
Abstract
Tumor necrosis factor related apoptosis inducing ligand (TRAIL) proved to be a promising new target for colorectal cancer treatment. Elevated expression of TRAIL protein in tumor cells distinguishes it from healthy cells, thereby delivering the drug at the specific site. Here, we formulated oxaliplatin immunohybrid nanoparticles (OIHNPs) to deliver oxaliplatin and anti-TRAIL for colorectal cancer treatment in xenograft tumor models. The polymeric chitosan layer binds to the lipid film with the mixture of phospholipids by an ultra sound method followed by conjugating with thiolated antibody using DSPE-PEG-mal3400, resulting in the formation of OIHNPs. The polymer layer helps in more encapsulation of the drug (71 ± 0.09%) with appreciable particle size (95 ± 0.01 nm), and lipid layer prevents degradation of the drug in serum by preventing nanoparticle aggregation. OIHNPs have shown a 4-fold decrease in the IC50 value compared to oxaliplatin in HT-29 cells by the MTT assay. These immuno-nanoparticles represent the successful uptake and internalization of oxaliplatin in HT-29 cells rather than in MCF-7 cells determined by triple fluorescence method. Apoptotic activity in vitro of OIHNPs was determined by the change in the mitochondria membrane potential that further elevates its anti-tumor property. Furthermore, the conjugated nanoparticles can effectively deliver the drug to the tumor sites, which can be attributed to its ability in reducing tumor mass and tumor volume in xenograft tumor models in vivo along with sustaining its release in vitro. These findings indicated that the oxaliplatin immuno-hybrid nanoparticles would be a promising nano-sized active targeted formulation for colorectal-tumor targeted therapy.
Introduction
Colorectal is the second leading cause of death among cancers showing significant morbidity and mortality (Dhawale et al., Citation2010). This lists cancer among the most dreaded diseases in the world causing ∼6 millions death per year. Various treatment strategies were employed and followed but to limit success mainly due to the physico-chemical properties of the cancer drugs. The different therapeutic strategies for cancer treatment currently in practice include chemotherapy, radiation, surgery, immuno therapy and hormonal therapy (Ramanathan et al., Citation2003).
Various diseases occur in the colon region apart from the colorectal cancer such as Crohns disease, ulcerative colitis and amoebiasis (Jain et al., Citation2007, Citation2010). So drug targeting and localization at the target site plays an important role in the design of a dosage form. Chemotherapy comes along as a most followed treatment strategy that induces cell death in cancer cells upon employment of anti-neoplastic agents. Still it was deprived of its chemotherapeutic efficiency due to adverse effects caused to healthy cells. The reason behind this lack of activity is caused by lack of site specificity of anti-cancer drugs thereby causing improper bio-distribution. Along with this, the other limitations of chemotherapy include multidrug resistance, gradual development of resistance along with the requirement of a high dose to induce the desired effect (Colotta, Citation2008; Vivek et al., Citation2014). The above limitations forces the way for an increase in the dose size for the lack of drug specificity there by causing undesired toxic effects to healthy cells (Krishnaiah & Satyanarayana, Citation2001; Michor et al., Citation2005). This situation paved the way for the formulators to develop a drug delivery system with a novel approach so as to target tumors specifically and deliver the drug inside the tumor instead of localization of the drug outside the tumor. Previous researches conclude the possibility of drug delivery into the tissues rather than through the tissues (Ciftci & Groves, Citation1996).
Among the current lot of strategies employed, nanotechnology comes as one of the promising approach in the treatment of cancer pertaining to its ability in enhancing the drug delivery inside the tumor there by helping in achieving intended therapeutic effect and decrease in the adverse effects to healthy cells (Feng & Chien, Citation2003; Feng et al., Citation2007; Jia et al., Citation2009; Lehár et al., Citation2009; Mitsiades et al., Citation2011; Woodcock et al., Citation2011). The nanoparticles also help in reducing the particle size along with their targeting ability that allows the drug to enter easily into the tumors. Nanoparticulate drug delivery also offers various benefits such as delivering high drug concentration at the target site, controlled release of the drug, prevent drug from enzymatic degradation, prevent covalent conjugation of the drugs directly to the therapeutic agents (Cirstoiu-Hapca et al., Citation2010). Recent researches have been conducted on liposomes and polymeric nanoparticles for various cancer treatments; however, they suffer from minor drawbacks. So, now-a-days the research is focused on the more feasible lipid polymer hybrid nanoparticles by incorporating the positive attributes of both of them. Three distinct functional compartments together comprise of lipid polymer hybrid nanoparticles. First one of them is the polymeric core that helps in incorporating drugs with high drug load, second of them is the lipid layer which prevents the drug retention into the polymer core and the third one is hydrophilic polymeric stealth layer that helps the lipid polymer hybrid nanoparticles in attaining stability along with enhancing the in vivo circulation life time (Chan et al., Citation2009; Liu et al., Citation2010; Sengupta et al., Citation2005; Thevenot et al., Citation2007; Zhang et al., Citation2008).
Oxaliplatin (oxalato(transl-1,2-diaminocyclohexane) belongs to the third generation anti-tumor compound and is at present widely used as the first line therapy along with 5-fluorouracil and Irinotecan for the treatment of colorectal cancer. Oxaliplatin differs from other platinum compounds among its properties mostly attributed to the presence of an oxalate “leaving group” and the diamino cyclohexane (DACH) carrier ligand, which are responsible, at least in part, for its unique properties. Replacement of an amine group by DACH in cisplatin differs oxaliplatin from it. Presence of oxalate group that helps in non-enzymatic transformation into reactive compound in plasma by its displacement supports our rationale in selecting oxaliplatin due to it’s better pharmacokinetics profile compared with cisplatin. Oxaliplatin alone is therapeutically active, but still it possesses various side effects such as peripheral distal neurotoxicity and acute dysesthesias that limits its clinical activity. Oxaliplatin also lacks site specific nature which causes biodistribution to healthy tissues that causes side effects. So there is a need for development of a dosage form with enhanced targeting to the tumor site and drug release inside the tumor rather than localization. So active targeting of oxaliplatin minimizes its limitations by intracellular accumulation of the drug only into the tumor tissues by receptor-mediated endocytosis. (Jia et al., Citation2009; Krishnaiah & Satyanarayana, Citation2001; Lehár et al., Citation2009; Michor et al., Citation2005; Mitsiades et al., Citation2011; Tummala et al., Citation2014a,Citationb).
TRAIL (tumor necrosis factor-related apoptosis-inducing ligand or Apo-2L or CD-253) and TRAIL death receptor antibodies are clearly distinguished and established themselves in the class of successful biologically targeted anti-tumor proteins. The main reason behind selecting the TRAIL is its ability to target only human cancer cells there by excluding healthy cells (Holoch & Griffith, Citation2009; Walczak et al., Citation1999). This feature of TRAIL helps in targeting individual cancer cells as it is expressed in cancer cells only there by interfering with TRAIL induced apoptosis. Generally TRAIL-R1 and TRAIL-R2 are death receptors that are referred to as DR4 and DR5, respectively. The main property of these receptors is their ability to transduce death signals to the intracellular apoptosis machinery. This property of TRAIL allows us to select it as a targeting ligand along with its cytotoxicity activity there by helping the drug in a synergistic way (LeBlanc & Ashkenazi, Citation2003).
We have employed biocompatible hybrid nanoparticles containing both lipid and polymer to deliver oxaliplatin and TRAIL antibody along with imparting stability and site specificity. All the component materials used were approved by USFDA for their clinical use. The nanoparticles formulated were composed with a polymer core in which oxaliplatin is encapsulated. The outer lipid layer consisting of different lipids helps in preventing unfavorable aggregation due to the negative charge of the lipids. Also PEG on the lipid shell further protects the drug from serum degradation (Avgoustakis, Citation2004; Zhang et al., Citation2007). Then the hybrid nanoparticles were conjugated with thiolated anti-TRAIL covalently by thiol-maleimide conjugation. The prepared immuno nanopaticles were characterized and evaluated for their therapeutic activity in vitro in HT-29 cell lines and in vivo in xenograft tumor models.
Materials and methods
Materials
Oxaliplatin was obtained as a gift sample from Cipla pharmaceuticals, India. Soyalecithin, DSPE-PEG2000, DSPE-PEG-mal3400 were purchased from Sigma Aldrich, Mumbai, India. Anti-body CD-253/TRAIL which is used for conjugation was obtained from Santa Cruz Biotechnology, India. 2-(4-amidinophenyl)-6-indolecarbamidine dihydrochloride solution (DAPI) and actin tracker green were purchased from Beyotime Institute of Biotechnology, Japan. Chitosan (purified viscosity grade 50 cps; molecular weight [MW] 150 kDa; deacetylation degree 85%) that was used as the polymer, MTT (3-(4,5-dimethylthiazolyl- 2)-2, 5-di-phenyltetrazolium bromide) and polyvinyl alcohol (PVA), which was used as surfactant, were purchased from Sigma Aldrich, Mumbai, India. All cell culture medium and cell lines were obtained from the National Institute of Health Sciences (NIH), India. Acetonitrile (high performance liquid chromatography [HPLC] grade) was purchased from Himedia leading biosciences company, India. De-ionized Milli Q water was used for the preparation of all the samples.
Preparation of polymeric nanoparticles
The idea behind using chitosan is due to its biocompatibility in drug delivery in vivo and in vitro along with clinical use safety approval by USFDA. The above-prepared nanoparticles were weighed accurately for 20 mg and dissolved in 2 ml of acetic acid and 1 ml of water (in which Oxaliplatin is dissolved) followed by centrifugation. Mixture was later sonicated for emulsification for 5 min leading to the preparation of water in oil emulsion (W/O). This was further emulsified by addition of 2 ml of 1.5% PVA and again sonicated for 10 min followed by adding it to 8 ml of 0.5% PVA with necessary mechanical agitation by stirring forming water in oil in water emulsion (OPNs) (W/O/W). Excess solvent was further removed through evaporation by vacuum. Then the emulsion was subjected to centrifugation (SIGMA, Germany) for 30 min at 12 000 rpm (Zhao et al., Citation2015)
Preparation of lipid film for coating
A mixture of phospholipids were selected for the preparation of the lipid film that can coat the polymeric nanoparticles and the outer PEG layer can be conjugated to the CD-253/TRAIL protein, which expresses majorly in tumor cells of colorectal cancer. Cholesterol, soyalecithin, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[maleimide(polyethylene glycol)-3400] (DSPE-PEG-3400-mal) and 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[methoxy (polyet-hylene glycol)-2000] (DSPE-PEG-2000) were selected as lipids. The lipids were selected in a ratio of 15:50:3:15 mass ratios followed by dissolving them in dichloromethane. Rotary vacuum evaporator was utilized to develop a thin lipid film in a round bottom flask attributed to the reduction in pressure (Li et al., Citation2015).
Prepartion of hybrid nanoparticles and immuno hybrid nanoparticles
Then the prepared polymeric emulsion was added to the lipid film leading to the coating of the lipid film around the polymeric nanoparticles by ultrasound method resulted in forming lipid polymer hybrid nanoparticles of oxaliplatin (OHNPs) by sonicating them for 15 min. DSPE-PEG-3400 mal was added again for helping its conjugation with CD-253 antibody for surface functionalization (Li et al., Citation2015).
In total, 600 μg of thiolated anti-CD253 (using Traut’s reagent) and OHNPs micelle solution containing DSPE-PEG-MAL (15 mg/ml) were kept at 4 °C incubated overnight. The mixture was further then neutralized with 1 M NaOH to pH 7.5 so that it helps in increasing the bonding to sulfur 1000 times faster compared with amine there by helping in forming sulfur bonds. It was then allowed to keep it for 1 d followed by centrifugation them for 15 min at 15 000 rpm and washing them thrice with distilled water before collecting oxaliplatin immuno hybrid nanoparticles (OIHNPs). The thiolated antibody used was already characterized for its thiol group at 3000 cm−1 in FTIR Spectrophotometer (FTIR 8400 S, Shimadzu).
Characterization of nanoparticles
FTIR studies
Infrared spectra matching approach was used for detection of any possible chemical interaction between the drug, lipid and polymer. A physical mixture of drug, lipid and polymer was prepared and mixed with a suitable quantity of potassium bromide (KBr). This mixture was compressed to form a transparent pellet using a hydraulic press at 15 tons pressures. It was scanned from 4000 to 400 cm−1 in a FTIR spectrophotometer (FTIR 8400 S, Shimadzu, Kyoto, Japan). The IR spectrum of the physical mixtures was compared with those of pure drug, lipid, polymer and peak matching was done to detect any appearance or disappearance of peaks. FTIR studies were also done for hybrid immuno nanoparticles for their preliminary confirmation of attachment of antibody to the nanoparticles.
Differential scanning calorimetry
The morphous of oxaliplatin in OPNPs and OHNPs was studied using a Thermal Analysis System TA-60 WS (Shimidazu Co., Ltd., Kyoto, Japan) in order to evaluate lipid crystallinity. An empty pan was used as a reference, samples were analyzed in standard aluminum sample pans under a nitrogen purge. A heating rate of 10 °C/min was employed in the 30–300 °C temperature range. Thermograms were obtained for oxaliplatin, lipids, polymers, physical mixtures and oxaliplatin hybrid nanoparticles (OHNPs) (Jain et al., Citation2010).
Particle size and zeta potential analysis
The mean particle size was determined by dynamic light scattering (DLS) with a NicompTM 380 (Precision System Science Co., Ltd., USA). The zeta potential (ξ) was determined using Malvern zetasizer. All samples were diluted with double distilled water to a suitable scattering intensity prior to the measurements. Particle size and zeta potential was obtained for different batches formulated using various ratios of lipid.
