3022C, Wolf Laboratories, North Yorkshire, UK) at 40 °C for 20 min to form a thin film on the wall of the flask, and kept in a desiccator under vacuum for 2 h to ensure total removal of trace solvents. Ten milliliters of simulated nasal electrolyte solution pH 5.5 were then added, and complete hydration of the dry film was performed by rotation at 60 rpm for 1 h at room temperature. The resulting vesicles were sonicated for 10 min in a bath sonicator (Sonix TV ss-series ultrasonicator, Sonix IV Ultrasonic Cleaning Systems, North Charleston, SC) to reduce the size of the vesicles. Different EAs (in several ratios) were used for the preparation of transfersomes, the composition of these formulations is shown in .
Table 1. Composition, drug entrapment efficiency and vesicles diameter of carvedilol-loaded transfersomes.
Characterization of carvedilol vesicles
Determination of carvedilol entrapment efficiency (EE%)
The EE% of transfersomes was determined after separation of the unentrapped drug by centrifugation of the transfersomal suspension using cooling centrifuge (SIGMA 3-30K, Steinheim Germany) at 14 000 rpm for 3 h at 4 °C, then washed and recentrifuged to ensure complete removal of the unentrapped drug. The amount of entrapped carvedilol was determined by lysis of the separated vesicles using a mixture of Triton X-100:ethanol:propylene glycol 40%:15%:45% (v:v:v), heating to 60 °C and cooling the solution to room temperature then centrifugation of the solution at 4000 rpm for 30 min and then analyzing the drug content by the HPLC method (Haggag et al., Citation2010). The analyses were carried out with a liquid chromatograph (Agilent 1260 Infinity, Waldbronn, Germany), equipped with Agilent 1260 Infinity Diode array detector VL (Agilent Technologies, Walnut Creek, CA) (G 1315D), Agilent 1260 Infinity preparative pump (G 1361A), Agilent 1260 Infinity thermo stated column compartment (G 1316A), and Agilent 1260 Infinity preparative Auto sampler (G 2260A). Separation and quantification were carried out on C18 column (ZORBAX Eclipse Plus, Agilent Technologies, Walnut Creek, CA) (25 cm × 4.6 mm i.d., a particle size of 5 µm). The mobile phase was a mixture of acetonitrile–0.05 M disodium hydrogen phosphate (50:50, v/v), pH 4.0. The mobile phase was filtered by passing through a 0.45-μm membrane filter, degassed and delivered at a flow rate of 1 ml/min. Detection was performed at a wavelength of 242 nm at ambient temperature. Carvedilol EE% was determined using the following formula:
Determination of vesicle size
The size and size distribution of drug vesicles was determined by the dynamic light scattering method (Guinedi et al., Citation2005) using a Malvern Mastersizer (Zetasizer Nano ZS, Malvern Instruments, Malvern, UK). The samples were diluted with distilled water before measurement.
In vitro release study of transfersomes
In vitro release studies were performed using vertical diffusion Franz cells with an effective diffusion area of 5 cm2. Transfersomal suspension of different formulations equivalent to 3 mg drug was placed in the donor compartment. A 50 ml of simulated nasal electrolyte solution (SNES) pH 5.5 containing 20% propylene glycol was used as receptor medium to ensure a sink condition (Aboelwafa et al., Citation2010).
The SNES was composed of 7.45 mg/ml NaCl, 1.29 mg/ml KCl, and 0.32 mg/ml CaCl2·2H2O and pH adjusted at 5.5 (Cheng et al., Citation2002; Callens et al., Citation2003). The receptor compartment was maintained at 37 ± 0.5 °C and stirred by a magnetic bar at 100 rpm. The donor compartment was separated from the receptor compartment by cellulose dialyzing membrane with molecular weight cutoff of 12 000 kDa which was soaked in the receptor medium overnight. At predetermined time intervals (0.5, 1, 2, 3, 4, 5, 6, 7, and 8 h), 1 ml aliquots were withdrawn from the sampling port and were replaced with an equal volume of fresh medium to maintain constant volume. The samples were filtered through 0.45 µm pore filter and analyzed by the HPLC method as stated above and the drug release percent was determined using the following equation:
where Mi and Mt are the initial amount of drug encapsulated in the transfersomes and the amount of drug released at time t, respectively. All the experimental procedure was repeated three times. The release of the free drug solution (3 mg carvedilol dissolved in 2 ml ethanol) was also investigated in the same way.
The data obtained from the release studies were kinetically analyzed and the order of drug release from the different formulations was determined. Zero- and first-order kinetics as well as the Higuchi diffusion model were employed and the correlation coefficient values (R2) were determined.
Ex vivo permeability study
The freshly excised sheep nasal mucosa except the septum part was collected in phosphate buffer saline (PBS), pH 6.4. The membrane was kept in PBS pH 6.4 for 30 min to equilibrate (Gavini et al., Citation2005). The superior nasal concha was identified and separated from the nasal membrane. The excised superior nasal membrane was then mounted on Franz diffusion cell. Franz diffusion cell used for ex vivo diffusion studies had a surface area of 5 cm2. The temperature of the receiver chamber containing 50 ml of diffusion media PBS pH 6.4 containing 20% propylene glycol was controlled at 37 ± 0.5 °C under continuous stirring with magnetic bar at 100 rpm, in a way that the nasal membrane surface just flushes the diffusion fluid.
Different volumes of transfersomal suspension carrying fixed weights of carvedilol (3 mg) were placed in the donor compartment of Franz diffusion cell. Samples from the receptor compartment were withdrawn at predetermined time intervals (0.5, 1, 2, 3, 4, 6, 8, 10, 12, and 24 h) then the receptor chamber was compensated with equal volumes of fresh medium. The withdrawn samples were filtered through 0.45 µm pore filter and finally analyzed by the HPLC method as mentioned before.
The permeation data of carvedilol was graphically plotted as the cumulative amount of drug permeated per unit area as a function of time and the permeation parameters could be provided. The cumulative drug in receptor compartment after 24 h was defined as Qcum (μg cm−2). The permeability coefficient Kp (cm h−1) of carvedilol form each formulation can be calculated by dividing the slope of the straight line portion of the curve by concentration of drug originally added. The lag time could be determined from the X-intercept of the linear portion of the graph.
Statistical analysis
All data were presented as mean ± SD. The results were analyzed statistically by one-way ANOVA with subsequent multiple comparisons using the Tukey multiple comparisons test. Significance level was set at p < 0.05. All calculations were made using the computer program SPSS 16.0 (SPSS, Chicago, IL).
Confocal laser scanning microscopy (CLSM) study
Preparation of rhodamine B-labeled transfersomes
Transfersomes were prepared as described before with some modifications. A 10 mg/ml methanol stock solution of rhodamine was prepared. Five µl from this stock solution was diluted in the organic phase which contained the mixed lipids.
CLSM
CLSM was used to scan the fluorescence signal of labeled transfersomes T14 (containing SDC with ratio of 85:15% w/w PC:EA) and methanolic solution of rhodamine B in PBS pH 6.4 (Marttin et al., Citation1997; Ghirardelli et al., Citation1999). For CLSM study, the ex vivo permeation study was carried out as described above. After 2 h, the nasal mucosa was removed and washed with distilled water. The treated area was frozen at –20 °C, and then the frozen mucosa was sectioned with a cryostat into 20 µm slices and placed on glass cover slips. The samples were then microscopically examined without additional tissue processing using an inverted laser scanning confocal microscope LSM 710 (Carl Zeiss, Germany). The rhodamine was excited with a He/Ne laser (excitation = 524 nm) and light emission was detected using a bypass filter (emission = 580 nm).
Transmission electron microscopy (TEM)
A selected transfersomal formula T14 was chosen for morphological examination of the vesicles. A drop of the vesicular suspension was applied on a carbon-coated grid. The suspension was left for 2 min, to allow its absorption in the carbon film, and the excess liquid was drawn off with filter paper and uranyl acetate was used as a negative stain. Samples were examined by TEM (Jeol, Tokyo, Japan) operating at an accelerating voltage of 80 kV (Manconi et al., Citation2003, Citation2009).
Stability study of transfersomes
A selected transfersomal formula T14 was stored in a glass vial at 4 °C for up to 3 months. Samples from the selected formula were withdrawn at definite time intervals (30, 60, and 90 d) and characterized for the vesicle size and EE% (Nasr et al., Citation2008).
Tolerability and toxicity studies (histopathological studies)
Four male Wistar albino rats weighing 200–250 g received once daily nasal administration of 20 µl of the transfersomal vesicles (T14 after being incorporated into a mucoadhesive in situ gel base which consisted of 20% poloxamer 407, 10% poloxamer 188, and 0.5% Carbopol 971P) for 14 d. The rats were then sacrificed and their nasal septum with the epithelial cell membrane were taken (Tengamnuay et al., Citation2000; Zaki et al., Citation2007), fixed in 10% formaldehyde for 24 h, decalcified, washed with tap water and then dehydrated by ethyl alcohol. Specimens were cleared in xylene then embedded in paraffin at 56 °C in a hot air oven for 24 h. Tissue blocks of paraffin beeswax were then prepared for sectioning at 5 µm by slide microtone. The obtained tissue sections were collected on glass slides, deparaffinized, and stained by hematoxylin and eosin stains. Slides of untreated and treated tissues were examined using a light microscope (Bancroft & Gamble, Citation2007).
Pharmacokinetic studies
Animals
This study was approved by the local animal ethical committee of Beni Suef University. A group of six New Zealand White (NZW) male rabbits weighing 2.5 ± 0.17 kg were used in this study. After an overnight fasting, rabbits were dosed, held in restrainers during blood sampling and they were conscious throughout the duration of the experiments.
Study design
The study was assigned in fasted state with single-dose; three-treatment and three-period crossover design with 1-week wash out period was given before administration of another formulation.
Dosage and drug administration
Each rabbit received a dose of 1.0 mg/kg from each of the three formulations used in this study. The formulations were carvedilol coarse suspension (1 mg/ml) in purified water was given by oral route, in situ gelling transfersomal formula T14 (12.5 mg/ml) where 100 μl were administered in each nostril and carvedilol solution (1 mg/ml) prepared in sterile water for injection containing 1% w/v Tween 80 and 2% w/v ethanol and the solution was injected into marginal ear vein of each rabbit.
Sample collection
After administration of the different formulations, blood sample (2.0 ml) was collected at different time intervals (0.5, 1, 2, 4, 8, 12, and 24 h) from the marginal ear vein of the rabbits. Blood samples were collected in EDTA tubes to avoid clotting and samples were centrifuged at 3000 rpm for 15 min to obtain the plasma. The separated plasma tubes were stored at −20 °C until assayed.
Chromatographic conditions
Plasma samples were analyzed for carvedilol adopting a modified liquid chromatography tandem mass spectrometry (LC-MS/MS) method (Kim et al., Citation2010) which composed of a Shimadzu Prominence (Shimadzu, Japan) series LC system equipped with degasser (DGU-20A3) and auto-sampler (SIL-20 AC) using Luna C18 column (Phenomenex, Inc., Torrance, CA) (4.6 × 50 mm2; a particle size of 5 μm).
The mobile phase was a mixture of acetonitrile and 0.1% formic acid (50:50 %v/v) and was delivered at a flow rate of 1 ml/min into the mass spectrometer's electrospray ionization chamber and the injection volume was 20 μl. Quantitation was achieved by MS/MS detection in the positive ion mode using a MDS Sciex (Foster City, CA) API-3200 mass spectrometer. The column and auto-sampler tray were maintained at 25 °C. The ion spray voltage was set at 3.6 kV and nebulizer gas pressure was 35 psi. Multiple reaction monitoring (MRM) mode was employed for the quantification; m/z 407.2 → 100.2 for carvedilol. Fragmentor and collision energy were adjusted at 145 and 29 V, respectively. The gas temperature was 300 °C and the gas flow rate was 10 l/min. All data acquisition was controlled by Analyst software 1.4.2 (Applied Biosystems Inc., Foster City, CA).
Samples preparation for analysis
An aliquot of 1.5 ml of acetonitrile was added to 0.5 ml of the plasma samples. The samples were then vortexed for efficient mixing for 2 min, centrifuged at 3000 rpm for 10 min and the clear supernatant was injected into the LC apparatus. The carvedilol recovery (5–300 ng/ml) varied between 93.65 and 100.26%. The calibration line was linear between 5 and 300 ng/ml (r2 = 0.9986) (n = 8).
Data analysis
Pharmacokinetic characteristics from plasma data following administration of the three treatments were estimated for each rabbit using, WinNonlin® (version 1.5, Scientific Consulting, Inc., Rockville, MD). Non-compartmental analysis was used. Cmax (ng/ml) and tmax (h) were the observed maximal drug concentration and the time needed to reach this concentration, respectively. The area-under-the-curve, AUC0–24 (ng h/ml), was calculated using the trapezoidal rule from zero time to the last time of the blood sample. The AUC from zero to infinity, AUC0–∞ (ng h/ml), was calculated as AUC0–∞ = AUC0–24 + Ct/k, where Ct is the last measured concentration at time t, and k is the terminal elimination rate constant estimated by log-linear regression analysis on data visually assessed to be at terminal log-linear phase. Apparent terminal elimination half-life (t1/2) was calculated as t1/2 = 0.693/k.
Results and discussion
Carvedilol entrapment efficiency
The entrapment of drug occurs in both the bilayers and the aqueous compartment of the vesicles. When the lipid compartment and aqueous phase became saturated with the drug, the vesicles provided limited entrapment capacity (Lopes et al., Citation2004; Ning et al., Citation2005).
The amount of drug entrapped is affected by the hydrophilic or lipophilic properties of the drug, surface area, and preparation method of the vesicles (Barenholz, Citation2003; Nii & Ishii, Citation2005). On one hand, the drug EE% for all vesicles was in the range of 43.68–88.72% (). The ratio 95:5% (w/w) (PC:EA) showed optimum EE% (T1, T4, T7, T10, and T13). Transfersomes containing high concentrations of the investigated surfactants (T3, T6, T9, T12, and T15) showed low drug EE% whatever their type or hydrophilic lipophilic balance (HLB) values where the EE% decreased with increasing EA ratio from 5 to 15 then 25% w/w. Incorporation of low concentration of EA led to growth in vesicle size (Van den Bergh et al., Citation2001). On the other hand, additional increase in the concentration of EA may have led to pore formation in the bilayers. Mixed micelles coexisted with the transfersomes when EA concentration exceeded 15% w/w resulting in lower drug entrapment due to the rigidity and smaller size of these mixed micelles (Jain et al., Citation2003).
Upon comparing the ratio of maximum EE% (95:5%, w/w), Span 80 (T4) showed the highest EE% (88.72%) followed by span 20 (T1) (83.56%), Tween 80 (T10) (79.64%), SDC (T13) (78.26%), and finally Tween 20 (T7) (64.16%) (). These results may be explained on the basis of HLB values of these EAs. They are 4.3, 8.6, 15, 16.7, and 16.7 for Span 80, Span 20, Tween 80, SDC, and Tween 20, respectively. Based on these HLB values, the affinity for lipids was expected to be in the order of Span 60 > Span 20 > Tween 80 > SDC and Tween 20 which illustrated higher EE% with formulae containing Span 80 and Span 20 compared with those containing Tween 80, SDC and Tween 20.
Our results are in accordance with Salama et al. (Citation2012a) who reported low drug EE% in transfersomes containing high concentration of surfactants. They suggested that hydrophilic surfactants destabilized the lipid bilayers which lead to decreased EE%. Moreover, the higher HLB of these surfactants may lead to drug solubilization and diffusion in the aqueous medium during preparation. In contrast, a competition between the surfactant and drug may be occurred when less hydrophilic surfactants present at high concentration which led to exclusion of the drug from the bilayer (Fang et al., Citation2001).
Size of carvedilol vesicles
Mean diameter of the prepared carvedilol vesicles was in the range of 295–443 nm as shown in with polydispersity index ranging from 0.08 to 0.2 which indicates that most of the prepared transfersomes are mono-dispersed in the suspension which is a desirable property in such drug delivery system as it enhances stability and decreases the chance of aggregation of the formed particles.
On increasing of the surfactant concentration, the vesicle diameter for all carvedilol-loaded transfersomes was found to decrease (). Small sized-vesicles may be formed at high surfactant concentrations as a result of the larger amount of surfactant that covers the nanoparticles surface which lower the interfacial tension and allow smaller particles to be formed (Dora et al., Citation2010).
According to the effect of the EAs on vesicle size, they can be arranged in the following order: Tween 20 > Span 60 > Span 20 > Tween 80 > SDC. It was reported that the vesicle size is dependent on the molecular structure, HLB, and ionic nature of the surfactant used in their preparation. EAs with low HLB values usually give vesicles with small size which may be due to the decrease in surface energy with the increase in hydrophobicity (El Zaafarany et al., Citation2010). On one hand, these findings are in accordance with Yoshioka et al. (Citation1994) who studied the properties of niosomes prepared from different spans. In contrast, the presence of several ethylene oxide side chains in the EA molecule, such as in Tweens, provides higher steric repulsion in the continuous aqueous phase (Young et al., Citation2004). On the other hand, small vesicles could be obtained on incorporation of charged molecules (e.g. SDC) into the vesicle bilayer because of a steric repulsion between the charged molecules exposed from the outer layer of the vesicle membrane, which increases the vesicle membrane curvature and thus lessens the size of the vesicles (Mu & Zhong, Citation2006). SDC has been reported to result in smaller sized vesicles (Lee et al., Citation2005). In our study, T14-containing SDC as an EA in the ratio of 85:15% w/w (PC:EA) gave the smallest size while T7 containing Tween 20 as an EA in the ratio of 95:5% w/w (PC:EA) gave the largest particle size.
In vitro release study of transfersomes
The release profiles of carvedilol from the transfersomal formulations prepared as well as the control solution are illustrated in . The release rate of carvedilol from all transfersomes was lower than the corresponding control solution. This may be due to the ability of the prepared transfersomes to retard the release of carvedilol. In vitro drug release from transfersomes after 8 h first increased with increasing EA concentration (from 5 to 15% w/w) in the formulation and then decreased which was observed with all EAs. These findings suggested that a too low or high concentration of EA was not suitable for the delivery of carvedilol from transfersomes. Lower drug release at low EA concentrations may be explained by the presence of the lipid membranes in an ordered and less leaky form, which hindered drug release. Similarly, rigid mixed micelles are formed at high EA concentrations which also hindered drug release as a result of being less sensitive to a water activity gradient than transfersomes (El Maghraby et al., Citation2000; Hiruta et al., Citation2006). Therefore, the formulation containing the ratio with optimum release (85:15%, w/w) was selected to compare the release patterns from different EA-containing vesicles. The percentage drug release after 8 h was 42.75, 39.84, 47.34, 43.95, and 59.51% for Span 20, Span 80, Tween 20, Tween 80, and SDC transfersomes, respectively. The differences in molecular ordering caused by EAs may be a reasonable explanation for the observed variations in drug release. The release of carvedilol from transfersomes was in the following order: SDC > Tween 20 > Tween 80 > Span 20 > Span 60.
Figure 1. In vitro release profiles of carvedilol from its transfersomal formulae: (A) T1–T6, (B) T7–T12, and (C) T13–T15.
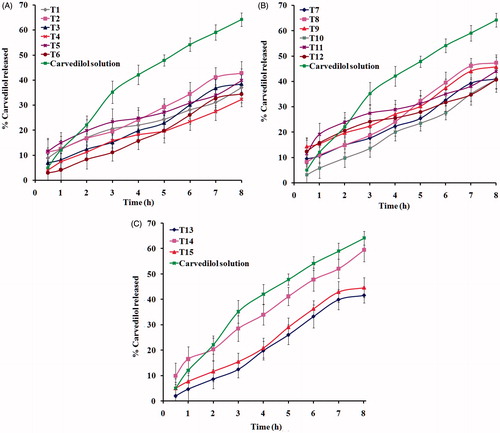
Formula T14-containing SDC in the concentration of 15 % w/w exhibited the highest values for release percentage. This may be due to increased solubility of the drug (Srivastava & Nagappa, Citation2005) as well as increased fluidity of the lipid bilayer at this EA concentration which leads to improved drug permeability and release (Fang et al., Citation2001; El Zaafarany et al., Citation2010). In addition, the small size of T14 vesicles led to an increase in their surface area; this would contribute to the high drug release.
Linear regression analysis of the release data revealed that carvedilol was released from the majority of transfersomes by a diffusion-controlled mechanism except formulae T7, T11, T13, and T15 which followed zero-order kinetics while carvedilol solution followed by first-order kinetics.
Ex vivo permeability study
The diffusion studies were performed on carvedilol-loaded transfersomes and carvedilol solution, the results obtained are presented in and the calculated permeation parameters for each formula along with carvedilol solution are listed in . The permeation parameters including Q24 (µg/cm2), lag time (min), and Kp (cm/h) were used to assess the permeation capability of the tested formulae against carvedilol solution.
Figure 2. Permeation profiles of carvedilol from transfersomal formulae: (A) T1–T6, (B) T7–T12, and (C) T13–T15.
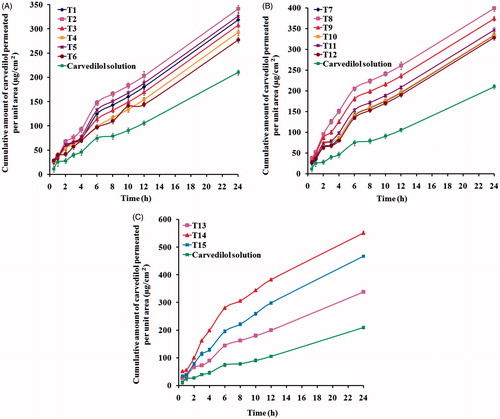
Table 2. Ex vivo permeation parameters of carvedilol-loaded transfersomes versus carvedilol solution.
It can be noticed from the results listed in that the developed transfersomes exhibited a significant higher (p < 0.05) nasal mucosa permeation compared with the control solution containing equivalent amount of carvedilol. Different mechanisms have been proposed for the enhanced delivery by transfersomes (Honeywell-Nguyen & Bouwstra, Citation2003; Honeywell-Nguyen et al., Citation2003). The first mechanism suggests that vesicles can act as drug carrier systems, whereby intact vesicles enter the nasal mucosa carrying loaded drug molecules (Cevc & Blume, Citation2004). The second mechanism suggests that vesicles can act as penetration enhancers, whereby vesicles bilayers open new pores in the paracellular tight junctions when enter the nasal mucosa (Mainardes et al., Citation2006), hence, increasing its fluidity. In addition, phospholipids have a high affinity for biological membranes which contribute to permeability enhancement of transfersomes.
The flexibility of the nanotransfersomes allows them to pass through the nasal mucosa more easily. The extremely high flexibility of the membrane permits nanotransfersomes to squeeze themselves even through pores much smaller than their own diameters (Ahad et al., Citation2012).
Based on the amount of drug permeated and permeability coefficient, transfersomes containing high (25% w/w) or low concentrations (5% w/w) of EAs were not beneficial in vesicular delivery, while transfersomes containing the middle concentration of EAs (15% w/w) exhibited enhanced delivery of carvedilol through the nasal mucosa. As mentioned previously, low concentration of EAs produces vesicles with large size, while at a high surfactant concentration, mixed micelles are formed which are reported to be less deformable in nature and less effective in drug delivery as compared with a transfersomal system. These results correlate well with in vitro drug release results. These findings are consistent with previous studies (El Maghraby et al., Citation2000; Jain et al., Citation2003; Hiruta et al., Citation2006).
