
Abstract
Context: Nasal route of drug administration is preferred more and more for the targeted delivery to the brain in current drug development scenario due to its ease of use, reliability, quick action, and lesser side effects. Those CNS drugs which have limited oral bioavailability due to pharmacokinetic consequences and brain barrier repulsion are getting onto this direction.
Objective: Quetiapine fumarate, an analogous to above and an antischizophrenic agent, is tested for its diffusion property with and without lipophilic carrier through sheep nasal membrane. Being a BCS class II' and high permeable candidate, it tends to crossover easily, so made up in a simple dispersion.
Materials and methods: To improve its diffusion rate, it was embedded into liposomal dispersion, which has proven that it has advanced efficiency for diffusion. For this, both the formulations were checked and compared for their diffusion profile, as it is an essential property for bioavailability through nasal route. Comparison was made on the basis of % drug diffusion within 6 h, rate, mechanism, profile, and coefficient.
Results: Liposomal dispersion has been proved superior with greater percentage diffusion of 32.61 ± 1.70 and very high permeability with a coefficient value of 4.1334 ± 0.7321 (× 10 − 5 cm/s). Diffusion profile comparison bearing dissimilarity of 18 and similarity of 74 indicated that the diffusion profiles of liposomal dispersions and simple dispersion were similar but not identical.
Conclusion: Liposomal diffusion supremacy was further sustained by in vivo, ciliotoxicity, and gamma scintigraphy studies.
Introduction
Schizophrenia is a syndrome of central nervous system (CNS) and categorized by the unexpected collapse of the thought processes and impulsive psychological depression. Because of rigorous permeability of blood–brain barrier (BBB), it is considered to be the key obstacle in the development of brain-targeted drug delivery (Pathan et al., Citation2009). Several endeavors have been demonstrated like administration of drug through nasal route (Patel et al., Citation2014) and/or equestrian with nanocarriers to overcome the concerns of the drug transportation across BBB (Abbott & Romero, Citation1996; Nivsarkar et al., Citation2014). The Olfactory region is of prime interest for the drug delivery to the brain in modernization as it bypasses the brain barrier (Frey, Citation2002). Nasal delivery is a convenient and effective system that provides a quick absorption of the drug into the systemic circulation (Kumar et al., Citation2014) which results into speedier onset of pharmacological activity, circumventing the hepatic first pass clearance enables reduction in the dose and least side effects (Ugwoke et al., Citation2001; Arora et al., Citation2002; Türker et al., Citation2004; Parmar et al., Citation2011).
Quetiapine fumarate (QTF) is used for the treatment of schizophrenia and also for the acute manic episodes associated with bipolar I disorder. Being a member of biopharmaceutical classification system (BCS) class II, QTF possesses high permeability and less solubility. QTF has limited oral bioavailability (7–9%) due to hepatic metabolism and elimination by BBB (Sweet man. 2007). Although various strategies have been applied to attain brain delivery of QTF, still requires further improvement (Kararli et al., Citation1992; Lohan et al., Citation2015). Several studies reported that lipophilic drugs are generally well absorbed from the nasal route with pharmacokinetic profiles (Ding et al., Citation2007), which are frequently indistinguishable to those acquired after an intravenous infusion with a bioavailability approaching 100% (Acharya et al., Citation2015).
Nanocarriers like emulsions, liposomes, and nanoparticles have been utilized largely for brain targeting because methods of preparation are simple and easy to scale-up (Garcia et al., Citation2005; Woensel et al., Citation2013). Liposomes have been established as a versatile carrier for brain delivery in vivo (Shi et al., Citation2001; Xie et al., Citation2005; Schnyder et al., Citation2005; Afergan et al., Citation2008) and proved to enhance bioavailability of numerous underprivileged agents into the brain (Vyas et al., Citation1995; Alsarra et al., Citation2008; Arumugam et al., Citation2008; Hamed et al., Citation2012)
The aim of the present study was to formulate, characterize, and evaluate physical, chemical, and biological factors of simple and liposomal dispersion of QTF both having simulated nasal fluid (SNF) of pH 6.8 as a dispersed phase. Furthermore, both the formulations were compared for diffusion profile by similarity and dissimilarity factors, diffusion rate kinetic profile, and mechanism as well as diffusion coefficient through sheep nasal mucosa. Finally, brain-targeted delivery attainment of the formulations is confirmed by gamma scintigraphy and in vivo studies.
Materials and method
Materials
QTF was obtained as a gift sample from Elite Pharmaceuticals, Ahmedabad, India. Egg phosphatidylcholine (EPC) was acquired as a generous gift specimen from Vav Life Sciences Pvt. Ltd., Mumbai, India. Cholesterol (CH), Tween 80, potassium dihydrogen ortho-phosphate, sodium hydroxide, chloroform, and methanol were purchased from Astron Chemicals, Ahmedabad, India. All the chemicals and solvents used in this study were of analytical grade.
Analytical method
QTF was analyzed for %EE, %CDR, and % permeation study by UV double-beam spectrophotometer (Shimadzu-1800, Tokyo, Japan) in SNF, pH 6.8 by generating a standard curve for the entire range from 5 to 25 μg/ml at 242 nm (Vincenzo et al., Citation2003; Sahu & Rana, Citation2011). The method used for the estimation of QTF in brain homogenate and plasma involved high-performance liquid chromatography (HPLC) analysis (Model LC) using a C18 column with UV detector. Mobile phase consisted of phosphate buffer (pH 3):acetonitrile:methanol (50:40:10) and the flow rate was 0.8 ml/min. The sample volume injected was 20 μl and the wavelength was 247 nm (Reddy et al., Citation2011).
Formulation development
Formulation of QTF simple dispersion
As QTF is insoluble in water, and is, in nano-crystalline state, in the bulk form, it is easy to prepare its simple dispersion in SNF pH 6.8 with high shear impact in colloidal mill. QTF was first passed through US sieve size 200 (0.074 mm) and then QTF (10%) and Tween 80 (1.25%) were incorporated into SNF pH 6.8 with continuous stirring in a 100 ml glass beaker. Prepared dispersion was then transferred to the colloidal mill and operated for 30 min at the rate of 50 ml/min.
Formulation of QTF liposomal dispersion
Liposomes of QTF were prepared by modified thin lipid film hydration technique using a rotary flask evaporator as described by the method of Bangham, Juliano, and Daoud (Senthilkumar et al., Citation2012). Process parameters like formula, solvent system, rehydration volume, vacuum, drying and hydration time, temperature, and flask rotation speed were optimized based on the morphology of film formed and particle size distribution and drug entrapment efficiency by preliminary screening. All these factors were analyzed for their effects on liposome formulation. The effect of one variable was learned by keeping other variables consistent (Rathod & Deshpande,. Citation2010; Shariat et al., Citation2014).
In the first step of the preparation, CH:EPC in 1:2 molar ratio and 38.35 mg of QTF (a molar ratio of 1) were dissolved in 10 ml of methanol:chloroform solution of 2:1 ratio. The flask was connected to the rotary evaporator and pivoted at 90 rpm speed for 120 min at 37 °C under vacuum (600 mmHg). Our preliminary study findings showed that liposome size, zeta potential, and drug leaching could be lowered at reduced temperature, which results in more percentage drug entrapment but the time required for drying was increased.
The organic solvent was gradually removed by this procedure such that a thin, smooth, and dry film of lipid was formed on the internal plane of the flask. The film was allowed to dry for 1 h under vacuum at temperature as mentioned previously. The dry lipid film was then slowly hydrated with an aqueous phase (10 ml SNF, pH 6.8) and the flask was rerotated at the above-mentioned speed for 30 min at 20 °C. Liposomal dispersion was kept overnight just below 20 °C to ensure complete hydration. The liposomal dispersion obtained was filled in glass ampoules and sonicated for 2 min at 80% amplitude and then was subjected to centrifugation at 5000 rpm, 8 °C for 5 min using Remi ultracentrifuge. Liposomal supernatant was separated from pellet of drug and debris and then stored in refrigerator (Liu et al., Citation2013).
Here, the drug was water insoluble so that it will not be in a molecular state and free drug will be separated at lower rpm. Similarly, insoluble fragments and crystals of cholesterol and EPC will settle down in the form of pellet. The numbers of sonication cycles were previously screened and fixed for two times. The time was fixed for only 2 min as above so that liposomal dispersion catches heat and drug leaches out which results into decreased entrapment efficiency. The amplitude of sonication was kept 80% as it increases sonochemical effect, which is accountable for fine particle formation (Santos et al., Citation2009).
Characterization, evaluation, and comparison
Microscopy
Morphology of both the dispersion systems was studied under Olympus binocular microscope (Olympus Inc., Center Valley, PA) with camera attachment having S-Viewer version 1.10.6.2 software (SA Inc., Cary, NC) to study their shape with a magnification of 15 × 100.
Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) of both the formulations were performed to determine the surface morphology, size, and shape of the formulation and to observe the aggregation property.
Particle size and surface charge
The particle size and the size distribution of the formulations were determined by Malvern zetasizer based on the laser light scattering principle. A Malvern laser light-scattering zetasizer equipped with an argon laser was utilized for evaluating the particle size and the size distribution. Light scattering was monitored at 90° angle and at 25 °C. The mean droplet size was ascertained from force, volume, and bimodal distribution expecting circular particles. The zeta potential is a notation of the strength of the dispersion systems and demonstrates charge present on the dispersion systems. Zeta potential of formulations was measured using Malvern zetasizer.
% Entrapment efficiency of liposomes
The entrapment efficiency was determined by the solvent sonication method (Tomoko & Fumiyoshi, Citation2005; Brgles et al., Citation2008). Briefly, the vesicles were broken by dissolving 1 g of liposomes (equivalent to 167.8 mg QTF) into 10 ml of methanol (theoretical yield 16.78 mg/ml) and sonicated for 5 min to release the drug, which was then estimated for the drug content by diluting up to 100 times (Laouini et al., Citation2012). The percent drug entrapped (PDE) was then calculated by referring initial amount added. Alternatively, the clear supernatant can be used for the determination of free drug at 242 nm spectrophotometrically (He et al., Citation2012). The percentage of drug unentrapped can be calculated from this amount.
Ex vivo % drug diffusion (%DD)
Ex vivo drug diffusion study was carried out using freshly obtained sheep nasal mucosal epithelium from slaughter house (Arumugam et al., Citation2008; Jung et al., Citation2009; Acharya et al., Citation2015; Lohan et al., Citation2015). Mucosal membrane (0.2 mm thick, 10 mm diameter and 78.5 mm2 area) was fixed in between receptor and donor compartment of the Franz diffusion cell. Two sets of three cells were used for the formulations, one set for liposomal dispersion and another set for simple dispersion of QTF. The acceptor compartment was filled with 10 ml SNF, pH 6.8, and the donor compartment was filled with 1 ml 5% w/v dispersion. The temperature in diffusion chamber was maintained at 37 °C using thermostatic water bath. The samples were withdrawn at times 1, 2, 3, 4, 5, and 6 h and fresh SNF was added to make the required volume. The samples were filtered through 0.45 μm nylon filter membrane and analyzed using a spectrophotometer for the drug content at 242 nm. Results were expressed as an amount permeated or percent diffusion or ex vivo brain availability.
Diffusion coefficient
Permeability coefficients (Dc) were calculated according to the following equation:
where Dc (cm/s) is the permeability coefficient, dQ/dt (μg/s) is the amount of drug diffused per unit of time calculated from the regression line of time points of sampling, A (cm2) is the surface area available for permeation, and C0 (μg/ml) is the initial drug concentration in the donor compartment.
Diffusion profile
Comparison was done by calculating dissimilarity f1 and similarity factor f2. Difference variable (f1) depicts the relative lapse between two diffusion profiles and approximately the percent inaccuracy between curves. The percent error is zero when the test and reference profiles were indistinguishable and increments relatively with the divergence between the two profiles. The diffusion closeness was evaluated using the FDA prescribed methodology (f2 comparability factor). The similarity factor is a logarithmic, complementary square root change of the sum of squared errors, and it is a measure of the similitude of two respective diffusion profiles.
Diffusion kinetics – pattern and mechanism
There are a few straight and non-direct kinetic models generally used to portray diffusion patterns and mechanisms from various systems and to compare test and reference dissolution profiles. Zero order, Higuchi and Hixson–Crowell from linear models, while the first order, Weibull, Korsmeyer–Peppas, and Hopfenberg from non-linear models, were checked in the present study. All model-dependent methods were based on a curve fitting formula. The method of Bamba (Bamba et al., Citation1979) was used for choosing the most proper model.
Gamma scintigraphy
For 99mTc-labeled solution, 0.4 ml solution of SNF pH 6.8 was infused into a 10 ml sterile vacuum vial. A freshly prepared solution of SnCl2·H2O 0.5 ml was added, immediately followed by the addition of 0.1 ml of elute containing 1 mCi 99mTc as sodium pertechnetate (NaTcO4). The mixture was warmed for 10 min in a boiling water bath, and then cooled to room temperature.
For 99mTc-labeled liposomal dispersion, above prepared solution was added into the thin film formed in a round flask at the stage of rehydration instead of SNF pH 6.8. After rehydration of lipid film by the solution of sodium pertechnetate, sonication was done to reduce the size of lamella. Liposomes were separated by centrifugation, and redispersed by the addition of SNF pH 6.8.
Albino mice weighing 20–25 g were used after overnight fasting. Two mice were employed in the study. About 25 μl liposomal dispersion containing 99mTc (equivalent to 250 μCi) in SNF pH 6.8 (equivalent to 12 mCi/kg body weight) was administered intranasally to the first mouse, while in the second mouse, SNF pH 6.8 solution containing 99mTc was administered intranasally of the same strength. Dose for mice was calculated by the same method as described for in vivo study considering 1 mCi/kg dose of human (Hildebrandt et al., Citation2008).
In vivo study
All the animal studies were properly designed and performed, which were reviewed and approved by an institutional animal ethics committee. Albino mice weighing 25 g were used after overnight fasting. Human equivalent dose for mice was calculated by body surface area as well as weight method (Freireich, Citation1966). General dose for 60 kg healthy human is 400 mg, so human equivalent dose (HED) is 6.67 mg/kg. So, dose for mice weighing 20–25 g is about 2 mg.
Two groups of mice were employed in the study and measurements were made using three (n = 3) mice at each time point for each formulation. About 10 μl QTF dispersion containing 2 mg QTF in SNF pH 6.8 was administered intranasally to the first group, while in the second group, QTF 100 μl liposomes containing 2 mg QTF was administered intranasally. For this study, mice were tied up in supine condition and nasal administration was done by micropipette. Mice were sacrificed after every 1 h interval of administration by stunning, blood was collected from carotid artery after decapitation; intact brains were excised from the skull and homogenized immediately at 5000 rpm for 10 min under chilled condition. Brain homogenate was made acidic by adding 1 ml of phosphate buffer (pH 3) and the drug was extracted two times by adding a mixture of 0.6 ml chloroform and 0.4 ml of methanol. The organic layer was separated after centrifugation at 5000 RPM for 10 min at 4 °C and then evaporated under vacuum. The residue was resuspended in 1.6 ml acetonitrile and 0.4 ml HPLC grade methanol and then analyzed by HPLC method.
Nasal ciliotoxicity study
After diffusion study, nasal mucosa was unmounted from diffusion cell and stained by safranin and eosin, and then analyzed under Olympus microscope (Olympus Inc., Center Valley, PA) and compared with untreated nasal mucosal layer to check for any conformational changes.
Results and discussion
Analytical method
The UV overlay absorption spectra were taken and the standard calibration curve was made for the whole range from 5 to 25 μg/ml. Linearity was detected between 5 and 25 μg/ml at 242 nm in SNF pH 6.8. The equation derived from standard calibration curve was y= 0.0079x−0.005 having a value of R2 = 0.9978. Similarly, in HPLC method, the standard retention peak of the drug was observed at 5 min with a peak area of 30 793 μv s for the concentration of 1 ppm.
Microscopy
The formation of spherical dispersion particles could be stipulated from the microscopic images of . SEM and TEM images further detail about particles size, shape, and dimensions. Liposomal microscopic images from indicated the formation of circular vesicles of phospholipid. SEM of the optimized liposomal formulation showed the intact, regular surface of completely dispersed liposomal spheres as shown in the figure. From TEM, thick and confined liposomal lamella had been identified.
Figure 1. (a) Microscopic image of Simple dispersion of QTF (1500×). (b) SEM of simple dispersion of QTF. (c) TEM of simple dispersion of QTF. (d) Microscopic image of liposomal dispersion of QTF (1500×). (e) SEM of liposomal dispersion of QTF. (f) TEM of liposomal dispersion of QTF.

Particle size and surface charge
Average particle size of simple dispersion was measured 139.6 nm as shown in with a zeta potential of 32.1 mV having intensity 100% which indicated that the system was moderately stable (Michel & Suzuki, Citation1985).
Figure 2. (a) Particle size distribution of simple dispersion. (b) Zeta potential distribution of simple dispersion. (c) Particle size distribution of liposomal dispersion. (d) Zeta potential distribution of liposomal dispersion.
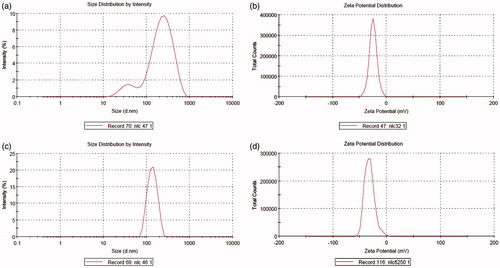
Average particle size of liposomal dispersion was measured as 152.2 nm as shown in with a zeta potential of 24.7 mV having intensity 100% which indicated that the system was moderately stable (Michel & Suzuki, Citation1985).
% Entrapment efficiency of liposomes
Entrapment efficiency obtained was 78.66 ± 3.42% with liposomes prepared with 1:2 (CH:EPC) molar ratio and two cycles of the sonication.
Ex vivo % drug diffusion (%DD)
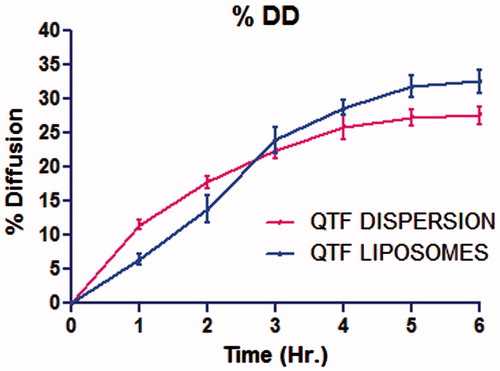
The drug diffusion was of 32.61 ± 1.70% for liposomal formulation containing 1:2 molar ratio of cholesterol:EPC at 6 h (). Higher EPC content resulted in increased drug diffusion due to its bipolar nature that facilitates drug diffusion across biological membrane (Chandaroy et al., Citation2002). Liposomes had profuse diffusion that could be correlated with diffusion across membrane by concentration gradient. For the simple dispersion, drug diffusion was of 27.56 ± 1.33% at 6 h, which was 83.23 ± 8.42% of liposomal dispersion.
Table 1. Ex vivo % drug diffusion (n = 3).
Diffusion is directly dependent on availability and partition coefficient of the drug, especially for nasal mucosa (Ehrhardt & Kim, Citation2007). The main cause of the difference of diffusion profile between simple and liposomal dispersion was the availability of drug at diffusion site and log p values. For simple dispersion, drug is directly available and, for liposomes, drug was slowly available as it releases from liposomes. Moreover, log p value of QTF was 2.8, while for liposomes, it was 4.55 (egg lecithin) (Heng et al., Citation2014), which reflects in different diffusion rates.
In simple dispersion, drug was completely available at diffusion site, so its diffusion was directly dependent on concentration gradient. As shown in the graph of , drug diffusion rate was higher at the initiation and gradually decreases to get stable diffusion. For diffusion through liposomal dispersion, drug first needs to be released from liposomal sac and then diffuse. Some liposomes directly fused to plasma membrane of the cell, resulted into sudden release, and even increased diffusion through membrane. From the graph, drug diffusion rate was less at the initiation due to the duration of drug release from liposomes, and gradually increased to get faster diffusion due to liposomal fusion with biological membrane of sheep nasal mucosa.
Figure 3. % Drug diffusion of simple and liposomal dispersion.
Diffusion coefficient
Diffusion coefficient for simple dispersion was derived as 3.0917 ± 0.3702 (× 10 − 5 cm/s) and liposomal dispersion was derived as 4.1334 ± 0.7321 (× 10 − 5 cm/s) which indicates high permeability of quetiapine in both the forms. As the coefficient is higher, diffusion is higher and as the coefficient is lower, diffusion is lower. Comparing both the formulations, liposomes possess about 1.34 times higher permeation rate than that of simple dispersion. This is due to permeation enhancing effect of cholesterol and lecithin present in liposomal formulation.
Diffusion profile
The difference factor f1 was measured as 18 and the similarity factor f2 was measured as 74 considering simple dispersion as the reference and liposomal dispersion as the test formulation and by taking all 6-time points of 6-h study for diffusion. Dissimilarity of 18 (<50) and similarity of 74 (50–100) indicated that the diffusion profiles of liposomal dispersions and simple dispersion were similar but not identical.
Diffusion kinetic studies – diffusion pattern and mechanism
Maximum goodness of fit value R2 was observed as 0.9941 for liposomal dispersion for Hopfenberg and Korsmeyer–Peppas than that of any other dissolution model. The Hopfenberg model suggested that drug discharged from surface eroding polymers as long as the surface region stays steady during the erosion process. Further, that could be validated by performing Korsmeyer–Peppas modeling and finding value of diffusion exponent. The value of n was found to be 1.561, indicating that the transport mechanism of the drug diffusion follows super case II. Super case II mechanism mostly refers to degradation of polymeric chain and abnormal transport (non-Fickian) refers to a combination of both diffusion and erosion-controlled drug discharge and autonomous of concentration gradient.
Maximum goodness of fit value R2 was observed as 0.9854 for simple dispersion for the Higuchi model which is indicative of rapid diffusion of drug through a membrane following Fick’s first law. For simple dispersion, n value was derived as 0.457 indicating that drug absorption through cell barrier follows Fickain diffusion. It means drug is diffused on the basis of concentration gradient which is indicative of simple single-phase phenomenon.
Generally, diffusion from liposomal lamella of lipophilic drugs follows zero order and Fickian mechanism for transporting that diffusion by concentration gradient in isotonic solution. SNF is slightly hypotonic to body fluids that resulted in its swelling and rupturing of liposomal sac. Moreover, at the body temperature of 37 °C, EPC melts down, hence, liposomes were eroded.
Gamma scintigraphy
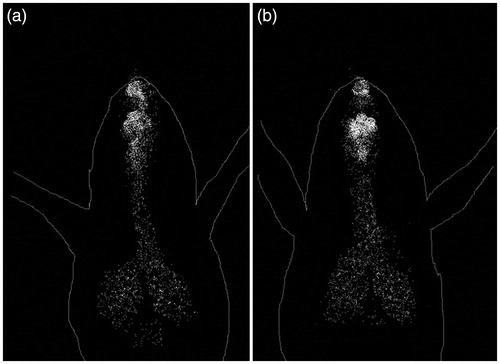
The scintigraphy pictures in mice after intranasal administrations are demonstrated in . This obviously exhibits significant radioactivity aggregation in the brain after intranasal administration for a desired period and these results were in great concurrence with the past discoveries (Patil et al., Citation2010). Results of gamma scintigraphy study revealed that liposomal dispersion shows more accumulated 99mTc into the brain in comparison with simple dispersion. Elevated 99mTc accumulation into the brain by liposomes confirms its efficiency of targeted delivery. Moderate amount of 99mTc into the lungs and respiratory tract through nasal route administration was observed as breathing by mice during nasal administration drags the dispersion into those tissues. This study clearly states that accumulation of 99mTc was more in brain of intranasally given liposomes than that of the simple dispersion.
Figure 4. Gamma scintigraphy of (a) simple and (b) liposomal dispersion in mice.
In vivo study
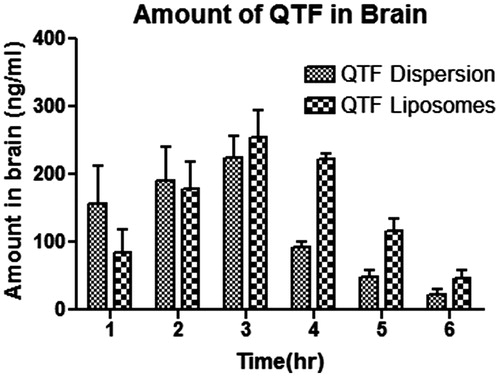
The QTF amount for both the formulations indicated that the amount of drug into the brain for liposomes administered by intranasal route was significantly higher (p < 0.05, n = 3) in comparison with simple dispersion. Initially, the amount obtained by liposome was lesser than that obtained by the simple dispersion. Nevertheless, after that, in the second hour, the amount obtained for both dispersions was almost similar. In addition, at the peak, liposomes exceeded the availability of QTF into the mice brain at the third hour. Till that, QTF level increased at a constant rate by dispersion, while for liposome, QTF level significantly increased. Oppositely, after 3 h, QTF level decrement occurred at slower rate by liposomes, while for dispersion, QTF level progressively decreased. These results support the existence of an alternative brain entry pathway for QTF by formulating in liposome by nasal route as indicated by higher drug uptake and bioavailability obtained after intranasal administration of QTF liposome in comparison with QTF dispersion administered ().
Figure 5. Amount of QTF in brain homogenate (ng) of simple and liposomal dispersion (n = 3).
Nasal ciliotoxicity study
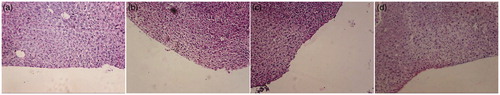
Nasal ciliotoxicity studies () revealed that nasal mucosa treated with SNF pH 6.8 (negative control ) showed intact epithelium layer without any necrosis while nasal mucosa treated with isopropyl alcohol (positive control mucociliary toxic agent ) showed complete destruction of epithelium layer, necrosis and even the deeper tissue parts were also destroyed. QTF simple and liposomal dispersion prepared in our studies () did not exhibit any toxicity as no change could be noticed in the gross morphology and histology of the nasal mucosa.
Figure 6. Nasal histopathological images of (a) negative control. (b) QTF simple dispersion. (c) QTF liposomal dispersion. (d) Positive control.
Conclusion
All the methods appear to be applicable and useful in comparing diffusion profiles of both the formulations. QTF simple and liposomal dispersion were successfully prepared in SNF pH 6.8, characterized and compared for diffusion through sheep nasal mucosa by ex vivo % drug diffusion within 6 h. Liposomal dispersion determined as 32.61 ± 1.70 led the simple dispersion with a measurement of 27.56 ± 1.33 for percent drug diffusion at 6 h. For diffusion coefficient, liposomal dispersion was measured as 4.1334 ± 0.7321 (×10 − 5 cm/s), which was again higher than that of simple dispersion with a measurement of 3.0917 ± 0.3702 (× 10 − 5 cm/s). Dissimilarity of 18 and similarity of 74 indicated that the diffusion profiles of liposomal dispersions and simple dispersion were similar but not identical. Liposomal dispersion was found to follow the Hopfenberg model and non-Fickian super case II anomalous transport, on the other hand, simple dispersion was found to follow Higuchi model and Fickain diffusion mechanism. Higher quetiapine amount with better diffusion rate established liposomal dispersion superior for brain drug delivery than colloidal dispersion through nasal route. Results of in vivo, ciliotoxicity, and gamma scintigraphy studies on mice proved liposomal formulation appropriate for administration through nasal route for brain delivery.
Acknowledgements
The authors would like to thank L. J. Institute of Pharmacy, LJK trust, Ahmedabad and Department of Pharmaceutical Sciences, Saurashtra University, Rajkot, for providing the needed lab facilities. The authors also thank Infocus Diagnostic Center, Nuclear Medicine Department, Mithakhali, Ahmedabad for their kind support in mice gamma scintigraphy. The authors express their gratitude to Vav Life sciences Pvt. Ltd., Mumbai, for the generous gift of Egg Phosphatidylcholine.
Declaration of interest
The authors report that they have no conflicts of interest.
References
- Acharya SP, Pundarikakshudu K, Upadhyay P, et al. (2015). Development of phenytoin intranasal microemulsion for treatment of epilepsy. J Pharm Invest 45:375–84
- Abbott NJ, Romero IA. (1996). Transporting therapeutics across the blood brain barrier. Mol Med Today 2:106–13
- Afergan E, Epstein H, Dahan R, et al. (2008). Delivery of serotonin to the brain by monocytes following phagocytosis of liposomes. J Control Release 132:84–90
- Alsarra IA, Hamed AY, Alanazi FK. (2008). Acyclovir liposomes for intranasal systemic delivery: development and pharmacokinetics evaluation. Drug Deliv 15:313–21
- Arora P, Sharma S, Gary SS. (2002). Permeability issues in nasal drug delivery. Drug Discov Today 7:967–75
- Arumugam K, Subramanian GS, Mallayasamy SR, et al. (2008). A study of rivastigmine liposomes for delivery into the brain through intranasal route. Acta Pharm 58:287–97
- Bamba M, Puisieux F, Marty JP, Carstensen JT. (1979). Release mechanisms in gel forming sustained release preparation. Int J Pharm 2:307–15
- Brgles M, Jurasin D, Sikirić MD, et al. (2008). Entrapment of ovalbumin into liposomes-factors affecting entrapment efficiency, liposome size, and zeta potential. J Liposome Res 18:235–48
- Chandaroy P, Sen A, Alexandridis S, Hui SW. (2002). Utilizing temperature-sensitive association of Pluronic F-127 with lipid bilayers to control liposome cell adhesion. Biochim Biophys Acta 1559:32–42
- Ding WX, Qi XR, Fu Q, Piao HS. (2007). Pharmacokinetics and pharmacodynamics of sterylglucoside-modified liposomes for levonorgestrel delivery via nasal route. Drug Deliv 14:101–4
- Ehrhardt C, Kim KJ. (2007). Drug absorption studies: in situ, in vitro and in silico models. New York, USA: Springer Science & Business Media
- Freireich EJ. (1966). Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep 50:219–44
- Frey WH. (2002). Intranasal delivery: bypassing the blood–brain barrier to deliver therapeutic agents to the brain and spinal cord. Drug Deliv Technol 2:46–9
- Garcia EG, Andrieux K, Gilb S, Couvreur P. (2005). Colloidal carriers and blood-brain barrier (BBB) translocation: a way to deliver drugs to the brain? Int J Pharm 298:274–92
- Hamed SA, Mahmoud AA, Kamel AO, et al. (2012). Phospholipid based colloidal poloxamer–nanocubic vesicles for brain targeting via the nasal route. Colloids Surf B: Biointerfaces 100:146–54
- He L, Wang Y, Li Z, et al. (2012). Determination of entrapment efficiency and transmembrane activation energy of gefitinib liposomes. Asian J Pharm Sci 7:251–8
- Heng KY, Kei TY, Singh KJ, et al. (2014). Handbook of cosmeceutical excipients and their safeties. Cambridge, UK: Elsevier
- Hildebrandt IJ, Su H, Weber WA. (2008). Anesthesia and other considerations for in vivo imaging of small animals. ILAR J 49:17–26
- Jung SH, Seong H, Cho SH, et al. (2009). Polyethylene glycolcomplexed cationic liposome for enhanced cellular uptake and anticancer activity. Int J Pharm 382:254–61
- Kararli TT, Needham TE, Griffin M, et al. (1992). Oral delivery of a renin inhibitor compound using emulsion formulations. Pharm Res 9:888–93
- Kumar A, Pandey AN, Jain SK. (2014). Nasal-nanotechnology: revolution for efficient therapeutics delivery. Drug Deliv. [Epub ahead of print]. doi:10.3109/10717544.2014.920431
- Laouini A, Jaafar MC, Limayem BI, et al. (2012). Preparation, characterization and applications of liposomes: state of the art. J Colloid Sci Biotechnol 1:147–68
- Liu JJ, Nazzal S, Chang TS, Tsai T. (2013). Preparation and characterization of cosmeceutical liposomes loaded with avobenzone and arbutin. J Cosmet Sci 64:9–17
- Lohan S, Sharma S, Murthy RR. (2015). Formulation and evaluation of solid lipid nanoparticles of QTF and Quetiapine Hemifumarate for brain delivery in rat model. Pharm Nanotechnol 1:239–47
- Michel AD, Suzuki K. (1985). Zeta potential of colloids in water and waste water. ASTM Standard D 95:4187–92
- Nivsarkar M, Pathak R, Dash RP, Misra M. (2014). Role of mucoadhesive polymers in enhancing delivery of nimodipine microemulsion to brain via intranasal route. Acta Pharm Sin B 4:151–60
- Parmar H, Bhandari A, Shah D. (2011). Recent techniques in nasal drug delivery: a review. Int J Drug Dev Res 3:99–106
- Patel RB, Patel MR, Bhatt KK, et al. (2014). Evaluation of brain targeting efficiency of intranasal microemulsion containing olanzapine: pharmacodynamic and pharmacokinetic consideration. Drug Deliv. [Epub ahead of print]. DOI: 10.3109/10717544.2014.912694
- Pathan SASZI, Zaidi MA, Talegaonkar S, Vohra D. (2009). CNS drug delivery systems: novel approaches, recent patents on drug delivery & formulation, vol. 3. Oak Park (IL): Bentham Science Publishers Ltd
- Patil S, Babbar A, Mathur R, et al. (2010). Mucoadhesive chitosan microspheres of carvedilol for nasal administration. J Drug Target 18:321–31
- Rathod S, Deshpande SG. (2010). Design and evaluation of liposomal formulation of pilocarpine nitrate. Indian J Pharm Sci 72:155–60
- Reddy SP, Satyanarayana P, Verma KK, et al. (2011). Novel reverse phase HPLC Method development and validation of Quetiapine Fumatrate in bulk and tablet dosage form. Int J Pharm Int Res 1:95–9
- Sahu D, Rana AC. (2011). Development of analytical method for quetiapine fumarate by UV spectrophotometry. IJRAP 2:588–91
- Santos HM, Lodeiro C, Martinez JLC. (2009). 1-The power of ultrasound. In: Capelo-Martínez J-L, eds. Ultrasound in chemistry: analytical applications. Weinheim: Wiley-VCH Verlag GmbH & Co. KGaA, 1–16
- Schnyder S, Krahenbuhl J, Drewe J, Huwyler S. (2005). Targeting of daunomycin using biotinylated immunoliposomes: pharmacokinetics, tissue distribution and in vitro pharmacological effects. J Drug Target 13:325–35
- Senthilkumar KL, Ezhilmuthu RP, Praveen P. (2012). Preparation and characterization of nabumetone liposomes. Int J Life Sci Bt Pharm Res 1:81–6
- Shariat S, Badiee A, Jaafari MR, Mortazavi SA. (2014). Optimization of a method to prepare liposomes containing HER2/Neu-derived peptide as a vaccine delivery system for breast cancer. Iran J Pharm Res 13:15–25
- Shi N, Zhang Y, Zhu C, et al. (2001). