
Abstract
The aim of this work was to investigate the transdermal gel loaded with tadalafil, a practically insoluble selective phosphodiesterase-5 inhibitor (PDE5) in order to improve the solubility and bioavailability. The solubility of tadalafil in mixed solution of hydroxypropyl-β-cyclodextrin (HPCD), polyethylene glycol (PEG) 400 and tween 80 (T2 solution) was 260.8 ± 4.3 µg/mL and that of tadalafil in modified T2 (M-T2) solution, which tadalafil was dissolved in 20% (w/v) HPCD at first and then mixture solutions of PEG 400 and tween 80 were added, was increased to 344.9 ± 30.6 µg/mL. Four gel formulae were prepared, subsequently in vitro and in vivo skin permeation studies were carried out. Interestingly, tadalafil gel in M-T2 and oleic acid (OA) (F3) could promote the percutaneous absorption of tadalafil by 179.4% in vitro and increase AUC by 223% in vivo compared with tadalafil gel in the absence of M-T2 and OA (F1). Also, there was a finding that tadalafil gel in M-T2 and OA did not cause dermal irritations in an experimental animal.
Introduction
Tadalafil is a recently approved phosphodiesterase-5 (PDE5) inhibitor which was officially born in 2000 and then approved by the FDA in 2003, indicated for the treatment of erectile dysfunction. In comparison to other PDE5 inhibitor, it is the most potent and at least 9000 times more selective than other PDE5 inhibitor. Compared with sildenafil and vardenafil, tadalafil is much less inhibitory for PDE6, consequently, tadalafil has less than 0.1% occurrence of vision abnormalities (Brock et al., Citation2002; Carson et al., Citation2005). Due to its therapeutic window of 36 h (long duration of action) and minimum potential to cause vision abnormalities, tadalafil has gained wide clinical acceptance for the treatment of erectile dysfunction even in difficult-to-treat cases (Coward & Carson, Citation2008). However, tadalafil with low-solubility and high-permeability is classified as Class II drug within the FDA biopharmaceutical classification system (Chavda et al., Citation2010), which leads to its poor dissolution in the gastrointestinal tract, resulting in variable bioavailability (Lobenberg & Amidon, Citation2000). Tadalafil has the slowest absorption of the available PDE5 inhibitors with a mean of 2 h to reach its maximum concentration, compared with about 50 min for sildenafil and vardenafil (Briganti et al., Citation2005).
Poor solubility and hence poor bioavailability often result in limited or irreproducible clinical response of the drug. Therefore, various techniques have been used in attempt to enhance a bioavailability as alternative formulations which include solid dispersion, micronization, lipid-based formulations, liquid solid compacts, and complexation (Saharan et al., Citation2009).
However, to our knowledge, there is little work done in the field of transdermal delivery system (TDDS) of tadalafil. TDDS have introduced as an attractive alternative to traditional oral and hypodermic delivery, as they overcome the limitation of first-pass liver metabolism encountered by oral administration and have a lot of advantages such as safe, painless, and easy to use, in contrast to intravenous injection (El Maghraby et al., Citation2009).
On the contrary, several analgesic preparations are available in the market with gel, cream, ointment, lotion or patch as well as tablet or capsule. But, tadalafil is only available in the form of tablets. There is no marketed transdermal formulation of tadalafil till date. The effectiveness of transdermal drug delivery depends on the drug’s ability to penetrate the skin sufficiently to reach therapeutic level (Dos Anjos & Alonso, Citation2008). The stratum corneum is the outermost epidermal layer that is composed of corneocytes surrounded by a multilamellar lipid matrix and is a rate limiting factor against many drugs (Bouwstra & Honeywell-Nguyen, Citation2002). Corneocytes are keratin filled dead cells containing an insoluble layer, which reduces absorption of drugs into the cells. To overcome this skin barrier problem, the most widely implemented approach is the use of chemical penetration enhancers, which ideally alter the physicochemical nature of the stratum corneum safely and reversibly to facilitate the drug’s delivery through the skin (Verma & Pathak, Citation2010).