In the present study, formula T14 (containing SDC as an EA in the concentration of 15% w/w) exhibited the highest permeability coefficient, highest amount of permeated drug, and the shortest lag time; these results could be attributed to smallest particle size which gave the highest surface area for the diffusion of the drug. The lowest permeation parameters were obtained in formula T4-containing Span 60 as an EA in the concentration of 5% w/w which may be a result of less hydrophilic nature of Span 60 that decreased the formation of transient hydrophilic holes, thus, reducing the amphiphilic property of the bilayers responsible for membrane fluidity (El Zaafarany et al., Citation2010).
CLSM study
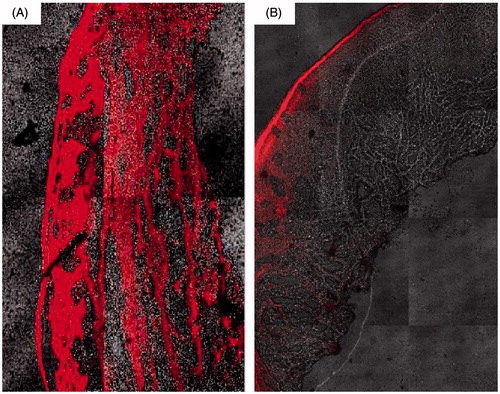
Transfersomal formula T14 was selected for this study as it represents optimum formula due to its acceptably high EE%, smallest particle size, and the best permeation parameters among all other formulae. The extent of vesicular penetration was measured by CLSM after the application of methanolic PBS of rhodamine B and transfersomal formula T14 labeled with rhodamine B. CLSM study results revealed that transfersomal formula T14 was highly penetrated through different layers of nasal mucosa with a high fluorescence intensity and homogeneous distribution as compared with solution of rhodamine B, evidencing the nasal potential of transfersomes as a penetration enhancer, . These results correlate well with ex vivo permeation study.
Figure 3. Confocal images of cross-sections of nasal mucosa after application of (A) rhodamine-loaded transfersomes (T14) and (B) rhodamine solution.
Morphology of carvedilol vesicles
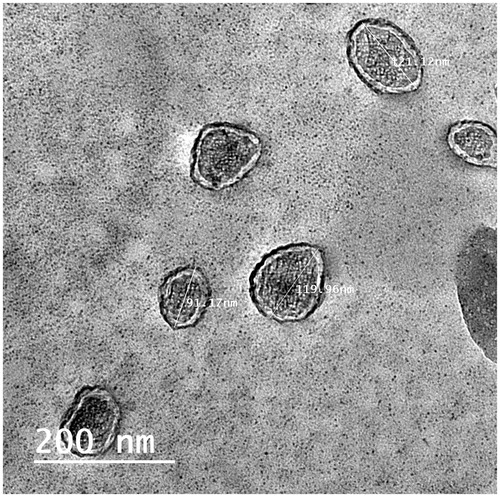
Transmission electron micrograph of formula T14 is shown in . It shows the outline and core of the well-identified ellipsoidal vesicles. They are small unilamellar vesicles with a homogenous size distribution. The vesicles were smooth and showed bilayers' structure of their membrane. They seem to be mono-dispersed with no aggregation or drug crystals.
Figure 4. Transmission electron micrograph of formula T14.
Stability study of transfersomes
Drug EE% as well as particle size of T14 was evaluated after 30, 60, and 90 d. It can be observed from that formula T14 exhibited a decrease in EE% w/w and a slight increase in vesicle diameter which was found to be insignificant.
Figure 5. Effect of storage on the amount of carvedilol retained and vesicle size of transfersomal formula T14.
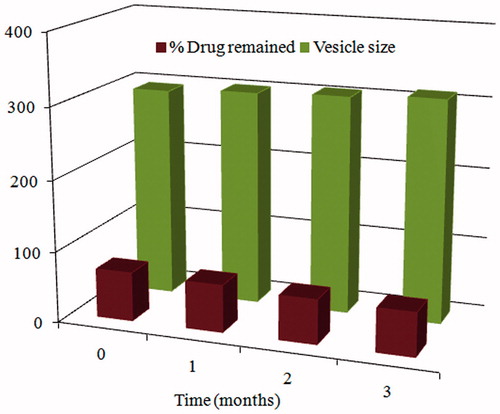
This small increase in size upon storage could be attributed to the lower tendency of SDC-containing vesicles to aggregate due to the weak cohesive forces between the vesicles.
Tolerability and toxicity studies (histopathological studies)
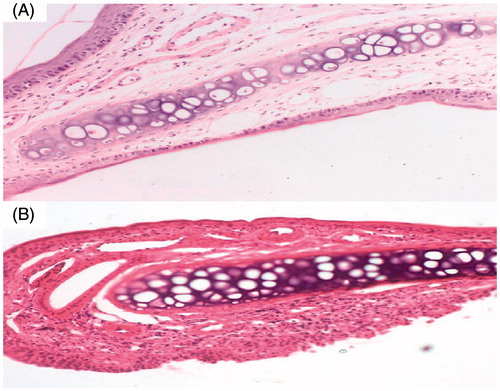
Transfersomes are composed of phospholipids and surfactants, it is necessary to consider potential nasal mucosal irritation of these vesicular formulations. shows light photomicrographs taken from anterior cross-sections of rat nasal cavity following 14 d exposure to in situ gelling transfersomal formula T14. Examination showed that none of the severe signs such as appearance of necrosis, sloughing of epithelial cells, or hemorrhage was detected in any of the rats. However, moderate degenerative changes in olfactory epithelium associated with moderate mononuclear cell infiltrations in the lamina propria were observed.