Drug content and entrapment efficiency
Nanoparticles (20 mg) after freeze-drying were added to their specific solvent (10 ml) to facilitate the coat of the nanoparticles to get dissolved. The suspension formed was subjected to evaporation causing removal of the solvent prior to filtration. Then the residue was washed and diluted appropriately with phosphate buffer of pH 6.8 to determine drug content and entrapment efficiency. Samples were measured at an absorbance of 264 nm in double beam UV spectrophotometer. Drug content loading and entrapment efficiency of oxaliplatin in nanoparticles were determined by the following equations (Muller et al., Citation2000):
Scanning electron microscopy
Scanning electron microscopy (SEM) was used to verify uniformity of particle shape and size. Freeze-dried nanoparticles were resuspended in distilled water and were later dropped onto a silicon grid and dried under room temperature. The nanoparticle suspension was vacuum coated with gold for 3 min. The surface morphology of the samples was observed under a scanning electron microscope (JEOL–JAPAN) operated at 15-keV pulse at different resolutions (Mohideen et al., Citation2013).
In vitro release study
Dialysis test was performed for in vitro release studies. Phosphate buffer solution of pH 7.4 and acetate buffer of pH 4.5 were used. 100 ml was poured into a closed glass vessel as the dissolution medium. Total volume of 5 ml of OIHNPs, OPNPs, OHNPs were applied to a dialysis bag (MW cut-off 3000–6000 KDa) and placed into the glass vessel. Incubator shaker was used to shake the samples horizontally at 37 °C and 100 rpm. One millilitre of the sample was taken which was replaced with fresh buffer at definite time intervals and filtered through a 0.22 μm filter membranes. The filtrate was measured using HPLC. The samples were taken at various time intervals till 48 h in order to evaluate its sustained release property. The experiments were conducted in triplicates so as to minimize the error. Cumulative in vitro drug release was found for both hybrid nanoparticles and immuno hybrid nanoparticles of oxaliplatin.
Measure of stability in serum
Plain nanoparticles (PNPs), OHNPs and OIHNPs were investigated for their stability in serum by dispersing them in 10% FBS. DLS technique was then employed for effective determination of changes in the nanoparticles size (Li et al., Citation2015).
In vitro cytotoxicity assay
MTT assay was performed for OIHNPs, unconjugated nanoparticles and pure oxaliplatin. HT-29 cells were seeded onto 96-well plates at a seeding density of 1 × 104 cells/well. It was then left overnight with incubation for attachment of cells to the plate. In each well, the culture medium was replaced with 100 μl of medium with serial dilutions of the samples for treatment. All the treatment samples were given by concentration of 0.5–10 μm and were left for 48 h. 10 μl of MTT (5 mg/ml in phosphate buffer saline) was added at precise time intervals, which was followed by incubation for 4 h and removal of the culture solution. In total, 100 μl of DMSO was then added to each of the well separately in order to dissolve the formazan crystals while shaking the plates vigorously. Then the absorbance was determined for each well on a microplate reader (Thermo scientific, Pittsburgh, PA) at a wavelength of 570 nm. The experiment was conducted in triplicate in parallel. IC50 is the concentration that can inhibit 50% of cell growth was calculated by SPSS software (Version 19.0). Along with that relative cell viability was calculated for different concentrations for all the samples, which was compared with the untreated control.
Mitochondrial membrane potential
HT-29 cells were seeded in 6-well plates and were subjected to trypsinization followed by suspending them in phosphate buffer consisting of 10 μg/ml of JC-1 (Sigma Aldrich, St. Louis, MO). The cells were incubated for 15 min at 37 °C in the incubator before centrifugation in order to remove the supernatant. This was followed by suspending them in PBS. The cells which were identified as green was considered to have exhibited change in the membrane potential (color attributed to the emission of green fluorescence from JC-1 monomers).
Cellular uptake and internalization
HT-29 cells and MCF-7 cells were seeded separately at 1 × 104 cells/well into 96-well black plates (Corning Costar Corp, Corning, NY) followed by culturing them for 24 h to determine the cellular uptake and internalization. The cells were allowed to reach 85% confluence for addition of Nile red at a concentration of 0.45 μm at various time points followed by incubation. Further attachment of any nanoparticles was prevented during removal of nanoparticle suspension by washing them with PBS. RIPA lysis buffer was used to lyse the cells and were subjected to the determination of fluorescence intensity of Nile red nanoparticles with spectral scanning multimode reader (Varioskan Flash 3001; Thermo Scientific Inc., Rockford, IL) at an excitation wavelength at 552 nm and emission wavelength at 636 nm (Zhang et al., Citation2015).
Eight-well cover-glass chamber was taken and HT-29, cells were seeded into it followed by incubation for 24 h at 37 °C for enhancing cell adhesion and even spread. The above prepared Nile red nanoparticles at a concentration of 200 μg/ml were dispersed in the cell culture medium followed by washing them for 2 h after incubation. Then triple fluorescence method was employed by washing three times with immunostaining wash buffer, and stained with Actin-Tracker Green (Beyotime Biotechnology, Shanghai, People’s Republic of China) for 1 h. Then DAPI solution was added and allowed for 15 min which can stain the cell nucleus followed by washing thrice with PBS before observing them under CLSM (Zeiss LSM 710; Carl Zeiss Meditec AG).
Xenograft tumor model in nude mice and in vivo treatment with chemotherapeutic agents
Male BABL/C nu/nu mice were selected, obtained and maintained well under pathogen and hygiene limited conditions upon approval from animal ethical committee. HT-29 cell line was selected as it is a human colonic adenocarcinoma cell line and was grown as a monolayer by adequately supplementing 10% bovine serum at 37 °C in humidified atmosphere comprising 5% CO2. Then observation of growth in tumor cells was performed followed by harvesting and incubating with 0.25% trypsin-EDTA solution. Upon harvesting, xenograft cells (1 × 107 cells) were subcuteaneously (s.c) injected into the right flank of male nude mice. It was allowed to grow for at least three weeks followed by dissecting them and mechanical mincing. Then tumor pieces with 3 mm3 size have been transplanted subcutaneously by trocar needle into 25 animals and waited till they reach 33–38 mm3. Animals were grouped into five groups each consisting of five animals. A 10 μm/kg of drug (of all formulations) was injected through the tail vein every 2 d. Digital vernier callipers has been used for the determination of tumor size every 3 d and tumor volume was also calculated very precisely by using the formula (Li et al., Citation2015; Radulovic et al., Citation1991)
where a denotes long axis and b denotes the short axis.
Further mice with tumor implants were euthanized after 2 weeks followed by excising them and weighing.
Statistical analysis
Statistical analysis was performed using minitab (version 16). ANOVA was mainly used to evaluate statistical differences between different mean values. All data were considered statistically significant at p < 0.05 and were represented as mean ± SD.