Recently, it was reported that we prepared the pharmaceutical composition (T2) consisting of 15% PEG 400, 2.5% tween 80, and 20% HPCD, subsequently T2 showed the solubility enhancement and skin permeability of zaltoprofen (Baek et al., Citation2013). In the present study, we investigated whether tadalafil gel containing the combination of pharmaceutical compositions (PEG400, tween 80, and HPCD, called as T2) and oleic acid (OA) could promote the percutaneous absorption of tadalafil.
Materials and methods
Materials
Tadalafil was obtained from Korea United Pharm. Inc. (Seoul, Korea). 2-Hydroxypropyl-β-cyclodextrin (HPCD) was purchased from Sigma-Aldrich (Steinheim, Germany). Polyethylene glycol (PEG) 400, tween 80, OA, and triethanolamine were purchased from Samchun chemical Co. Ltd (Pyungtaek, Korea). HPLC grade acetonitrile was from JT Baker (Phillipsburg, NJ). All the solvents were of analytical grade and used without further purification.
Solubility determination
An excess amount of tadalafil was added to microcentrifuge tubes containing 1 mL of various solvents such as D.W., 20% (w/v) HPCD solution, 15% (w/v) PEG solution, 2.5% (w/v) tween 80 solution, which was called as T2 and T2 solution or solution without one component from T2. In case of modified T2 (M-T2), tadalafil was dissolved in 20% (w/v) HPCD at first and then mixture solutions of PEG and tween 80 were added. The samples were put on an end-to-end labquake rotator (Barnstead Thermolyne, Sparks, NV) at 50 rpm at an ambient temperature for 72 h in order to achieve equilibrium. The samples were filtered with a 0.45 µm membrane filter (Whatman, Dismic-25, Whatman Ltd., Tokyo, Japan) and the absorbance of the filtrate was measured by HPLC. All solubility determinations were performed in triplicate. Also, the octanol/water partition coefficient of tadalafil was determined by the shake-flask method (De Mello et al., Citation2004). About 1.0 mg of tadalafil was weighed and dissolved in the same volume of 1-octanol and water. The microcentrifuge tubes were stoppered to prevent solvent evaporation. The flasks were then shaken horizontally at ambient temperature on an end-to-end labquake rotator (Barnstead Thermolyne, Sparks, NV) at 8 rpm for 72 h and then allowed to stand for another 30 min. The mixtures were then centrifuged for 15 min at 3000 rpm. Each phase was analyzed by HPLC. Each determination was performed at least three times and the mean values were used to produce the log p values.
Preparation of tadalafil gel
The formulations and composition of tadalafil gel are shown in . Briefly, 500 mg of carbomer 940 was dispersed into distilled water under stirring to form homogeneous solution. Twenty-five milligrams of tadalafil were dissolved with T2 solutions and then carefully added to the carbomer solution with continuous stirring until uniformly dispersed. Other penetration enhancer was also added simultaneously. Then distilled water was added to adjust a final volume of 50 mL. The gelation was achieved by the addition of triethanolamine. All 0.05% tadalafil gels were stored at room temperature prior to use.
Table 1. Composition of tadalafil gel.
HPLC analysis
HPLC analysis of tadalafil from buffer and rat plasma was performed with slight modification (Farthing et al., Citation2010). The mobile phase composed of acetonitrile and water (30:70, v/v) was filtered (0.45 µm), degassed, and delivered at a flow rate of 1.2 mL/min. The column temperature was maintained at 25 °C, injection volume was 20 µL and tadalafil was detected at 285 nm.
A stock standard solution of tadalafil was prepared in ethanol and calibration samples were prepared by spiking blank plasma samples with this stock solution to obtain final concentrations of 1, 2, 5, 10, and 15 µg/mL. All solutions were stored at 4 °C before analysis. Quality control (QC) samples containing various concentrations of tadalafil (1, 2, and 10 µg/mL) were prepared in a similar manner. Intra-day precision was determined by obtaining three concentrations of tadalafil in QC samples, three times on the same day. Inter-day precision was determined by successive concentration determinations over a 3 d period. Precision is indicated as the coefficient of variation (RSD). Accuracy was calculated from the mean value of the observed concentrations and the theoretical concentrations.
In vitro skin permeation study
Dorsal skin was excised from experimental rat, whose hair had been previously removed. Adhering fat and other visceral tissue was removed carefully. The excised rat skin was stored at −20 °C prior to use. Franz-type diffusion cells with an effective diffusion area of 1.81 cm2 with the diameter of 16 mm and a receptor volume of 11.8 mL were used to assess in vitro drug permeation. The receptor compartment was kept at 37 °C. The receptor fluid was selected as pH 7.4 phosphate buffer solution containing 1% tween 80 and stirred continuously with magnetic stirrer at 500 rpm. Each formulation (1.0 g) was placed in the donor compartment. Permeation experiments were carried out until 24 h after application. Samples were taken from the receiver compartment at scheduled time intervals (0, 1, 2, 4, 8, 12, and 24 h) and immediately replaced with the same volume of fresh receptor fluid. The amount of tadalafil in the samples was determined by HPLC. This animal experiment procedure was approved by the Animal Ethics Board of Chungnam National University.
Mathematical modeling regarding release profile
In our study, the drug release rates were evaluated according to the simplified Higuchi diffusion equation (Equation1(1) ), depicting the drug release from one side of a semisolid layer in which the drug is dissolved
(1)
where q is the amount of drug released into the receptor medium per unit area of exposure, C0 is the initial drug concentration in vehicle, D is the apparent diffusion coefficient of drug, and t is the time elapsed since the start of drug release. In case of passive diffusion, the steady-state flux through unit area of a membrane is given by Fick’s law
(2)
where J is the flux per unit area, P represents the permeability coefficient and Cd and Cr are the concentrations in the donor and receptor solutions, respectively. In case, sink conditions are maintained on the receptor side, (Cd − Cr) is replaced by Cd
(3)
The permeability coefficient, P, is the constant for a given drug under the same experimental condition. There should be a linear relationship between the flux and the donor concentration.
Cytotoxicity study
HaCaT and Pam212 cell lines for cytotoxicity study were utilized. The murine epidermal cell line (PAM212) was kindly donated by Dr. B. K. Kang (Chonnam National University, Gwangju, Korea) and the immortalized human keratinocyte cell line (HaCaT) was from Dr. S. K. Kim (Chungnam National University, Daejeon, Korea). Cells were grown in DMEM supplemented with 10% fetal calf serum, 2 mM l-glutamine, 100 units/mL penicillin, and 100 µg/mL streptomycin in 5% CO2 atmosphere at 37 °C. The cells were exposed to a series of concentrations (0.001–10 µM) of tadalafil solution (dissolved in DMSO) and tadalafil gel, where the final concentration of DMSO in the culture medium was below 0.5%. After a 72 h incubation of the cells (70% confluent) with tadalafil solution or gel, cytotoxicity was determined by MTT assay according to the manufacturer’s protocol. Briefly, after incubation of cells with vehicles of gel, MTT (3-(4,5-dimethylthoazol-2yl)-2,5-diphenyl-2H-tetrazolium bromide) was added to each well and incubated for 2 h at 37 °C. The crystals of viable cells were solubilized in isopropanol. The absorbance was determined at 570 nm in a microplate reader (sunrise, Tecan, Grödig, Austria). Cell viability was expressed as the percent absorbance exposed to vehicles of gel relative to absorbance measured for cells that were not exposed to any material.