Figure 6. Light photomicrographs of (A) untreated rat epithelium (B) rat epithelium treated with in situ gelling of carvedilol-loaded transfersomal formula T14.
Pharmacokinetic studies
The LC–MS/MS method was used to assess pharmacokinetic parameters of carvedilol in rabbit plasma to understand the in vivo behavior of the in situ gelling transfersomal formula T14 compared with intravenous solution or oral coarse suspension. The LC–MS/MS assay was validated and confirmed good linearity from 5 to 300 ng/ml with acceptable intra-day and inter-day reproducibilities. The mean plasma drug concentration–time profiles after administration of the different formulations are illustrated in and the corresponding pharmacokinetic parameters are shown in .
Figure 7. Mean carvedilol concentrations in plasma of rabbits after administration of IV solution, in situ gelling transfersomal formula T14 and oral suspension.
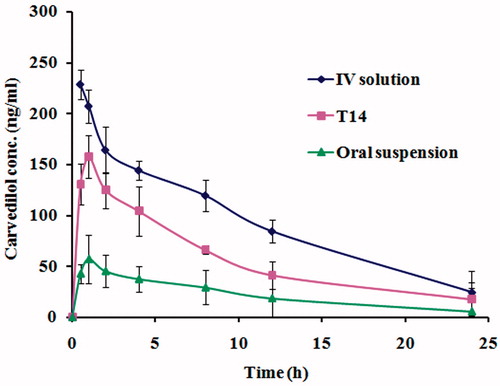
Table 3. Pharmacokinetic parameters for carvedilol in rabbit plasma following administration of intravenous solution, intranasal in situ gelling transfersomal formula T14 and oral suspension.
The mean Cmax and tmax estimates for the in situ gelling transfersomal formula T14 were 157.47 ± 3.45 ng/ml and 1 h, respectively, while the mean Cmax and tmax for the oral suspension were 57.02 ± 4.38 ng/ml and 1 h, respectively. Although T14 exhibited slow release pattern of carvedilol, the in vivo studies showed a low value for tmax. This may be due to interaction of vesicle membrane with serum proteins (opsonins) which induces its lysis and subsequent release of the encapsulated drug into blood circulation (Ishida et al., Citation2002). Additionally, both high density lipoproteins (HDL) and low density lipoproteins (LDL) destabilize vesicle membrane lipid contents, resulting in release of the drug from the vesicles (Allen, Citation1981; Shahrokh et al., Citation1982; Senior, Citation1987; Comiskey & Heath, Citation1990). These findings are in accordance with previously reported results (Salama et al., Citation2012a,Citationb). The results showed that the Cmax of carvedilol in plasma after intranasal administration of in situ gelling T14 was 2.8 times that of oral suspension (p < 0.05). Moreover, the AUC0–∞ values were 2496.86, 1582.97, and 601.21 ng h/ml for the IV solution, the nasal in situ gelling formula T14, and the oral carvedilol suspension, respectively. The absolute drug bioavailability for T14 was higher than that for the oral suspension (63.4% versus 24.1%). The high values of Cmax, AUC0–∞, and absolute bioavailability of the in situ gelling transfersomal vesicles compared with that of the oral suspension might be attributed to avoidance of first pass effect and increase in permeability due to nanotransfersomal formulation which acts as a penetration enhancer.
Conclusion
In this study, transfersomal vesicles containing SDC with a lipid to surfactant ratio of 85:15 w/w can be considered as an efficient nanocarrier for nasal delivery of carvedilol. The developed nanotransfersomal vesicles exhibited absolute bioavailability of 63.4% via the intranasal route which is considered as a remarkable enhancement in the bioavailability of carvedilol as compared with oral drug formulation. Hence, transfersomes could be considered as a valuable mean for the non-invasive nasal delivery of drugs with poor oral bioavailability.
Acknowledgements
The authors would like to thank Prof. Dr. Mahmoud Al-Begawy and Dr. El-Shaymaa Al-Nahass, Department of Pathology, Faculty of Veterinary Medicine, Beni Suef University for helping in histopathological studies and CLSM studies. We also are grateful to Dr. Reda Abd Allah, Department of Anatomy and Embryology, Faculty of Veterinary Medicine, Beni Suef University for helping in trimming nasal mucosa for ex vivo permeability studies and Dr. Ossama M. Sayed, Department of Pharmaceutics and Industrial Pharmacy, Faculty of Pharmacy, Beni Suef University for helping in analysis of plasma samples.
Declaration of interest
The authors report that they have no conflicts of interests.
