
Abstract
The present systematic study focused to investigate the oleic acid derivative of branched polyethylenimine (bPEI-OA)-functionalized proliposomes for improving the oral delivery of extract of Ginkgo biloba (GbE). The GbE proliposomes were prepared by a spray drying method at varying ratios of egg yolk phosphatidylcholine and cholesterol, and the optimized formulation was tailored with bPEI-OA to obtain bPEI-OA-functionalized proliposomes. The formulations were characterized for particle size, zeta potential, and entrapment efficiency. The release of GbE from proliposomes exhibited a sustained release. And the release rate was regulated by changing the amount of bPEI-OA on the proliposomes. The physical state characterization studies showed some interactions between GbE and other materials, such as hydrogen bonds and van der Waals forces during the process of preparation of proliposomes. The in situ single-pass perfusion and oral bioavailability studies were performed in rats. The significant increase in absorption constant (Ka) and apparent permeability coefficient (Papp) from bPEI-OA-functionalized proliposomes indicated the importance of positive charge for effective uptake across the gastrointestinal tract. The oral bioavailability of bPEI-OA-functionalized proliposomes was remarkable enhanced in comparison with control and conventional proliposomes. The bPEI-OA-functionalized proliposomes showed great potential of improving oral absorption of GbE as a suitable carrier.
Introduction
Oral delivery has always been the most preferred route of drug administration due to its advantages of ease to carry and take, safety, and good patient compliance. However, many herbal and chemical synthetic drugs with poor oral bioavailability are restricted by low solubility, poor permeability across cell membranes, enzymatic degradation in the gastrointestinal (GI) tract, and high hepatic metabolism (Zhang et al., Citation2015b). Various nanocarrier systems have been developed for improving the oral bioavailability, such as liposomes (Sun et al., Citation2010), micelles (Ouahab et al., Citation2012), solid lipid nanoparticles (SLN) (Hosny & Aljaeid, Citation2014), and self-nanoemulsifying drug delivery system (SNEDDS) (Tang et al., Citation2008), nanocrystal (Du et al., Citation2013), and porous absorbent particles (Zhang et al., Citation2014, Citation2015a; Wang et al., Citation2015). Among them, liposomes have been broadly researched and applied as drug carriers since 1965 (Karn et al., Citation2013). The similarity between lipid bilayers of liposomes and biomembranes contributes to the bioadhesive and biocompatible properties of liposomes. Hence, the oral absorption of liposomal drugs can be enhanced by adhering to the surface of the GI tract. Nevertheless, the success of oral liposomes is limited by some physicochemical stability problems including drug leakage, aggregation, sedimentation, phospholipid susceptibility to hydrolysis, and oxidation (Hiremath et al., Citation2009). The solid proliposomes system, consisting of a mixture of phospholipids, drugs, and other materials can solve the stability concerns. Proliposomes are defined as dry, free-flowing powder products, which can disperse to form a liposome suspension upon the addition of water (Yanamandra et al., Citation2014). Several studies have been reported that the adoption of oral proliposomes could enhance oral bioavailability for low soluble drugs (Xiao et al., Citation2006; Xu et al., Citation2009; Bobbala & Veerareddy, Citation2012).
Extract of Ginkgo biloba (GbE), an extract of Ginkgo biloba tree leaves, has become one of the most widely used herbal medicines all over the world (Rao et al., Citation2014). GbE is commonly used for the treatment of cardiovascular disease and age-related mental deterioration (Chen et al., Citation2013). The most of positive effects are based on the bioactive components of GbE, such as flavonoid glycosides (quercetin, kaempferol, and isorhamnetin) and terpene lactones (bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C) while the clinical efficacy of GbE products is limited by poor oral bioavailabilities of flavonoid glycosides (Singh et al., Citation2008). In our previous study, a formulation of GbE proliposomes containing sodium deoxycholate has been developed and significantly enhanced drug absorption in the GI tract (Zheng et al., Citation2015). Compared to GbE, the relative bioavailabilities (RA) of quercetin, kaempferol, isorhmnetin, ginkgolide A, ginkgolide B, and ginkgolide C from GbE proliposomes were 2.45, 2.11, 2.64, 2.03, 3.33, and 2.94, respectively. The previous study mainly focused on the development of GbE proliposomes and the factor of bile salt (sodium deoxycholate) in improving the oral delivery.
Polyethylenimine (PEI, 25 kDa), a polymer with positive charge density and high molecular weight, possesses high transfection activity and high cytotoxicity (Yang et al., Citation2015). Several studies have been reported that hydrophobic derivatives of PEI, conjugating lipophilic substances to PEI, could dramatically reduce cytotoxicity, and increase gene delivery effectively (Kim et al., Citation2001; Xie et al., Citation2013). However, there are few reports on proliposomes containing hydrophobic derivatives of PEI for improving the oral absorption of drugs in the GI tract. Hydrophobic derivatives of PEI, amphiphilic components, could be embedded into the lipid bilayers to control the release rate of drug, and enhance the adhesion between proliposomes containing (positive charged) and intestinal epithelial cells (negative charged) in the GI tract.
In this study, an oleic acid (OA) derivative of branched PEI (bPEI-OA) was systhesized. The conventional GbE proliposomes and the bPEI-OA-functionalized proliposomes were prepared by a spray drying method and characterized. Subsequently the physical state characterization was performed to confirm the possible interaction between the formulations materials if any. In situ single-pass perfusion and oral bioavailability studies were conducted in rats to investigate the influence of bPEI-OA on the oral absorption of GbE.
Materials and methods
Materials
GbE was a gift from Yangtze River Pharmaceutical Group (Jiangsu, China), and its contents of flavonoid glycosides (quercetin, kaempferol, and isorhamnetin) and terpene lactones (bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C) in GbE were about 5.4%, 3.2%, 2%, 3.2%, 2.6%, 1.6%, and 1.4%, respectively. Egg yolk phosphatidylcholine (>80% purity, EPC) was obtained from Tywei Pharmaceutica Co. Ltd (Shanghai, China). Cholesterol (≥99% purity), mannitol (≥98% purity), branched polyethylenimine (25 kDa, bPEI), oleoyl chloride (≥99% purity, OC), triethylamine (TEA), dichloromethane (≥99% purity), diethyl ether (≥99% purity), and ethyl acetate (99.9% purity) were purchased from Sigma-Aldrich Co. (St. Louis, MO). All other chemicals used were of analytical grade, and solvents were of HPLC grade.
Animals
Male Wistar rats (240 ± 5 g, n = 27) were purchased from the Laboratory Animal Center of Jilin University (Changchun, China). All the rats were housed at a temperature of 25 ± 2 °C, under 12-h light/dark cycles with free access to water and food for 1 week before the experiment. The experimental protocol was approved by the Laboratory Animal Center of Jilin University (license No. SCXK-(JI) 2011-0003).
Synthesis of bPEI-OA
The amphiphilic component, bPEI-OA, was synthesized by conjugating OC to bPEI by N-acylation (Teng et al., Citation2012). In brief, 50 mg of bPEI and 5 μl of TEA were dissolved in 4 ml dichloromethane under N2 at room temperature. 75 mg of OC was dissolved in 4 ml dichloromethane and then slowly added into the PEI solution under stirring. The reaction processed for 24 h under N2 at room temperature. The precipitation was obtained using an excess volume of diethyl ether followed by washed three times. The product bPEI-OA was collected after drying under vacuum.
Preparation of proliposome powders
The proliposome powders were prepared by a spray drying method (Zheng et al., Citation2015). The lipid composition of the formulations is depicted in . In brief, EPC and cholesterol were dissolved in 5 ml ethanol as a lipid phase. 200 mg of GbE was dissolved in a mixture of 50 ml deionized water and 5 ml ethanol as an aqueous phase. The lipid phase was injected into the magnetically stirred aqueous phase at 60 ± 1 °C. The resultant solution was spray dried using a spray dryer (SP-1500, Shanghai Shuyi Tech. Co., Ltd, Shanghai, China) to obtain proliposome powders after adding 1500 mg of mannitol as a protective carrier. The obtained proliposome powders were stored in a desiccator at room temperature.
Table 1. Composition of GbE proliposomes.
The bPEI-OA-functionalized proliposome powders were prepared by adding bPEI-OA (various mol% of EPC) into the lipid phase of the proliposome formulation II (EPC: cholesterol molar ratio of 2:1), and processed as described above.
UHPLC-MS/MS analysis of flavonoid glycosides and terpene lactones
For quantitative analysis of flavonoid glycosides (quercetin, kaempferol, and isorhamnetin) and terpene lactones (bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C), a Waters Xevo TQ-S triple quadruple mass spectrometer (MS/MS) (Waters, Milford, MA) connecting with a Waters Acquity UHPLC system was used.
The mass spectrometer was operated in positive electrospray ionization (ESI+) mode. The optimized instrumental parameters for mass spectral acquisition were as follows: capillary voltage at 3.0 kV; source voltage at 60 V; source temperature at 150 °C; desolvation temperature at 500 °C; cone gas flow at 150 L/h; and desolvation gas flow at 1000 L/h. The optimized conditions of precursor to product ion pairs performed in multiple-reaction monitoring (MRM) mode are described in . Mass Lynx software version 4.1 was used to control all parameters of UHPLC and MS/MS.
Table 2. The information for MRM parameters used for the flavonoid glycosides and terpene lactones.
Chromatographic separations of prepared samples were obtained using a Waters Acquity BEH C18 column (2.1 mm × 50 mm, 1.7 μm) with a 0.2 μm filter. Acetonitrile (containing 0.01% formic acid) (A) and water (containing 0.01% formic acid) (B) were used as the mobile phase, and a gradient program was set as follows: 0–0.6 min, 5%–30% A; 0.6–3 min, 30%–80% A; 3–4 min, 80%–90% A; 4–4.1 min, 90%–5% A; 4.1–6 min, 5%–5% A. The analysis was performed at 40 °C and at a flow rate of 0.4 ml/min.
The samples obtained in the study of encapsulation efficiency (EE), drug release, in situ single-pass perfusion, and oral bioavailability were prepared as follows. In brief, 50 μl of sample was treated with 50 μl of 4 M hydrochloric acid (containing 10 mM ascorbic acid) and 50 μl of internal standard (IS) (5 ng/ml taxifolin), and vortexed for 10 s. The resulting mixture was then acid hydrolyzed at 80 °C for 30 min in a water bath. After cooled immediately to room temperature, 600 μl of ethyl acetate was added and vortexed for 1 min. After centrifugation at 12 000 rpm for 10 min, the supernatant was dried at 37 °C under nitrogen. The residue was reconstituted with 100 μl of acetonitrile, followed by centrifugation at 12 000 rpm for 10 min. 2 μl of the resulting supernatant was used for further UHPLC-MS/MS analysis. The quantification analysis of quercetin, kaempferol, isorhamnetin, bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C was performed by establishing standard curves with excellent linearity (R2 > 0.995). The amounts of flavonoid glycosides and terpene lactones were calculated using the following equations:
Where mf is the amount of flavonoid glycosides, mQCT, mKMF, and mISR are the amount of quercetin, kaempferol, and isorhamnetin, respectively, mt is the amount of terpene lactones, mBBL, mGLA, mGLB, and mGLC are the amount of bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C, respectively.
Characterization of GbE proliposomes
Morphology
The surface morphology of proliposomes was evaluated using a scanning electron microscope (SEM) (JSM-6700F, JEOL, Akishima, Tokyo, Japan) (Chen et al., Citation2014). The liposomal suspension was reconstituted by hydrating proliposome powders with deionized water followed by shaking manually for 2 min. A thin film of samples was made on a silicon slice by dripping a drop of liposomal suspension and naturally dried. Samples were made electrically conductive by coating with a thin layer of gold. SEM photographs were taken at 3.0 kV accelerating voltage.
Particle size and zeta potential
The particle size and zeta potential of the reconstituted liposomes were measured by using a Nicomp Zeta Potential/Particle Sizer (Model 380 ZLS, Santa Barbara, CA) (Kosloski et al., Citation2011). Each sample was diluted to a suitable concentration with deionized water, and analysis was performed at 23 °C with a fixed detection angle of 90°.
Encapsulation efficiency
The EE of the reconstituted liposomes, defined as the percentage of flavonoid glycosides and terpene lactones encapsulated in the liposomal vesicles, was determined by the ultracentrifugation method (Jin et al., Citation2013). An aliquot of reconstituted liposomes was ultracentrifuged (Optima MAX-XP Ultracentrifuge, Beckman Coulter Inc., Pasadena, CA) at 100 000 rpm and at 4 °C for 1 h. After ultracentrifugation, the amounts of flavonoid glycosides and terpene lactones in the supernatant, defined as the unencapsulated drug, were analyzed using the UHPLC-MS/MS method. Further, the amounts of flavonoid glycosides and terpene lactones in the equal volume of reconstituted liposomes, defined as the total drug, were directly analyzed by UHPLC-MS/MS. The experiment was performed in triplicate, and the EEs of flavonoid glycosides and terpene lactones were calculated using the following equations:
where EEf is the encapsulation efficiency of flavonoid glycosides, EEt is the encapsulation efficiency of terpene lactones, muf is the amount of unencapsulated flavonoid glycosides, mtf is the total amount of flavonoid glycosides, mut is the amount of unencapsulated terpene lactones, and mtt is the total amount of terpene lactones.
In vitro release study
The release behavior of flavonoid glycosides and terpene lactones from the reconstituted liposomes were evaluated in different kinds of release medium (hydrochloric acid solution at pH 1.2, and phosphate buffer saline at pH 6.8) using the dialysis method (molecular weight cut-off 12 000–14 000 Da). Control (GbE dissolved in 20% v/v ethanol solution) or liposomal suspension (equivalent to 5 mg of GbE) were added into the dialysis bag and were kept in 500 ml of release medium. The release system was constantly stirred and the temperature was kept at 37 ± 0.5 °C throughout the study. Aliquots of 1 ml release medium were withdrawn at fixed time points, and equal volumes of fresh release medium were added back. The samples were analyzed by UHPLC-MS/MS.
Physical state characterization
Differential scanning calorimetry (DSC)
The physical state of proliposomes was evaluated using a differential scanning calorimeter (Mettler DSC STARe System, Mettler-Toledo, Germany) (Singh et al., Citation2014). The DSC analysis of GbE, cholesterol, mannitol, proliposome powders, and relevant physical mixture were performed by scanning in the temperature range of 25–300 °C at a rate of 10 °C/min under a nitrogen gas flow of 50 ml/min. A second empty aluminum pan was used as the reference. DSC thermograms of samples were recorded for the comparison.
Fourier transform infrared spectroscopy (FT-IR)
The interaction between GbE and other materials in proliposomes was evaluated using a FT-IR spectrophotometer (IR Prestige-21, SHIMADZU, Tokyo, Japan). The FT-IR analysis of GbE, cholesterol, mannitol, proliposome powders, and relevant physical mixture were performed by scanning from 400 to 4000 cm−1 at a spectral resolution of 4 cm−1 using the conventional KBr pellet method (Jayant et al., Citation2009). A background spectrum was recorded using the pure KBr under identical conditions. FT-IR spectra of samples were recorded for the comparison.
In situ single-pass perfusion study
Before perfusion experiment, rats were fasted for over 12 h and had free access to water. Nine rats were randomly divided into three groups. Under anesthesia by intraperitoneal pentobarbital sodium (40 mg/kg body weight), a gentle midline incision was made on the abdomen after fixing rats on a heated plate to maintain body temperature. The small intestine segment was exposed, and the duodenum of approximately 10 cm was cannulated with silicone tubes on two ends and attached to a peristaltic pump (BT100-2 J, LongerPump Ltd., Baoding, China) followed by ligation with silk suture. Some pieces of degreasing cotton wetted with normal saline (37 °C) were covered on the entire surgical area for keeping moist. The duodenum segment was gently flushed with normal saline at 37 °C for 20 min until the effluent was clear. The inlet was then perfused with Krebs-Rings solution containing a known concentration of drug at 0.2 ml/min for 30 min to assure steady state conditions (Sun et al., Citation2014). Eight samples were collected from the outlet using pre-weighed tubes at intervals of 15 min for 2 h. Then the samples were weighed and centrifuged at 5000 rpm for 10 min. At the end of the experiment, the length and circumference of the perfused duodenum segment were measured. The inlet drug solution and outlet samples were analyzed by UHPLC-MS/MS. The absorption constant (Ka) and apparent permeability coefficient (Papp) were calculated using the following equations:
where min is the weight of the inlet drug solution, mout is the weight of the outlet sample, pin is the density of the inlet drug solution, pout is the density of the outlet sample, Cin is the concentration (ng/ml) of the drug in the inlet drug solution, Cout is the concentration (ng/ml) of the drug in the outlet sample, Vin is the volume of the inlet drug solution, Vout is the volume of the outlet sample, Q is the flow rate (ml/min) of the inlet drug solution, and V, l, and r are the volume, length, and radius of the perfused duodenum segment, respectively.
Oral bioavailability study
Before pharmacokinetic experiment, rats were fasted for over 12 h and had free access to water. Eighteen rats were randomly divided into three groups. Control group was administered a single oral suspension of GbE, and the two tested groups were administered single oral suspension of formulation II or VI, respectively. The oral dose was set at 25 mg/kg. Blood samples of approximately 0.5 ml were collected from the fossa orbital vein into 1.5 ml heparinized centrifuge tubes before drug administration and at 0.083, 0.167, 0.333, 0.5, 1, 2, 4, 6, 8, 10, 12, and 24 h post-dosing, and were centrifuged at 10 000 rpm for 10 min. Plasma samples were collected into new centrifuge tubes and stored at −20 °C until UHPLC-MS/MS analysis.
The pharmacokinetic parameters includes maximum plasma concentration (Cmax), time of the maximum concentrations (Tmax), elimination half-time (T1/2), area under plasma level-time curve (AUC0−t), and elimination rate constant (K) were calculated through non-compartmental model analysis using the Drug and Statistics 3.0 program (DAS, College of Life Sciences at Jilin university, Changchun, China).
Statistical analysis
All the data were expressed as mean ± standard deviation (SD). The identification of significant differences between different groups was carried out with t-test. p < 0.05 was considered statistically significant.
Results and discussion
Preparation and characterization of proliposome powders
GbE proliposome powders were prepared successfully in the present study. Several methods have been reported for the preparation of proliposomes, including film deposition on carrier method (Gupta et al., Citation2008), freeze-drying method (Guan et al., Citation2015), spray drying method (Rojanarat et al., Citation2012), fluidized bed method (Chen & Alli, Citation1987), solid dispersion and effervescent method (Fu et al., Citation2013), and solvent evaporation method (Hiremath et al., Citation2009). Because of its advantages of simple process, low cost, easy operation, and easy industrialization, the spray drying method was chosen for the preparation of GbE proliposome powders.
The particle sizes, zeta potentials, and EEs of various proliposome formulations were studied. As shown in , the mean particle sizes of the reconstituted liposomes for formulation I to VII were found to be in the range of 100–230 nm. The particle sizes of formulation V to VII were smaller than that of formulation II, and the particle size descended with an increase of amount of bPEI-OA. The results suggested that bPEI-OA has an influence on the particle size of liposomes. The value of polydispersity index (PDI) used for the evaluation of size distribution was within an acceptable limit (<0.3). The zeta potentials of the reconstituted liposomes (formulation I to IV) were between −5.7 and −3.8 mV. The existence of acidic phospholipids in EPC could be the reason for the feeble charge of formulation I to IV, and proliposomes without bPEI-OA were considered to be neutral. The surface charge of proliposomes affected the drug uptake by biological membrane due to electrostatic interaction. The bPEI-OA synthesis in low, middle, and high amounts were added into formulation II to prepare positive charged proliposomes (formulation V to VII). With the increasing amount of bPEI-OA, the zeta potentials of formulation V to VII were + 12.3 ± 2.7, +25.2 ± 3.2, and +37.6 ± 4.5 mV, respectively. The EEs of flavonoid glycosides and terpene lactones from proliposome formulations were within 71∼94%, and 77∼97%, respectively. With a decrease of the ratio of EPC to cholesterol, the EEs of flavonoid glycosides and terpene lactones both ascended at first and descended at last. The effect of ratio of EPC to cholesterol on EE indicated that cholesterol could increase EE and reduce the drug leakage at an appropriate concentration. The EEs of formulation III and IV were lower than formulation II due to the negative effect of higher concentration of cholesterol on reconstituted liposomes, which has been well documented in literature (Janga et al., Citation2012). Importantly, the bPEI-OA synthesis, as an amphiphilic compound, could decrease the particle size and increase the EE of proliposome formulation ().
Table 3. Characterization of GbE proliposome formulations.
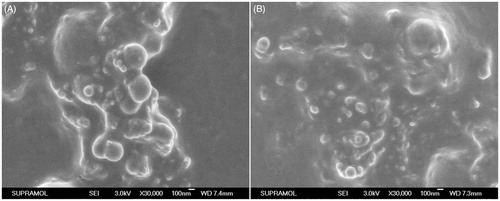
The surface morphology of proliposome powders was examined by SEM, and the micrographs are shown in . The surface morphology of formulation II in was similar to that of formulation VI from . Some spherical particles seemed to be deposited on the illegible surface of mannitol in both of formulations.
Figure 1. Scanning electron micrographs of proliposome powders (A) formulation II and (B) formulation VI both at ×20 000 maginfication.
In vitro release study
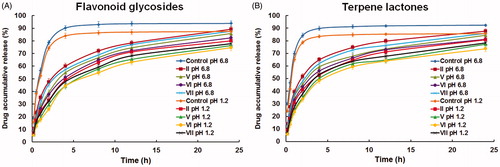
shows the in vitro release profiles of flavonoid glycosides and terpene lactones from proliposome formulations in hydrochloric acid (pH 1.2) and phosphate buffer saline (pH 6.8), respectively. The release of flavonoid glycosides and terpene lactones from control drug solution proceeded at a high rate, with approximately 80% release at pH 1.2 and approximately 90% release at pH 6.8 at 4 h. The release of flavonoid glycosides and terpene lactones from proliposome formulations appeared to be much slower, with less than 60% release for flavonoid glycosides and less than 70% for terpene lactones. The release of flavonoid glycosides and terpene lactones from control drug solution and proliposome formulations were both influenced by the pH of media. And, it was apparent that the release of flavonoid glycosides and terpene lactones were faster from formulation II compared to other formulations (V to VII) due to the effect of bPEI-OA. Among the formulations with bPEI-OA, the release rate of formulation VI was the slowest, which was supported by the results of EE. The slow release could be derived from high EE. Hence, formulation VI (proliposomes containing middle amount of bPEI-OA) was chosen for the following studies, including DSC, FT-IR, in situ single-pass perfusion, and oral bioavailability study.
Figure 2. In vitro release profiles of (A) flavonoid glycosides and (B) terpene lactones from proliposome formulations in hydrochloric acid (pH 1.2) and phosphate buffer saline (pH 6.8) (mean ± SD, n = 3).
Physical state characterization
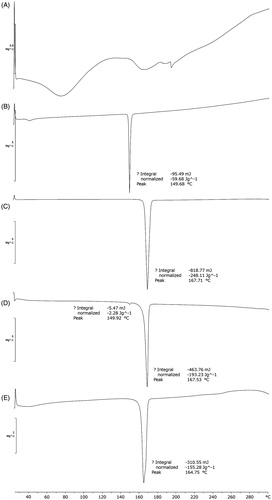
As shown in , several endothermic peaks could be seen in the DSC thermogram of GbE because GbE is a mixture of compounds. The DSC thermogram of physical mixture showed two endothermic peaks at 149.92 and 167.53 °C, which had the same peak temperature with that of cholesterol (149.68 °C) and mannitol (167.71 °C), indicating the additive effect. shows that the DSC thermogram of proliposome powders had a single and broad endothermic peak. Compared to physical mixture, the intrinsic endothermic peak of cholesterol was disappeared and the endothermic peak of mannitol was shifted to 164.75 °C, indicating that there were probably some interactions between GbE and materials, such as hydrogen bonds and van der Waals forces during the process of preparation of proliposome powders (Tan et al., Citation2012).
Figure 3. DSC thermograms of (A) GbE, (B) cholesterol, (C) mannitol, (D) physical mixture, (E) proliposome powders (formulation VI).
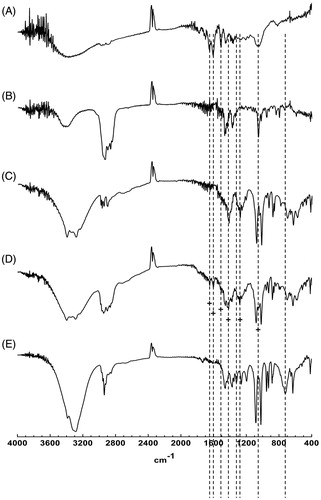
As shown in , the physical state characterization of GbE, materials, and proliposome powders were further confirmed by FT-IR. shows the characteristic absorption peaks of GbE were present at 1649.14, 1604.77, and 1508.33 cm−1. The characteristic absorption peak of cholesterol was present at 1056.99 cm−1. The characteristic absorption peaks of mannitol were seen at 1417.68 and 1280.73 cm−1. The six peaks highlighted by dash lines with plus signs could be seen in the spectra of physical mixture, demonstrating that the spectra profiles of physical mixture were overlaid by that of GbE and formulation ingredients. The characteristic absorption peaks mentioned above were shielded or changed in the spectra of proliposome powders, and two new peaks at 1321.24 and 723.31 cm−1 indicated by dash lines were not seen in that of physical mixture. The difference between FT-IR spectra of proliposome powders (formulation VI) and physical mixture were consistent with the results of DSC.
Figure 4. FT-IR spectra of (A) GbE, (B) cholesterol, (C) mannitol, (D) physical mixture, (E) proliposome powders (formulation VI).
In situ single-pass perfusion study
shows the in situ intestinal absorption parameters of seven components from control, formulation II and VI, respectively. Compared to control, the significant enhancement in Ka and Papp of quercetin, kaempferol, isorhmnetin, ginkgolide A, ginkgolide B, and ginkgolide C from proliposome formulation II (p < 0.05) and formulation VI (p < 0.001). And, the values of Ka and Papp of quercetin, kaempferol, isorhmnetin, ginkgolide A, ginkgolide B, and ginkgolide C from formulation VI significantly increased (p < 0.05) with respect to formulation II, except for bilobalide. Comparing the Papp of formulation II and VI with that of control, the enhancement ratios (ER) of proliposome formulations were obtained. The values of ER of quercetin, kaempferol, isorhmnetin, bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C were greater than 1 indicated that the potential of proliposomes for improved oral delivery. Compared to the conventional proliposomes (formulation II), the bPEI-OA-functionalized proliposomes (formulation VI) had a better absorption indicated that the importance of positive charge for effective uptake across the GI tract.
Table 4. In situ intestinal absorption parameters of seven components from control, formulation II and VI, respectively (mean ± SD, n = 3).
Oral bioavailability study
shows the concentration-time profiles of seven components in rat plasma following oral administration of control, formulation II and VI. The pharmacokinetic parameters are present in . The values of Cmax and AUC0–t of quercetin, kaempferol, isorhmnetin, ginkgolide A, ginkgolide B, and ginkgolide C for proliposomes (formulation II and VI) were significantly higher than those of control (p < 0.01). However, the Cmax and AUC0–t of bilobalide showed no obvious difference. Compared to control, the values of Tmax and T1/2 of quercetin, kaempferol, isorhmnetin, bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C in proliposomes showed a tendency to increase. A significantly higher values of K of quercetin, kaempferol, isorhmnetin, ginkgolide A, ginkgolide B, and ginkgolide C from proliposomes than control suggested proliposome formulations could decrease the elimination rate of GbE.
Figure 5. Concentration-time profiles of seven components in rat plasma following oral administration of control, formulation II and VI, respectively (mean ± SD, n = 6).
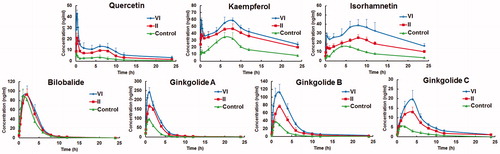
Table 5. Pharmacokinetic parameters of seven components in rats following oral administration of control, formulation II and VI, respectively (mean ± SD, n = 6).
In , a higher RA of quercetin, kaempferol, isorhmnetin, bilobalide, ginkgolide A, ginkgolide B, and ginkgolide C from proliposome formulations were shown by comparing the AUC0–t of formulation II and VI with that of control. Among the three formulations tested, the order of RA was VI > II > control. The mechanisms of improved oral bioavailability from proliposomes have been discussed in our previous study (Zheng et al., Citation2015). Importantly, formulation VI significantly (p < 0.01) enhanced the oral absorption of quercetin, kaempferol, isorhmnetin, ginkgolide A, ginkgolide B, and ginkgolide C in contrast with formulation II. In agreement with the result of in situ intestinal perfusion study, the enhancement indicated that the importance of bPEI-OA for the improved oral bioavailability of GbE. The positive charge of proliposome formulation was derived from the existence of bPEI-OA. The negative charge on the surface of intestinal epithelial cells is attributed to the negatively charged proteins and the selective active ion pumps in the outer membrane (El-Shabouri, Citation2002; Velpula et al., Citation2013). Hence, a large number of positive charged liposomes were absorbed on the surface of intestinal epithelial cells by electrostatic interaction and then uptaked by endocytosis.
Conclusion
In this study, bPEI-OA-functionalized proliposomes have been explored for the enhanced oral bioavailability of GbE. The GbE proliposomes were prepared by a spray drying method at varying ratio of EPC and cholesterol. The optimized proliposomes formulation was tailored with bPEI-OA and the bPEI-OA-functionalized proliposomes were characterized. The in situ intestinal perfusion and oral bioavailability studies indicated that the importance of bPEI-OA for the improved oral bioavailability of GbE. In summary, bPEI-OA-functionalized proliposomes would have a great potential to improve the oral absorption of drugs with poor bioavailability. However, the deeper mechanism of absorption needs further studies.
Declaration of interest
The authors report no declarations of interest.
References
- Bobbala SKR, Veerareddy PR. (2012). Formulation, evaluation, and pharmacokinetics of isradipine proliposomes for oral delivery. J Liposome Res 22:285–94
- Chen CM, Alli D. (1987). Use of fluidized bed in proliposome manufacturing. J Pharm Sci 76:419
- Chen F, Li L, Xu F, et al. (2013). Systemic and cerebral exposure to and pharmacokinetics of flavonols and terpene lactones after dosing standardized Ginkgo biloba leaf extracts to rats via different routes of administration. Br J Pharmacol 170:440–57
- Chen J, Ding BB, Wang TY, et al. (2014). Facile synthesis of uniform ZnxCd1-xS alloyed hollow nanospheres for improved photocatalytic activities. J Mater Sci-Mater Electron 25:4103–9
- Du B, Shen GP, Wang DD, et al. (2013). Development and characterization of glimepiride nanocrystal formulation and evaluation of its pharmacokinetic in rats. Drug Deliv 20:25–33
- El-Shabouri MH. (2002). Positively charged nanoparticles for improving the oral bioavailability of cyclosporin-A. Int J Pharm 249:101–8
- Fu Q, Fu HL, Huan L, et al. (2013). Preparation of cefquinome sulfate proliposome and its pharmacokinetics in rabbit. Iran J Pharm Res 12:611–21
- Guan P, Lu Y, Qi J, et al. (2015). Solidification of liposomes by freeze-drying: the importance of incorporating gelatin as interior support on enhanced physical stability. Int J Pharm 478:655–64
- Gupta V, Barupal AK, Ramteke S. (2008). Formulation development and in vitro characterization of proliposomes for topical delivery of aceclofenac. Indian J Pharm Sci 70:768–75
- Hiremath PS, Soppimath KS, Betageri GV. (2009). Proliposomes of exemestane for improved oral delivery: formulation and in vitro evaluation using PAMPA, Caco-2 and rat intestine. Int J Pharm 380:96–104
- Hosny KM, Aljaeid BM. (2014). Sildenafil citrate as oral solid lipid nanoparticles: a novel formula with higher bioavailability and sustained action for treatment of erectile dysfunction. Expert Opin Drug Deliv 11:1015–22
- Janga KY, Jukanti R, Velpula A, et al. (2012). Bioavailability enhancement of zaleplon via proliposomes: role of surface charge. Eur J Pharm Biopharm 80:347–57
- Jayant RD, McShane MJ, Srivastava R. (2009). Polyelectrolyte-coated alginate microspheres as drug delivery carriers for dexamethasone release. Drug Deliv 16:331–40
- Jin Y, Wen JY, Garg S, et al. (2013). Development of a novel niosomal system for oral delivery of Ginkgo biloba extract. Int J Nanomed 8:421–30
- Karn PR, Cho W, Hwang SJ. (2013). Liposomal drug products and recent advances in the synthesis of supercritical fluid-mediated liposomes. Nanomedicine 8:1529–48
- Kim S, Choi JS, Jang HS, et al. (2001). Hydrophobic modification of polyethyleneimine for gene transfectants. Bull Korean Chem Soc 22:1069–75
- Kosloski MP, Peng A, Varma PR, et al. (2011). Immunogenicity and pharmacokinetic studies of recombinant factor VIII containing lipid cochleates. Drug Deliv 18:246–54
- Ouahab A, Shen Y, Tu JS. (2012). Novel oral delivery system of indomethacin by solidified mPEG-PDLLA micelles: in vivo study. Drug Deliv 19:232–7
- Rao Z, Qin HY, Wei YH, et al. (2014). Development of a dynamic multiple reaction monitoring method for determination of digoxin and six active components of Ginkgo biloba leaf extract in rat plasma. J Chromatogr B 959:27–35
- Rojanarat W, Nakpheng T, Thawithong E, et al. (2012). Inhaled pyrazinamide proliposome for targeting alveolar macrophages. Drug Deliv 19:334–45
- Singh B, Kaur P, Singh GRD, Ahuja PS. (2008). Biology and chemistry of Ginkgo biloba. Fitoterapia 79:401–18
- Singh C, Bhatt TD, Gill MS, Suresh S. (2014). Novel rifampicin-phospholipid complex for tubercular therapy: synthesis, physicochemical characterization and in-vivo evaluation. Int J Pharm 460:220–7
- Sun JK, Deng YG, Wang SL, et al. (2010). Liposomes incorporating sodium deoxycholate for hexamethylmelamine (HMM) oral delivery: development, characterization, and in vivo evaluation. Drug Deliv 17:164–70
- Sun YH, He X, Yang XL, et al. (2014). Absorption characteristics of the total alkaloids from Mahonia bealei in an in situ single-pass intestinal perfusion assay. Chin J Nat Med 12:554–60
- Tan QY, Liu S, Chen XL, et al. (2012). Design and evaluation of a novel evodiamine-phospholipid complex for improved oral bioavailability. AAPS PharmSciTech 13:534–47
- Tang JL, Sun J, Cui FD, et al. (2008). Self-emulsifying drug delivery systems for improving oral absorption of Ginkgo biloba extracts. Drug Deliv 15:477–84
- Teng LS, Xie J, Teng LR, Lee RJ. (2012). Enhanced siRNA delivery using oleic acid derivative of polyethylenimine. Anticancer Res 32:1267–71
- Velpula A, Jukanti R, Janga KY, et al. (2013). Proliposome powders for enhanced intestinal absorption and bioavailability of raloxifene hydrochloride: effect of surface charge. Drug Dev Ind Pharm 39:1895–906
- Wang Y, Han N, Zhao QF, et al. (2015). Redox-responsive mesoporous silica as carriers for controlled drug delivery: a comparative study based on silica and PEG gatekeepers. Eur J Pharm Sci 72:12–20
- Xiao YY, Song YM, Chen ZP, Ping QN. (2006). Preparation of silymarin proliposome: a new way to increase oral bioavailability of silymarin in beagle dogs. Int J Pharm 319:162–8
- Xie J, Teng L, Yang Z, et al. (2013). A polyethylenimine-linoleic acid conjugate for antisense oligonucleotide delivery. BioMed Res Int 2013:710502
- Xu HT, He L, Nie SF, et al. (2009). Optimized preparation of vinpocetine proliposomes by a novel method and in vivo evaluation of its pharmacokinetics in New Zealand rabbits. J Control Release 140:61–8
- Yanamandra S, Venkatesan N, Kadajji VG, et al. (2014). Proliposomes as a drug delivery system to decrease the hepatic first-pass metabolism: case study using a model drug. Eur J Pharm Sci 64:26–36
- Yang S, Guo ZH, Yang XW, et al. (2015). Enhanced survivin siRNA delivery using cationic liposome incorporating fatty acid-modified polyethylenimine. Chem Res Chin Univ 31:401–5
- Zhang LH, Zhu WF, Lin QS, et al. (2015a). Hydroxypropyl-beta-cyclodextrin functionalized calcium carbonate microparticles as a potential carrier for enhancing oral delivery of water-insoluble drugs. Int J Nanomed 10:3291–302
- Zhang YZ, Che EX, Zhang M, et al. (2014). Increasing the dissolution rate and oral bioavailability of the poorly water-soluble drug valsartan using novel hierarchical porous carbon monoliths. Int J Pharm 473:375–83
- Zhang YZ, Zhao QF, Zhu WF, et al. (2015b). Synthesis and evaluation of mesoporous carbon/lipid bilayer nanocomposites for improved oral delivery of the poorly water-soluble drug, nimodipine. Pharm Res 32:2372–83
- Zheng B, Teng L, Xing G, et al. (2015). Proliposomes containing a bile salt for oral delivery of Ginkgo biloba extract: formulation optimization, characterization, oral bioavailability and tissue distribution in rats. Eur J Pharm Sci 77:254–64