Skin irritation test
All animal studies were conducted in accordance with the “Guiding Principles in the Use of Animals in Toxicology” adopted by the Society of Toxicology (Orlando, FL) and the experimental protocols were approved by the Animal Care Committee of Chungnam National University. Skin irritation test was used on experimental rat to evaluate the irritation potential of tadalafil gel formulations (Bachhav & Pratravale, Citation2010; Ganeshpurkar et al., Citation2014). The back of the rat was shaved 24 h prior to the formulation application. Formulation was applied on the hair-free skin of rats by uniform spreading over an area of 9 cm2. The skin surface was observed for any visible change such as erythema (redness) after 24, 48, and 72 h of the formulation application. The mean erythemal scores were recorded depending on the degree of erythema: no erythema = 0, slight erythema (barely perceptible light pink) = 1, moderate erythema (dark pink) = 2, moderate to severe erythema (light red) = 3 and severe erythema (extreme redness) = 4.
In vivo studies
Male Sprague–Dawley (SD) rats weighing 160–200 g were purchased from the Samtako (Chungbuk, Korea). The rats were given commercial rat chow diet (No. 322-7-1) purchased from the Superfeed Co. (Gangwon, Korea) and tap water ad libitum. The animals were housed three per cage in laminar flow that were maintained at 22 ± 2 °C, and 50–60% relative humidity. The animals were kept in these facilities for at least 1 week prior to the experiment and were fasted for at least 24 h before commencing the experiments. Nine SD rats were employed in the in vivo study. Dorsal hairs were removed by a hair clipper and an area of 9 cm2 without hair was constituted. About 10 g of tadalafil gels (20 mg/kg tadalafil) were administered homogenously over this area. Blood samples (0.5 mL) were collected from the orbital vein before drug administration (0 h) and at the end of 1, 2, 4, 8, 12, and 24 h following percutaneous application of tadalafil gel. Tadalafil was extracted from the rat plasma sample using a modification of the procedure (Onishi et al., Citation2005). One hundred µL of the plasma sample was placed into a 15 mL centrifuge tube, and 100 µL of 1 N HCl was added. They were then extracted with 5 mL of ether. After vortexing for 10 min, the mixture was centrifuged at 4000 rpm for 15 min, the organic phase was transferred to the other tubes, and evaporated to dryness using a vacuum system. The residues were dissolved in 400 µL of acetonitrile and 20 µL aliquots were injected into the HPLC system for analysis.
Pharmacokinetic analysis
Non-compartmental pharmacokinetic analysis was carried out using the WinNonlin Standard Edition Version 2.1 (Pharsight, Mountain View, CA). The maximum plasma concentration (Cmax) and the time to reach the maximum plasma concentration (Tmax) were obtained from the experimental data. The area-under-the-plasma concentration curve as a function of time (AUC0–t) from time zero to the time of the last measured concentration (Clast) was calculated using the linear trapezoidal rule.
Statistical analysis
Student’s t-test was used to compare two different groups of samples. A p value <0.05 was considered significant.
Results and discussion
Solubility test
In order to select the solvent or vehicle to prepare the transdermal formulations of tadalafil, the solubility against various solvents was determined. Tadalafil was practically insoluble in water and its solubility in water was 1.2 ± 0.7 µg/mL. Also, the partition coefficient value (log P) corresponding to the quotient between aqueous and 1-octanol concentration of the molecule was 1.7 ± 0.2, suggesting a hydrophobic molecule. Tadalafil in HPCD and tween 80 mixed solutions showed the solubility of 237.3 ± 8.9 µg/mL. Tadalafil in PEG 400 and tween 80 mixed solutions exhibited that of 153.5 ± 3.4 µg/mL, tadalafil in HPCD and PEG 400 mixed solutions showed that of 220.5 ± 12.5 µg/mL. Tadalafil in HPCD solution alone represented the solubility of 255.7 ± 18.9 µg/mL. The solubility of tadalafil in mixed solution of HPCD, PEG 400, and tween 80 (T2 solution) was 260.8 ± 4.3 µg/mL. Interestingly, the solubility of tadalafil in M-T2 solution was increased to 344.9 ± 30.6 µg/mL (). It was suggested that HPCD played an important role in increasing the solubility of tadalafil from these results. There was a publication that large hydrophilic substituents (e.g., hydroxypropyl groups) at the entrance of the cyclodextrin cavity enhance the solubility of the cyclodextrin itself, but at the same time, they reduce the depth that ibuprofen can reach into the cavity. It yields the highest solubility of ibuprofen (Cagno et al., Citation2011).