References
- Aboelwafa AA, El-Setouhy DA, Elmeshad AN. (2010). Comparative study on the effects of some polyoxyethylene alkyl ether and sorbitan fatty acid ester surfactants on the performance of transdermal carvedilol proniosomal gel using experimental design. AAPS PharmSciTech 11:1591–602
- Aburahma MH. (2014). Bile salts-containing vesicles: promising pharmaceutical carriers for oral delivery of poorly water-soluble drugs and peptide/protein-based therapeutics or vaccines. Drug Deliv 12:1–21
- Ahad A, Aqil M, Kohli K, et al. (2012). Formulation and optimization of nanotransfersomes using experimental design technique for accentuated transdermal delivery of valsartan. Nanomedicine 8:237–49
- Al-Ghananeem AM, Sandefer EP, Doll WJ, et al. (2008). Gamma scintigraphy for testing bioequivalence: A case study on two cromolyn sodium nasal spray preparations. Int J Pharm 357:70–6
- Allen TM. (1981). A study of phospholipid interactions between high-density lipoproteins and small unilamellar vesicles. Biochim Biophys Acta 640:385–97
- Alsarra IA, Hamed AY, Alanazi FK. (2008). Acyclovir liposomes for intranasal systemic delivery: development and pharmacokinetics evaluation. Drug Deliv 15:313–21
- Arumugam K, Subramanian GS, Mallayasamy SR, et al. (2008). A study of rivastigmine liposomes for delivery into the brain through intranasal route. Acta Pharm 58:287–97
- Bancroft DJ, Gamble M. (2007). Theory and practice of histological techniques, 7th ed. USA: Churchill Livingstone
- Bangham AD, Standish MM, Watkins JC. (1965). Diffusion of univalent ions across the lamellae of swollen phospholipids. J Mol Biol 13:238–52
- Barenholz Y. (2003). Relevancy of drug loading to liposomal formulation therapeutic efficacy. J Liposome Res 13:1–8
- Callens C, Ceulemans J, Ludwing A, et al. (2003). Rheological study on mucoadhesivity of some nasal powder formulations. Eur J Pharm Biopharm 55:323–8
- Cevc G, Blume G. (2003). Biological activity and characteristics of triamcinolone-acetonide formulated with the self-regulating drug carriers, Transfersomes. Biochim Biophys Acta 1614:156–64
- Cevc G, Blume G. (2004). Hydrocortisone and dexamethasone in very deformable drug carriers have increased biological potency, prolonged effect, and reduced therapeutic dosage. Biochim Biophys Acta 1663:61–73
- Chavanpatil MD, Vavia PR. (2004). The influence of absorption enhancers on nasal absorption of acyclovir. Eur J Pharm Biopharm 57:483–7
- Cheng Y, Watts P, Hinchcliffe M, et al. (2002). Development of a novel nasal nicotine formulation comprising an optimal pulsatile and sustained plasma nicotine profile for smoking cessation. J Control Release 79:243–54
- Comiskey SJ, Heath TD. (1990). Serum-induced leakage of negatively charged liposomes at nanomolar lipid concentrations. Biochemistry 29:3626–31
- Ding WX, Qi XR, Fu Q, Piao HS. (2007). Pharmacokinetics and pharmacodynamics of sterylglucoside-modified liposomes for levonorgestrel delivery via nasal route. Drug Deliv 14:101–4
- Dora CP, Singh SK, Kumar S, et al. (2010). Development and characterization of nanoparticles of glibenclamide by solvent displacement method. Acta Pol Pharm 67:283–90
- Dubey V, Mishra D, Nahar M, Jain NK. (2008). Elastic liposomes mediated transdermal delivery of an anti-jet lag agent: preparation, characterization and in vitro human skin transport study. Curr Drug Deliv 5:199–206
- El Maghraby GM, Williams AC, Barry BW. (2000). Oestradiol skin delivery from ultradeformable liposomes: refinement of surfactant concentration. Int J Pharm 196:63–74
- El Zaafarany GM, Awad GA, Holayel SM, Mortada ND. (2010). Role of edge activators and surface charge in developing ultradeformable vesicles with enhanced skin delivery. Int J Pharm 397:164–72
- Fang JY, Yu SY, Wu PC, et al. (2001). In vitro skin permeation of estradiol from various proniosome formulations. Int J Pharm 215: 91–9
- Gavini E, Rassu G, Sanna V, et al. (2005). Mucoadhesive microspheres for nasal administration of an antiemetic drug, metoclopramide: in-vitro/ex-vivo studies. J Pharm Pharmacol 57:287–94
- Ghirardelli R, Bonasoro F, Porta C, Cremaschi D. (1999). Identification of particular epithelial areas and cells that transport polypeptide-coated nanoparticles in the nasal respiratory mucosa of the rabbit. Biochim Biophys Acta 1416:39–47
- Guinedi AS, Mortada ND, Mansour S, Hathout RM. (2005). Preparation and evaluation of reverse-phase evaporation and multilamellar niosomes as ophthalmic carriers of acetazolamide. Int J Pharm 306:71–82
- Gupta A, Aggarwal G, Singla S, Arora R. (2012). Transfersomes: a novel vesicular carrier for enhanced transdermal delivery of sertraline: development, characterization and performance evaluation. Sci Pharm 80:1061–80
- Haggag RS, Shaalan RA, Belal TS. (2010). Validated HPLC determination of the two fixed dose combinations (chlordiazepoxide hydrochloride and mebeverine hydrochloride; carvedilol and hydrochlorothiazide) in their tablets. J AOAC Int 93:1192–200
- Harris AS, Ohlin M, Svensson E, et al. (2006). Effect of viscosity on the pharmacokinetics and biological response to intranasal desmopressin. J Pharm Sci 78:470–1
- Hiruta Y, Hattori Y, Kawano K, et al. (2006). Novel ultra-deformable vesicles entrapped with bleomycin and enhanced to penetrate rat skin. J Control Release 113:146–54
- Honeywell-Nguyen PL, Arenja S, Bouwstra JA. (2003). Skin penetration and mechanisms of action in the delivery of the D2-agonist rotigotine from surfactant-based elastic vesicle formulations. Pharm Res 20:1619–25
- Honeywell-Nguyen PL, Bouwstra JA. (2003). The in vitro transport of pergolide from surfactant-based elastic vesicles through human skin: a suggested mechanism of action. J Control Release 86:145–56
- Ishida T, Harashima H, Kiwada H. (2002). Liposome clearance. Biosci Rep 22:197–224
- Jain S, Jain P, Umamaheshwari RB, Jain NK. (2003). Transfersomes – a novel vesicular carrier for enhanced transdermal delivery: development, characterization, and performance evaluation. Drug Dev Ind Pharm 29:1013–26
- Kim S, Lee HS, Lee HJ. (2010). Rapid and sensitive carvedilol assay in human plasma using a high-performance liquid chromatography with mass/mass spectrometer detection employed for a bioequivalence study. AJAC 1:135–43
- Kumar A, Pathak K, Bali V. (2012). Ultra-adaptable nanovesicular systems: a carrier for systemic delivery of therapeutic agents. Drug Discov Today 17:1233–41
- Lee EH, Kim A, Oh YK, Kim CK. (2005). Effect of edge activators on the formation and transfection efficiency of ultradeformable liposomes. Biomaterials 26:205–10
- Lopes LB, Scarpa MV, Silva GV, et al. (2004). Studies on the encapsulation of diclofenac in small unilamellar liposomes of soya phosphatidylcholine. Colloids Surf B Biointerfaces 39:151–8
- Mahajan HS, Gattani S. (2010). In situ gels of metoclopramide hydrochloride for intranasal delivery: in vitro evaluation and in vivo pharmacokinetic study in rabbits. Drug Deliv 17:19–27
- Mainardes RM, Urban MC, Cinto PO, et al. (2006). Liposomes and micro/nanoparticles as colloidal carriers for nasal drug delivery. Curr Drug Deliv 3:275–85
- Manconi M, Mura S, Sinico C, et al. (2009). Development and characterization of liposomes containing glycols as carriers for Diclofenac. Colloids Surf A: Physicochem Eng Asp 342:53–8
- Manconi M, Valenti D, Sinico C, et al. (2003). Niosomes as carriers for tretinoin: II. Influence of vesicular incorporation on tretinoin photostability. Int J Pharm 260:261–72
- Mao S, Chen J, Wei Z, et al. 2004. Intranasal administration of melatonin starch microspheres. Int J Pharm 272:37–43
- Marttin E, Verhoef JC, Chullander C, et al. (1997). Confocal laser scanning microscopic visualization of the transport of dextran after nasal administration to rats: effects of absorption enhancers. Pharm Res 14:631–7
- Mu X, Zhong Z. (2006). Preparation and properties of poly(vinly alcohol)-stabilized liposomes. Int J Pharm 318:55–61
- Muramatsu K, Maitani Y, Takayama K, Nagai T. (1999). The relationship between the rigidity of the liposomal membrane and the absorption of insulin after nasal administration of liposomes modified with an enhancer containing insulin in rabbits. Drug Dev Ind Pharm 25:1099–105
- Nasr M, Mansour S, Mortada ND, Elshamy AA. (2008). Vesicular aceclofenac systems: a comparative study between liposomes and niosomes. J Microencapsul 25:499–512
- Nema T, Jain A, Jain A, et al. (2013). Insulin delivery through nasal route using thiolated microspheres. Drug Deliv 20:210–15
- Nii T, Ishii F. (2005). Encapsulation efficiency of water-soluble and insoluble drugs in liposomes prepared by the microencapsulation vesicle method. Int J Pharm 298:198–205
- Ning MY, Guo YZ, Pan HZ, et al. (2005). Preparation and evaluation of proliposomes containing clotrimazole. Chem Pharm Bull 53:620–4
- Ozsoy Y, Gungor S, Cevher E. (2009). Nasal delivery of high molecular weight drugs. Molecules 14:3754–79
- Packer M, Fowler MB, Roecker EB, et al. (2002). Effect of carvedilol on the morbidity of patients with severe chronic heart failure: results of the carvedilol prospective randomized cumulative survival (COPERNICUS) study. Circulation 106:2194–9
- Quadir M, Zia H, Needham TE. (2000). Development and evaluation of nasal formulations of ketorolac. Drug Deliv 7:223–9
- Rajanikanth PS, Sankar C, Mishra B. (2003). Sodium alginate microspheres of metoprolol tartrate for intranasal systemic delivery: development and evaluation. Drug Deliv 10:21–8
- Salama HA, Mahmoud AA, Kamel AO, et al. (2012a). Brain delivery of olanzapine by intranasal administration of transfersomal vesicles. J Liposome Res 22:336–45
- Salama HA, Mahmoud AA, Kamel AO, et al. (2012b). Phospholipid based colloidal poloxamer–nanocubic vesicles for brain targeting via the nasal route. Colloids Surf B Biointerfaces 100:146–54
- Senior JH. (1987). Fate and behavior of liposomes in vivo: a review of controlling factors. Crit Rev Ther Drug Carrier Syst 3:123–93
- Shahrokh Z, Nichols AV. (1982). Particle size interconversion of human low density lipoproteins during incubation of plasma with phosphatidylcholine vesicles. Biochem Biophys Res Commun 108:888–95
- Srivastava RC, Nagappa AN. (2005). Application of surface activity in therapeutics, 1st ed., Amsterdam: Elsevier, North-Holland Biomedical Press
- Tengamnuay P, Sahamethapat A, Sailasuta A, Mitra AK. (2000). Chitosans as nasal absorption enhancers of peptides: comparison between free amine chitosans and soluble salts. Int J Pharm 197:53–67
- Thummel KE, Shen DD. (2001). Design and optimization of dosage regimens: pharmacokinetic data. In: Hardman JG, Limbirel LE, Gilman AG, eds. Goodman and Gilman's. The pharmacological basis of therapeutics, 10th ed., New York: McGraw Hill, 1936–40
- Van den Bergh BA, Wertz PW, Junginger HE, Bouwstra JA. (2001). Elasticity of vesicles assessed by electron spin resonance, electron microscopy and extrusion measurements. Int J Pharm 217:13–24
- Varshosaz J, Sadrai H, Heidari A. (2006). Nasal delivery of insulin using bioadhesive chitosan gels. Drug Deliv 13:1–38
- Vyas SP, Goswami SK, Singh R. (1995). Liposomes based nasal delivery system of nifedipine: development and characterization. Int J Pharm 118:23–30
- Yoshioka T, Sternberg B, Florence AT. (1994). Preparation and properties of vesicles (niosomes) of sorbitan monoesters (Span 20, 40, 60, and 80) and a sorbitan triester (Span 85). Int J Pharm 105:1–6
- Young JT, Johnston PK, Pace WG, Mishra KA. (2004). Phospholipid-stabilized nanoparticles of cyclosporine A by rapid expansion from supercritical to aqueous solution. AAPS PharmSciTech 5:E11
- Zaki NM, Awad GA, Mortada ND, Abd Elhady SS. (2007). Enhanced bioavailability of metoclopramide HCl by intranasal administration of a mucoadhesive in situ gel with modulated rheological and mucociliary transport properties. Eur J Pharm Sci 32:296–307