Results and discussion
FTIR studies
The FTIR analysis was performed for the pure drug oxaliplatin, chitosan, DSPE-PEG2000, Cholesterol, soya lecithin, which can be observed in . FTIR studies help to determine the compatibility between the drug and the excipients in the initial phases and also after formulation. FTIR spectra of oxaliplatin have shown – NH peak at 3507.67 cm−1 and C = O at 1711.88 cm−1 that can be seen in . The shift in the peaks was observed when the drug was taken as a physical mixture. Chitosan has shown C−O peak at 1100.34 cm−1, –CH at 2815.43 cm−1, −OH at 3210 cm−1 and –NH2 at 3511.12 cm−1. The lipid DSPE-Peg-2000 has shown characteristic peaks at 1121, 1710, 2891, 3100 cm−1 attributed to C−O, C = O, CH2 and –NH groups, respectively. Cholesterol has shown characteristic peaks at 3300, 2883, 2210 cm−1 attributed to –OH, CH2 and C = C groups, respectively. Soya lecithin has also shown characteristic peaks at 1721, 1125, 2781, 3311 cm−1 attributed to its C = O, C–O, CH2 and NH groups, respectively. Upon interpretation of the physical mixture, it was found to be characterized by peaks at their corresponding areas there by conforming its compatibility which can be clearly observed in .
Figure 1. FTIR spectra of (A) Oxaliplatin, (B) Chitosan, (C) Cholesterol, (D) Soyalecithin and (E) DSPE-PEG2000.
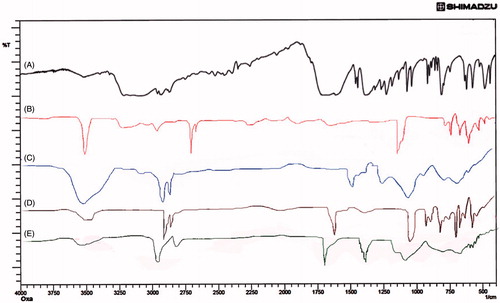
Figure 2. FTIR spectra of (F) Physical mixture (G) OIHNPs (oxaliplatin immuno nanoparticles with antibody coupled by thiol-maleimide conjugation.
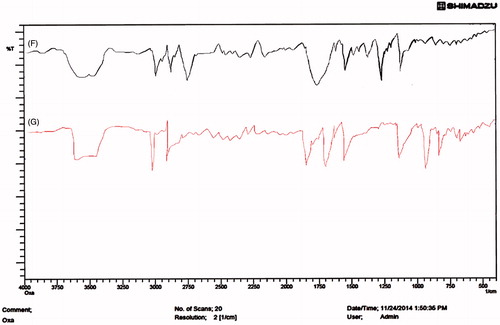
Shift of the –NH stretch from 3511.67 to 3528.23 cm−1 for physical mixture was observed. Shift of the C = O stretching from 1710 to 1721 cm−1 in physical mixture confirms the drug compatibility with the lipid both in physical mixture. Also the shift indicates the positive interaction of the drug with lipid and polymer, which also help in improving the drug entrapment as well as preventing the drug expulsion.
Differential scanning calorimetry studies
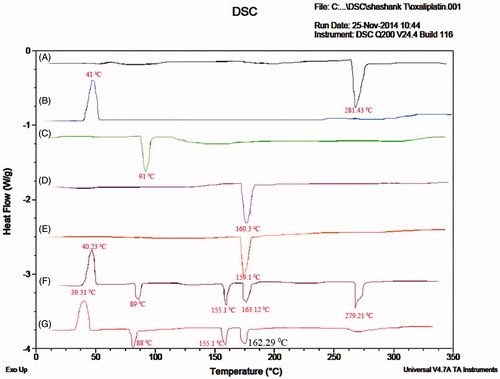
Differential scanning calorimetry (DSC) studies were performed for the drug, physical mixture of the drug, lipid, polymer and as well as drug lipid melts which are shown in . DSC thermogram of oxaliplatin has shown a peak at 281.43 °C. DSC thermogram of chitosan has shown a peak at 91 °C. DSC thermograms of cholesterol, soyalecithin, DSPE-PEG-2000 has shown peaks at 160.3, 155 and 41 °C, respectively. Physical mixture of drug, lipid and polymer has shown a peak at 283.21 °C corresponding to oxaliplatin and 89 °C corresponding to chitosan. Lipids have also shown drug peaks at their respective temperatures which indicate the compatibility of the polymer and the lipids with the drug in formulating hybrid nanoparticles. Drug lipid melts thermograms has shown a peak at 91 °C corresponding to the chitosan, but the peak at 280 °C was found decreased attributing to the fact that drug may get entrapped into the polymer lipid nanoparticles. The lipid peaks were found to be intact at the same temperature. Also the change in enthalpy for both the physical mixture and the drug lipid melts was observed and found it was more in case of drug lipid melts when compared with the physical mixture. Moreover it also helps in confirming the stability, as compatibility of the drug, polymer and the lipid helps in imparting stability to the prepared formulation.
Figure 3. DSC thermograms of (A) Oxaliplatin, (B) DSPE-PEG2000, (C) Chitosan, (D) Cholesterol, (E) Soyalecithin, (F) Physical mixture and (G) OHNPs.
Preparation and characterization of OIHNPs
Solvent emulsification method was used as it was optimized by us in our previous studies for the preparation of polymeric nanoparticles. The same method obtained better results for formulating oxaliplatin polymeric nanoparticles. Different ratios of drug: polymer (1:1, 1:2, 1:3, 1:4) was selected and the effect of the polymer ratio on the formulation was characterized using measurement of the particle size, entrapment efficiency and drug loading. The drug needs to get encapsulated properly inside the polymer in order to get released, so this makes this the most important parameter as improper entrapment of the drug leads to burst release and a decrease in therapeutic efficacy. We have observed decline in the particle size (95 ± 0.01 nm) with increase in the polymer coating, as proper coating of polymer with desired surfactant leads to increase in the effective surface area thereby decreasing the particle size as can be seen in . But as the polymer: drug ratio increases to 1:4, it causes an increase in the particle to 142 ± 0.09 nm size mainly attributed to the formation of thick polymer coat. The zeta potential was found to be stable with an increase in the polymer justifying its stability in preparing stable polymeric nanoparticles. The use of surfactant also helped polymeric nanoparticles in attaining appreciable stability. Also all batches prepared with different ratios of polymer were observed with PDI value <0.2, which indicates the dispersibility of the prepared formulation. Entrapment efficiency of PNPs also followed the same pattern with particle size, as increase in the polymer leads to higher entrapment, which correlates with previous researches on chitosan nanoparticles. EE of PNPs increased (71 ± 0.09%) till 1:3 drug: polymer ratio, and there after kept on decreasing (33 ± 0.01)% mainly due to the formation of more compact polymer coat at 1:4 ratio. So 1:3 ratio of drug: polymer can be optimized as proper entrapment leads to availability of intended therapeutic dose to achieve desirable therapeutic effect. Also drug loading was found to be satisfactory for this batch with 31 ± 0.05%. So, drug: polymer ratio of 1:3 was optimized for particle size (95 ± 0.01 nm), entrapment efficiency (71 ± 0.09%) and drug loading (31 ± 1.12%) and was further used for coating with the lipid film that can be seen in and .
Figure 4. Particle size evaluation of OPNPs by DLS.