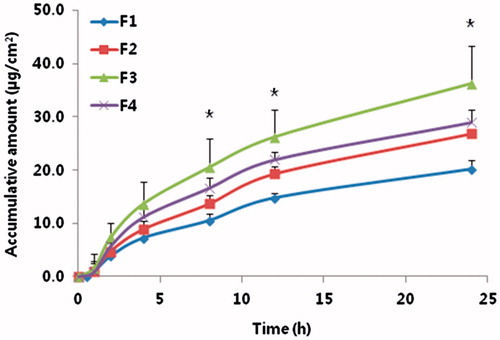
Figure 1. In vitro cumulative amount of tadalafil permeated (µg/cm2) through excised rat skin up to 24 h.
Precision and accuracy
HPLC with isocratic elution consisting of acetonitrile–water (30:70 v/v) yielded baseline resolution for tadalafil. Other endogenous plasma constituents interfered with tadalafil was confirmed by inspecting chromatograms derived from processed blank plasma samples. The retention time of tadalafil was 4.7 min, with no endogenous interference or matrix effect. Calibration curves were generated by plotting chromatographic peak area as a function of tadalafil concentration. Peak areas of tadalafil in rat plasma all displayed good linearity, described by the following regression line: y = 22.562x + 0.482 (where y is the peak area, and x is the tadalafil concentration in µg/mL). The intra-day and the inter-day precision determined from replicate analysis of QC samples were less than 2.4–3.9% and 1.8–2.4%, and the accuracy of the method ranged from 92.8 to 96.9% and 88.6 to 93.5%, as shown in . The results demonstrate that the values are within the acceptable (Guidance for the Validation of Analytical Methodology and Calibration of Equipment used for Testing of Illicit Drugs in Seized Materials and Biological Specimens, 2009) and the method is accurate and precise.
Table 2. The intra-day and inter-day precision and accuracy values for tadalafil in blank plasma.
In vitro skin permeation study
The cumulative permeated amount of tadalafil was increased linearly progressively with time (). In the presence of T2 and OA (F3), the permeation of tadalafil was remarkably enhanced compared with that of the control group (F1). The order of flux enhancement on tadalafil permeation from tadalafil gel was F3 > F4 > F2 > F1 (). It was suggested when OA was added, the synergic effect of permeation into skin was observed.
Figure 2. Cytotoxicity of tadalafil solution and gel against. (a) HaCaT cells and (b) Pam212 cells.
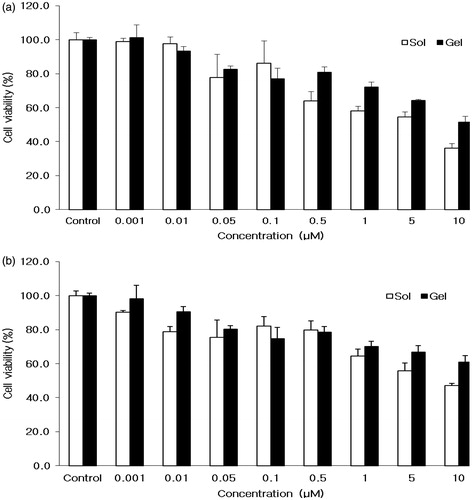
Table 3. Permeation parameters of tadalafil from the four different tadalafil gels through the rat skin.
OA is one of the fatty acids found abundantly in nature, including in the human skin. The use of OA as an absorption enhancer may be a useful tool to lessen the barrier function of the skin. Its capacity as a skin penetration enhancer has been widely recognized in various studies (Tanojo et al., Citation1999; Touitou et al., Citation2002; Da Silva et al., Citation2011). Specially, OA was found to be an effective enhancer for many NSAIDs (Takeuchi et al., Citation1992; Ogiso et al., Citation1995). OA can interact with stratum corneum lipids and disrupt their structures, increasing their fluidity and consequently increasing the flux (Ongpipattanakul et al., Citation1991). It has also been suggested that the major mechanism of enhancement of OA is an increase in permeation through the non-polar route, as it increases both diffusivity and partitioning (Naik et al., Citation1995).
Cytotoxicity study
To determine the cytotoxic activity of tadalafil gel formulation, tadalafil solution or tadalafil gel was tested against murine PAM212 and human HaCaT keratinocytes cells. After incubation for 72 h, cytotoxicity of vehicle with tadalafil to both PAM212 and HaCaT cells was observed according to its concentration up to 10 µM (). On one hand, the cell viability at 10 µM of tadalafil solution in HaCaT and PAM212 cells was 36.2 ± 2.8 and 47.1 ± 1.2%, respectively. On the other hand, the cell viability at 10 µM of tadalafil gel in HaCaT and PAM212 cells were 51.4 ± 3.8 and 60.9 ± 3.8%, respectively. With the increase in the concentration of tadalafil solution or tadalafil gel, the cell viability was decreased. Interestingly, the cell viability of tadalafil gel was higher than that of tadalafil solution in both cell lines due to DMSO. This result was due to absence of organic solvent in gel formulation (Nair et al., Citation1990).
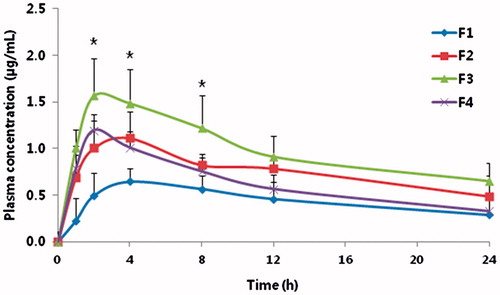
Figure 3. Mean plasma concentration–time profiles of tadalafil after transdermal administration of tadalafil gel.
Skin irritation test
Irritation in rat skins was visually evaluated after topical application of tadalafil gel formulation (F3) which showed the highest in vitro percutaneous absorption. F3 was devoid of any irritation potential and no edema formation was observed in any case. Irritation score (primary skin irritation index) for F3 was zero, which indicated its safety and acceptability for topical administration (data not shown).
In vivo study
shows the mean plasma concentration-time profiles of tadalafil from different tadalafil gels. Pharmacokinetic parameters calculated from the plasma drug concentration versus time profiles are listed in . AUC0–∞ of F2, F3, or F4 were increased slightly by 174, 223, or 201% compared with F1, respectively. Also, the Cmax of F2, F3, or F4 were increased by 157, 229, or 171% compared with F1, respectively. Consequently, the relative bioavailability of tadalafil in the rats treated with the tadalafil gel containing M-T2 (F2), containing M-T2, and OA (F3) and containing OA (F4) was higher than that of F1 (not containing T2 and OA). These results are correlated with in vitro permeation results. F3 showed 179.4% increase of permeation compared with F1 by in vitro permeation results and 223% increase of AUC compared with F1 by in vivo pharmacokinetic results. Interestingly, F2 and F4 showed 133.2 and 143.7% increase of permeation compared with F1 by in vitro permeation results but 174 and 201% increase of AUC compared with F1 by in vivo pharmacokinetic results, respectively.
Table 4. Mean pharmacokinetic parameters of tadalafil gels after transdermal delivery.