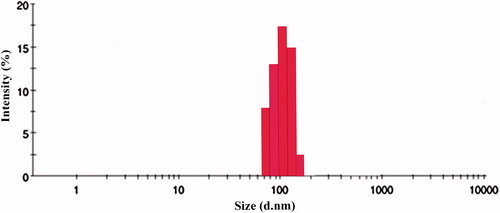
Table 1. Particle size, entrapment efficiency and drug-loading values of different drug: polymer ratios.
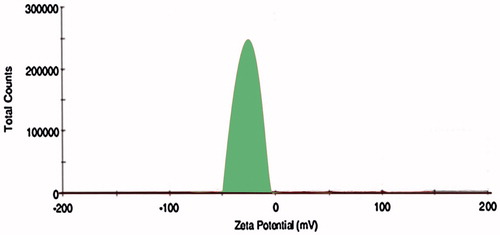
Then the effect of the lipid film coating on the polymeric nanoparticles was further evaluated for particle size and zeta potential. We have observed only minimal changes in the particle size with increasing in the lipid ratio from 0.01 to 0.20. There is increase in the particle size to 119 ± 0.10% after coating with lipid film, but increase in the lipid ratio does not increase it much. We found a decrease in the zeta potential on increase in the lipid: polymer ratio from 0.01:1 to 0.20:1 attributed to the negative zeta potential for the lipids. We observed an increase in the zeta potential to −19.5 mv, which conforms the attachment of lipid bilayer coat effectively on the surface of the polymeric nanoparticles. 0.10:1 ratio of lipid: polymer was selected as the optimized ratio due to its lower particle size and also zeta potential (−19.5 mv) conforming its stability.
Then the optimized batch was further covalently conjugated with CD-253 antibody with maleimiede-thiol coupling for surface functionalization for site specific targeting to the colon tumors. CD-253 also known as TRAIL/Apo-2L is over expressed mainly in tumor cells and not in healthy cells that enhances its therapeutic use. The prepared immuno hybrid nanoparticles were evaluated by FTIR for attachment of antibody to the lipids. Further drug encapsulation (67 ± 0.09%) and particle size (181 ± 0.01%) have been evaluated for OIHNPs and were found to be satisfactory, but we observed a little decline in the entrapment efficiency mainly due to the covalent attachments to the nanoparticles. Slight decrease in the entrapment efficiency was observed with OIHNPs compared with OPNPs which can attributed the covalent conjugation leading to the decrease ability of the drug in entering. Observation of DSC thermograms helps us to find one more attribute to the improvement of drug loading and entrapment efficiency as we found a decrease in crystallinity of the drug lipid formulation when compared alone with the drug which can be seen in . Once the drug starts losing its crystallinity there by generating imperfections with spaces that can accommodate the drug helps in improving drug loading. Particle size and zeta potential of OIHNPs can be seen in and .
Figure 5. Particle size evaluation of OIHNPs by DLS.
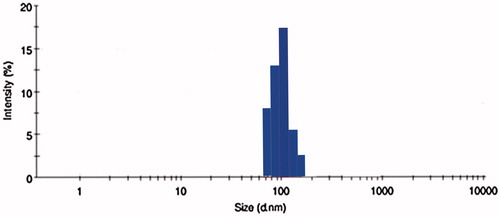
Figure 6. Determination of zetapotential of OIHNPs by Malvern zetasizer.
FTIR
The immuno hybrid nanoparticles were further characterized by FTIR for further conformation of the attachment of antibody to the DSPE-PEG-mal3000. Here the FTIR spectra were characterized by the absence of the thiol peak at 3000 cm−1 which generally is observed for thiolated antibody. Spectra have shown a peak at 911 cm−1attributing to the fact for the formation of S-O bonding between the hybrid nanoparticles and the thiolated antibody. Remaining significant peaks were present at their respective functional group regions. The peaks can be observed clearly in .
Scanning electron microscopy
Scanning electron micrographic image of OIHNPs was taken to evaluate the surface morphology. Careful examination of the image helps us in concluding the spherical morphology of nanoparticles with appreciable uniformity. This spherical shape of the nanoparticles also helps the formulation in sustaining its release. This can be seen in .
Figure 7. Scanning electron micrographic image of OIHNPs.

In vitro drug release studies
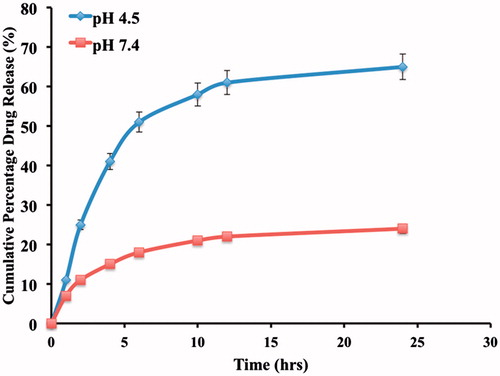
In vitro drug release studies of OHNPs showed biphasic pattern with initial burst release followed by sustained release at pH 4.5. The sustained release of the nanoparticles can be attributed to the smooth surface of the nanoparticles along with the release of the drug from the lipid core. The initial burst release can be attributed to the unentrapped drug present and also the drug that was absorbed followed it on the surface of the nanoparticles rather than inside the polymer. Drug release was found to be 65 ± 0.01% and 22 ± 0.21% within 24 h for pH 4.5 and 7.4, respectively, which is far more satisfactory as we developed this formulation for targeting rather than sustaining its release and shown in . Sustained release of the drug also acts as an added benefit to the formulation, which helps in decreasing the dosage frequency.
Figure 8. In vitro Cumulative drug release profile of OHNPs.
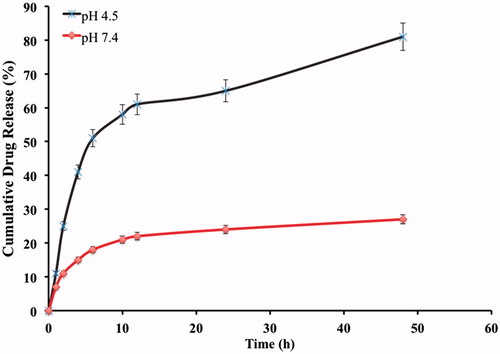
The drug release from the immuno hybrid nanoparticles (OIHNPs) was also observed at pH 4.5 and 7.4. As it was with the hybrid nanoparticles with out antibody, it has shown a biphasic pattern of drug release with an initial burst release attributed to the unentrapped drug and the drug absorbed on the surface. Initial burst phase was accompanied by much slower release for next 24 h conforming to the sustained release property of the drug entrapped inside the lipid rather than absorbed. The reason behind opting for different pH is explained by the fact that drug passes inside the tumors by receptor mediated endocytosis mainly by releasing the drug in early endosomes or secondary endosomes by pH controlled hydrolysis. Endosomes have a pH in the range from 5 to 7.2, whereas lysosomes ranges from pH 4 to 5 (Maya et al., Citation2013). Extracellular layer having pH close to basic causes limited swelling which inhibits the sudden diffusion of drug from the nanoparticles. But there is a minute decrease in the drug release observed in immuno nanoparticles when compared with the OHNPs, which can be due to the more highly conjugated complex matrix around the drug with the presence of CD-253/TRAIL antibody as opposed to the control. The cumulative drug release was found to be about 60 ± 0.01% at the end of 24 h and 81 ± 0.02% at the end of 48 h at pH 4.5. The cumulative drug release at pH 7.4 was found to be 14 ± 0.13% and 27 ± 0.07% at the end of 24 and 48 h, respectively. This can be seen from the .
Figure 9. In vitro Cumulative drug release profile of OIHNPs.
Measurement of stability in serum
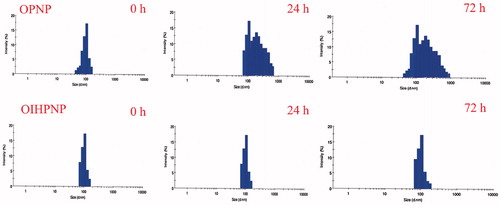
OIHNPS were investigated for their stability in serum by dissolving them in 10% of fetal bovine serum and determining their particle size by DLS technique to evaluate any particle size changes. Based on our observation, we did not find any significant change in the size distribution of OIHNPs contrary to PNPs which showed significant larger size over a span of 72 h. This justifies the behavior of external lipid shell of OIHNPs in serum. Also other reason for their appreciable stability in serum is due to their negative zeta potential that prevents it from protein adsorption and aggregation there by making its clinical application appreciable. This can be observed in .
Figure 10. Effect of serum on the size distribution of OPNPs and OIHNPs with time. OIHNPs showed minimal change in size distribution by DLS in serum after 72 h, where as OPNPs showed a significant change in the particle size at 24 and 72 h. Lipid coating around the OIHNPs protects the drug from degradation attributed to their negative surface charge.
In vitro cytotoxicity activity
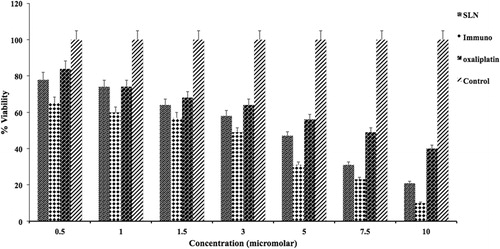
HT-29 cell lines were selected in order to evaluate the cell toxicity activity of formulated immuno nanoparticles, nanoparticles, oxaliplatin and untreated control. These can be observed in . Immuno hybrid nanoparticles (IOHNPs) had shown a considerable decrease in amount of cell viability when compared with the free oxaliplatin. Unconjugated hybrid nanoparticles (OHNPs) also showed a decrease in amount of cell viability but not as significant as OIHNPs. The reason attributed to the decrease in amount of cell viability of both conjugated nanoparticles is may be due to the better uptake of cells by receptor mediated endocytosis due to the attachment of ligand that was site specific in HT-29 cells there by allowing internalization of the drug. A 1.5-fold increase in cytotoxicity of immuno nanoparticles (at 2.5 μm nanoparticles equivalent to 0.63 μm of oxaliplatin) showed nearly 50% cell viability at the end of 48 h. The decrease in cell viability of OIHNPS, OHNPs and oxaliplatin at the end of 48 h when given in the concentration range of 0.5–10 μm was found to be 65–10, 79–21 and 84–42%, respectively. So we can conclude the predominance in the cytotoxic activity of the immuno nanoparticles when compared with others (p < 0.05). IC50 value (0.63 μm drug equivalent with 2.5 μm nanoparticles) of immuno hybrid nanoparticles has shown 4-folds decrease in IC50 value when compared with oxaliplatin. Also the increased cytotoxicity of immuno nanoparticles compared with the OHNPs shows the synergistic activity of immuno nanoparticles with preferential receptor mediated targeting. This concludes site specificity along with synergistic apoptotic activity attributed to the individual apoptotic pathways of TRAIL mediated death receptor activation and oxaliplatin general cytotoxic activity as it is a potent inhibitor of survivin (generally a downstream protein of apoptotic pathways). The IC50 values of the formulations and the drug were used for further studies. Although by observing the , it seems that both pure drug and OIHNPs showing nearly similar decrease in percentage cell viability, it should be noted that OIHNPs are the nanoparticles (2.5 μm) that are showing the same effect with 0.63 μm compared with the oxaliplatin pure drug activity at 2.9 μm.
Figure 11. In vitro cell viability of Oxaliplatin, OPNPs, OHNPs and OIHNPS.
Cellular uptake and internalization studies
Cellular uptake determination of OHNPs, OIHNPs was performed using TRAIL receptor overexpressed HT-29 colorectal cancer cell line and TRAIL-deficient MCF-7 breast cancer cell line. The rationale behind selecting Nile red as the stain is due to its lipophilicity that facilitates its smooth incorporation into the nanoparticles. CLSM microscopy was used for determination of the triple fluorescence-labeling experiments. Cellular distribution can be evaluated by differentiations in the fluorescence visualization as actin tracker dye stains actin filaments (green fluorescence), DAPI stains the nucleus (blue fluorescence) and Nile red stains nanoparticles (red fluorescence). In OIHNPs it can be clearly observed from the that the blue fluorescence from DAPI labeling the nucleus was circumvented by the Nile red nanoparticles (red fluorescence), which conforms the internalization of the nanoparticles into the tumor cells in HT-29 cells. Nevertheless, OIHNPs was found to be not internalized in MCF-7 cells which were TRAIL-negative determining its site-specific nature due to the attachment of specific ligand. OHNPs also showed significant internalization, but not as significant as OIHNPs as it can be clearly evident from the as most of the particles were present outside in HT-29 cells. OHNPs also showed a similar kind of effect of uptake and internalization in MCF-7 cells conforming its lack of ability in targeting specific tumor cells along with its decreased activity. This even elevates the significance of OIHNPs in delivering the drug to the target specifically along with its synergistic activity. Cellular uptake of immuno hybrid nanoparticles was increased due to the receptor mediated endocytosis promoting the entry of the nanoparticles into cells when TRAIL-hybrid nanoparticles meet the TRAIL receptor over expressed HT-29 cells.
Figure 12. Fluorescent images of hybrid nanoparticles loaded with three fluorescence dyes in cell. In vitro fluorescence images of HT-29 cells (TRAIL-positive) after incubation with OIHNPs-(Nile red)-DAPI-Actin green and OHNPs-(Nile red)-DAPI-Actin green for 1, 3, 5 or 7 h to measure intracellular drug release profiles. Initial first row (vertical) represents nuclei stained with DAPI, which was followed by an second row of actin filaments and third row of nile red nanoparticles that circumvents the nuclei in the fourth row (vertical).
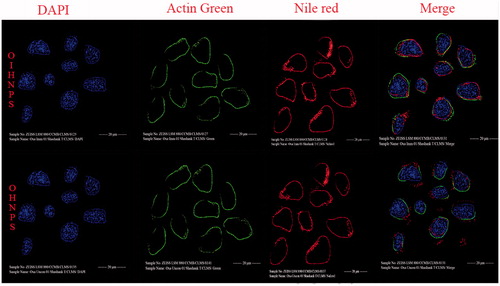
Figure 13. Fluorescent images of hybrid nanoparticles loaded with three fluorescence dyes in cell. In vitro fluorescence images of MCF-7 cells (TRAIL-negative) after incubation with OIHNPs-(Nile red)-DAPI-Actin green and OHNPs-(Nile red)-DAPI-Actin green for 1, 3, 5 or 7 h to measure intracellular drug release profiles. Initial first row (vertical) represents nuclei stained with DAPI, which was followed by an second row of actin filaments and third row of nile red nanoparticles that was present outside the nuclei in the fourth row (vertical).

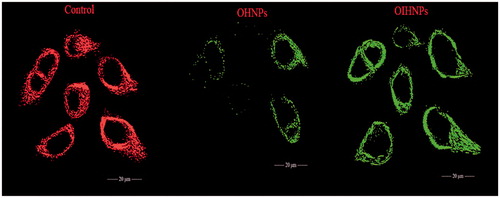
Evaluation of mitochondrial membrane potential change
The major step in the process of apoptosis is the liberation of cytochrome C through the outer mitochondrial membrane into the cytosol causing the change in the mitochondria membrane potential. Cytochrome C also unleashed the activation of caspase proteins, which are normally inhibited during the cancer, the activation of these pro-apoptotic proteins causes apoptosis. OHNPs and OIHNPs were evaluated for their change in membrane potential (ΔΨm) by JC-1 stain. Significant green emission was observed in case of OIHNPs and OHNPs has shown a partial green emission, which shows its lack of activity in inducing apoptosis comparable to the immuno nanoparticles. OIHNPs showed significant activity attributed to the synergistic activity caused due to both oxaliplatin and TRAIL receptor. These changes can be seen clearly in .
Figure 14. Depletion of mitochondrial membrane potential (ΔΨm) in HT-29 cells induced by OHNPs and OIHNPs. Cells were treated and stained with a mitochondria-selective dye JC-1 and analyzed. Controlled cells produce red fluorescence and green fluorescence (due to the depletion of mitochondrial membrane). Initial row (vertical) represents control that represents intact mitochondria membrane, second row represents partial depletion identified by JC-1 stain where as third row represents complete depletion as can be observed with full change in potential.
Inhibition of tumor growth
BALB/c nude mice with subcutaneous HT-29 xenografts were selected to determine the inhibition of tumor growth and randomly divided into five animals per group into five groups: saline, PNPs, OPNPs, OHPNPs and OIHNPs. When the tumor volume reached a predetermined size (30 mm3), treatment has been initiated and continued till 9 d and thereafter sacrificed at 15 d. We have observed vast growth of tumors in the saline group and plain nanoparticles (PNPs) without drug also did not show any effect on the tumor growth. Tumor growth in OIHNPs showed significantly slower comparable to OPNPs and OHPNPs, which further justify the use of CD-253 in attaining site specificity and synergistic activity (due to its apoptotic activity by activating death receptors). The tumors were sacrificed then after 15 d and were excised and weighed. The mean weight of the tumors was compared for all groups and it also showed lower tumor mass in OIHNPs. The tumor mass and volumes for OIHNPs and OHNPs can be seen in and .
Figure 15. Tumor growth curves with various oxaliplatin formulation treatments (saline, oxaliplatin, TRAIL, OPNP, OHNP, OIHNP). Data are presented as mean ± SD (n = 5), p < 0.05.
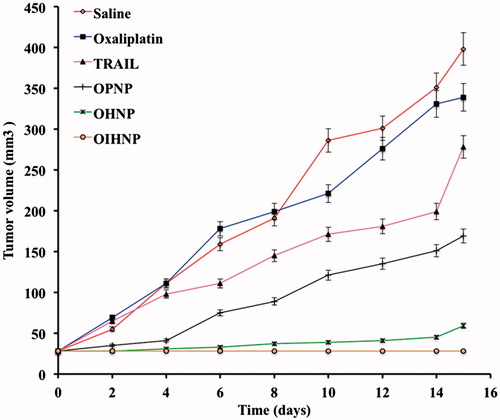
Figure 16. Measurement of tumor weights after excising the tumor xenografts by 15 d (n = 5) tained was due to the depletion of mitochondrial membrane.
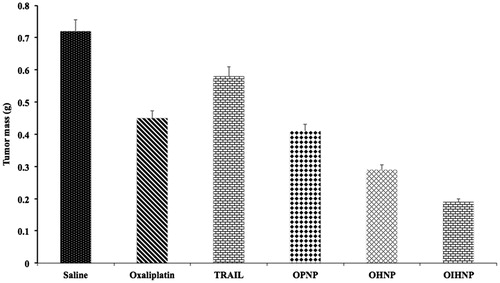
The reduction in tumor mass and volume justified its increased anti-tumor activity thereby enhancing its therapeutic effectiveness. Also it supports the in vitro cytotoxic activity shown by the OIHNPs conforming its clinical applicability. Synergistic activity was also observed due to the conjugation of CD-253 antibody that further acts as a boon for cancer patients along with its site specificity. Its site specificity allows the drug to reach the colon tumors rather than to the healthy cells by preventing the release of the drug in the systemic circulation.
Conclusion
Oxaliplatin which is used for the treatment of colorectal cancer mainly prescribed as one of the drugs in Folforinox regimen. In concluding the current study, biocompatible immuno lipid-polymer hybrid nanoparticles encapsulating oxaliplatin and covalently attached TRAIL antibody were fabricated for colorectal cancer treatment and investigated for their synergistic anti-tumor activity in subcutaneous and xenograft model. The current study was conducted with all FDA approved materials. OIHNPs are composed of polymeric core comprising of chitosan encapsulating oxaliplatin and an outer lipid layer consisting of soya lecithin, cholesterol and DSPE-PEG-2000 along with covalently attached Anti-TRAIL. The polymer layer helps in increasing the drug loading and also helps in releasing the drug in a sustained manner at the site of target. The lipid layer coated outside helps the formulation of attaining stability due to its negative surface charge. So, OHNPs showed better stability than OPNPs. The anti-TRAIL attached outside the lipid helps the formulation in delivering the drug to the specific site which can be evident from its enhanced therapeutic activity in treating colorectal cancer. These enhancements endow improved drug targeting ability and properties of the OIHNPs. OIHNPs can deliver both oxaliplatin and Anti-TRAIL there by exhibiting significant synergistic activity in vivo than OHNPs. OIHNPs also showed better apoptotic activity by internalizing the drug into the tumor cell rather than localizing it outside in HT-29 cells evaluated by triple fluorescence study. Synergistic activity of OIHNPs can be evident from its capability in reducing the tumor mass and volume significantly comparable to other with enhanced apoptotic activity. The current drug therapy can lay a more improved foundation for the development of immuno hybrid nanoparticles as an alternative to chemotherapy for the treatment of colorectal cancer.
Acknowledgements
I would like to dedicate my work to Dr M.N. Satish Kumar for his kind support in guiding me through out the research. I am very much thankful to Dr Ashish Wadhwani in supporting for my in vitro studies. Authors would like to thank Cipla pharmaceuticals, India for gifting oxaliplatin sample.
Declaration of interest
The authors report no declaration of interest.
References
- Avgoustakis, K. (2004). Pegylated poly (lactide) and poly (lactide-co-glycolide) nanoparticles: preparation, properties and possible applications in drug delivery. Curr Drug Deliv 1:321–33
- Chan JM, Zhang L, Yuet KP, et al. (2009). PLGA–lecithin–PEG core–shell nanoparticles for controlled drug delivery. Biomaterials 30:1627–34
- Ciftci, K, Groves MJ. (1996). Delivery of antitumor compounds to the rat colon: in vitro and in vivo evaluation. Int J Pharm 145:157–64
- Cirstoiu-Hapca A, Buchegger F, Lange N, et al. (2010). Benefit of anti-HER2-coated paclitaxel-loaded immuno-nanoparticles in the treatment of disseminated ovarian cancer: therapeutic efficacy and biodistribution in mice. J Control Release 144:324–31
- Colotta F. (2008). Anticancer drug discovery and development. Targeted therapies in cancer. New York: Springer, 128–43
- Dhawale S, Bankar A, Patro M. (2010). Formulation and evaluation porous microspheres of 5-fluorouracil for colon targeting. Int J Pharm Tech Res 2:1112–18
- Feng S-S, Chien S. (2003). Chemotherapeutic engineering: application and further development of chemical engineering principles for chemotherapy of cancer and other diseases. Chem Eng Sci 58:4087–114
- Feng S-S, Zhao L, Zhang Z, et al. (2007). Chemotherapeutic engineering: vitamin E TPGS-emulsified nanoparticles of biodegradable polymers realized sustainable paclitaxel chemotherapy for 168h in vivo. Chem Eng Sci 62:6641–8
- Holoch PA, Griffith TS. (2009). TNF-related apoptosis-inducing ligand (TRAIL): a new path to anti-cancer therapies. Eur J Pharmacol 625:63–72
- Jain A, Gupta Y, Jain SK. (2007). Perspectives of biodegradable natural polysaccharides for site-specific drug delivery to the colon. J Pharm Pharm Sci 10:86–128
- Jain A, Jain SK, Ganesh N, et al. (2010). Design and development of ligand-appended polysaccharidic nanoparticles for the delivery of oxaliplatin in colorectal cancer. Nanomed Nanotechnol Biol Med 6:179–90
- Jia J, Zhu F, Ma X, et al. (2009). Mechanisms of drug combinations: interaction and network perspectives. Nat Rev Drug Discov 8:111–28
- Krishnaiah Y, Satyanarayana S. (2001). Colon-specific drug delivery systems. Advances in Controlled and Novel Drug Delivery. New Delhi, India: CBS Publishers and Distributors, 89–119
- LeBlanc H, Ashkenazi A. (2003). Apo2L/TRAIL and its death and decoy receptors. Cell Death Different 10:66–75
- Lehár J, Krueger AS, Avery W, et al. (2009). Synergistic drug combinations tend to improve therapeutically relevant selectivity. Nat Biotechnol 27:659–66
- Li F, Zhao X, Wang H, et al. (2015). Multiple layer by layer lipid polymer hybrid nanoparticles for improved FOLFIRINOX chemotherapy in pancreatic tumor models. Adv Funct Mater 25:788–98
- Liu Y, Li K, Pan J, et al. (2010). Folic acid conjugated nanoparticles of mixed lipid monolayer shell and biodegradable polymer core for targeted delivery of Docetaxel. Biomaterials 31:330–8
- Maya S, Kumar LG, Sarmento B, et al. (2013). Cetuximab conjugated O-carboxymethyl chitosan nanoparticles for targeting EGFR overexpressing cancer cells. Carbohydr Polym 93:661–9
- Michor F, Iwasa Y, Lengauer C, Nowak MA. (2005). Dynamics of colorectal cancer. Semin Cancer Biol 15:484–93
- Mitsiades CS, Davies FE, Laubach JP, et al. (2011). Future directions of next-generation novel therapies, combination approaches, and the development of personalized medicine in myeloma. J Clin Oncol, 29:1916–23
- Mohideen B, Ezhilmuthu R, Dhanalekshmi U, Neelakanta Reddy P. (2013). Formulation and in-vitro characterization of gliclazide loaded polymeric nanoparticles. Int J Biol Pharm Res 4:533–40
- Muller RH, Mader K, Gohla S. (2000). Solid lipid nanoparticles (SLN) for controlled drug delivery-a review of the state of the art. Eur J Pharm Biopharm 50:161–77
- Radulovic S, Miller G, Schally AV. (1991). Inhibition of growth of HT-29 human colon cancer xenografts in nude mice by treatment with bombesin/gastrin releasing peptide antagonist (RC-3095). Cancer Res 51:6006–9
- Ramanathan RK, Clark JW, Kemeny NE, et al. (2003). Safety and toxicity analysis of oxaliplatin combined with fluorouracil or as a single agent in patients with previously treated advanced colorectal cancer. J Clin Oncol 21:2904–11
- Sengupta S, Eavarone D, Capila I, et al. (2005). Temporal targeting of tumour cells and neovasculature with a nanoscale delivery system. Nature 436:568–572
- Thevenot J, Troutier A-L, David L, et al. (2007). Steric stabilization of lipid/polymer particle assemblies by poly (ethylene glycol)-lipids. Biomacromolecules 8:3651–60
- Tummala S, Kumar MS, Gowthamarajan K, et al. (2014a). Preparation, physicochemical characterization and in vitro evaluation of oxaliplatin solid lipid nanoparticles for the treatment of colorectal cancer. Indo Am J Pharm Res 4:3579–87
- Tummala S, Kumar MS, Prakash A. (2014b). Formulation and characterization of 5-Fluorouracil enteric coated nanoparticles for sustained and localized release in treating colorectal cancer. Saudi Pharm J 23:308–14
- Vivek R, Thangam R, NipunBabu V, et al. (2014). Multifunctional HER2-antibody conjugated polymeric nanocarrier-based drug delivery system for multi-drug-resistant breast cancer therapy. ACS Appl Mater Interfaces 6:6469–80
- Walczak H, Miller RE, Ariail K, et al. (1999). Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo. Nat Med 5:157–63
- Woodcock J, Griffin JP, Behrman RE. (2011). Development of novel combination therapies. N Engl J Med 364:985–7
- Zhang L, Chan JM, Gu FX, et al. (2008). self-assembled lipid− polymer hybrid nanoparticles: a robust drug delivery platform. ACS Nano 2:1696–702
- Zhang L, Zhu D, Dong X, et al. (2015). Folate-modified lipid–polymer hybrid nanoparticles for targeted paclitaxel delivery. Int J Nanomed 10:2101
- Zhang S, Zhao B, Jiang H, et al. (2007). Cationic lipids and polymers mediated vectors for delivery of siRNA. J Control Release 123:1–10
- Zhao X, Li F, Li Y, et al. (2015). Co-delivery of HIF1α siRNA and gemcitabine via biocompatible lipid-polymer hybrid nanoparticles for effective treatment of pancreatic cancer. Biomaterials 46:13–25