Taken together, in vitro and in vivo permeation results of tadalafil had a correlation. Considered, surfactants can be localized inside the bio membrane and thereby enhance drug permeation mainly through interactions with intercellular lipids in the stratum corneum (SC) and the creation of a perturbed microenvironment among lipid alkyl chains and polar head groups (Wonglertnirant et al., Citation2012), PEG 400 has been reported to be an excellent solubilizer for many drugs (Chien & Lambert, Citation1975), HPCD has been reported to be a good solubilizer for many drugs and skin permeability enhancer (Felton et al., Citation2002; Nasongkla et al., Citation2003; Cho et al., Citation2006), tween 80 has also been reported to be a good solubilizer for many drugs and skin permeation enhancer (Shinha & Kaur, Citation2000; Som et al., Citation2012). Therefore, the enhancement of in vitro and in vivo skin permeation could explain by increase of solubility of tadalafil due to M-T2.
Conclusion
Transdermal drug delivery system of tadalafil was studied through the improvement of solubility of tadalafil using the mixed solution of HPCD, PEG 400, and tween 80. The AUC of tadalafil gel showing the highest in vitro permeation showed 223% increase compared with tadalafil gel only. Also, in vitro and in vivo permeation results of tadalafil had a correlation.
Declaration of interest
The authors report that they have no conflicts of interest. This research was supported by the Priority Research Centers Program (2009-0093815) through the National Research Foundation of Korea (NRF) funded by the Ministry of Education, Science and Technology.
References
- Bachhav YG, Patravale VB. (2010). Formulation of meloxicam gel for topical application: in vitro and in vivo evaluation. Acta Pharm 60:153–63
- Baek JS, Lim JH, Kang JS, et al. (2013). Enhanced transdermal drug delivery of zaltoprofen using a novel formulation. Int J Pharm 453:358–62
- Bouwstra JA, Honeywell-Nguyen PL. (2002). Skin structure and mode of actions of vehicles. Adv Drug Deliv Rev 54:S41–55
- Briganti A, Salonia A, Gallina A, et al. (2005). Drug insight: oral phosphodiesterase type 5 inhibitors for erectile dysfunction. Nat Clin Pract Urol 2:239–47
- Brock GB, McMahon CG, Chen KK, et al. (2002). Efficacy and safety of tadalafil for the treatment of erectile dysfunction: results of integrated analyses. J Urol 168:1332–6
- Cagno M, Stein PC, Skalko-Basnet N, et al. (2011). Solubilization of ibuprofen with β-cyclodextrin derivatives: energetic and structural studies. J Pharm Biomed Anal 55:446–51
- Carson C, Shabsigh R, Seqal S, et al. (2005). Trial Evaluating the Activity of Tadalafil for Erectile Dysfunction-United Stated (TREATED-US) Study Group. Efficacy, safety, and treatment satisfaction of tadalafil versus placebo in patients with erectile dysfunction evaluated at tertiary care academic centers. Urology 65:353–69
- Chavda HV, Patel CN, Anand IS. (2010). Biopharmaceutics classification system. Syst Rev 1:62–9
- Chien YW, Lambert HJ. (1975). Solubilization of steroids by multiple cosolvent systems. Chem Pharm Bull 23:1085–90
- Cho SH, Kim SY, Lee SI, Lee MY. (2006). Hydroxypropyl-β-cyclodetrin inclusion complexes for transdermal delivery: preparation, inclusion properties, stability, and release behavior. J Ind Eng Chem 12:50–9
- Coward RM, Carson CC. (2008). Tadalafil in the treatment of erectile dysfunction. Ther Clin Risk Manaq 4:1315–30
- Da Silva ER, de Freitas ZM, Gitirana LDEB, Ricci-Junior E. (2011). Improving the topical delivery of zinc phthalocyanine using oleic acid as a penetration enhancer: in vitro permeation and retention. Drug Dev Ind Pharm 37:569–75
- De Mello H, Echevarria A, Bernardino AM, et al. (2004). Antileishmanial pyrazolopyridine derivatives: synthesis and structure–activity relationship analysis. J Med Chem 47:5427–32
- Dos Anjos JLV, Alonso A. (2008). Terpenes increase the portioning and molecular dynamics of an amphipathic spin label in stratum corneum membranes. Int J Pharm 350:103–12
- El Maghraby GM, Alanazi FK, Alsarra IA. (2009). Transdermal delivery of tadalafil. I. Effect of vehicles on skin permeation. Drug Dev Ind Pharm 35:329–36
- Farthing CA, Farthing DE, Koka S, et al. (2010). A simple and sensitive HPLC fluorescence method for determination of tadalafil in mouse plasma. J Chromatogr B: Anal Technol Biomed Life Sci 878:2891–5
- Felton LA, Wiley CJ, Godwin DA. (2002). Influence of hydroxypropyl-beta-cyclodextrin on the transdermal permeation and skin accumulation of oxybenzone. Drug Dev Ind Pharm 28:1117–24
- Ganeshpurkar A, Vaishya P, Jain S, et al. (2014). Delivery of amphotericin B for effective treatment of candida albicans induced dermal mycosis in rats via emulgel system: formulation and evaluation. Indian J Dermatol 59:369–74
- Lobenberg R, Amidon GL. (2000). Modern bioavailabilty, bioequivalence and biopharmaceutics classification system. New scientific approaches to international regulatory standards. Eur J Pharm Biopharm 50:3–12
- Naik A, Pechtold LARM, Potts RO, Guy RH. (1995). Mechanism of oleic acid-induced skin penetration enhancement in vivo in humans. J Control Release 37:299–306
- Nair MP, Kronfol ZA, Schwartz SA. (1990). Effects of alcohol and nicotine on cytotoxic functions of human lymphocytes. Clin Immunol Immunopathol 54:395–409
- Nasongkla N, Wiedmann AF, Bruening A, et al. (2003). Enhancement of solubility and bioavailability of beta-lapachone using cyclodextrin inclusion complexes. Pharm Res 20:1623–33
- Ogiso T, Iwaki M, Paku T. (1995). Effect of various enhancers on transdermal penetration of indomethacin and urea, and relationship between penetration parameters and enhancement factors. J Pharm Sci 84:482–8
- Ongpipattanakul B, Burnette RR, Potts RO, Francoeur ML. (1991). Evidence that oleic acid exists in a separate phase within stratum corneum lipids. Pharm Res 8:350–4
- Onishi H, Takahashi M, Machida M. (2005). PLGA implant tablet of ketoprofen: comparison of in vitro and in vivo releases. Biol Pharm Bull 28:2011–15
- Saharan S, Kohil U, Lodha R, et al. (2009). Thrombotic microangiopathy associated with Plasmodium vivax malaria. Pediatr Nephrol 24:623–4
- Shinha VR, Kaur MP. (2000). Permeation enhancers for transdermal drug delivery. Drug Dev Ind Pharm 26:1131–40
- Som I, Bhatia K, Yasir M. (2012). Status of surfactants as penetration enhancers in transdermal drug delivery. J Pharm Bioallied Sci 4:2–9
- Takeuchi T, Yasukawa H, Uamaoka Y, et al. (1992). Effects of fatty acids, fatty amines and propylene glycol on rat stratum corneum lipids and proteins in vitro measured by Fourier transform infrared/attenuated total reflection (FT-IR/ATR) spectroscopy. Chem Pharm Bull 40:1887–92
- Tanojo H, Boelsma E, Junginger HE, et al. (1999). In vivo human skin permeability enhancement by oleic acid: a laser Doppler velocimetry study. J Control Release 58:97–104
- Touitou E, Godin B, Karl Y, et al. (2002). Oleic acid, a skin penetration enhancer, affects Langerhans cells and corneocytes. J Control Release 80:1–7
- Verma P, Pathak K. (2010). Therapeutic and cosmeceutical potential of ethosomes: an overview. J Adv Pharm Technol Res 1:274–82
- Wonglertnirant N, Ngawhirunpat T, Kumpugdee-Vollrath M. (2012). Evaluation of the mechanism of skin enhancing surfactants on the biomembrane of shed snake skin. Biol Pharm Bull 35:523–31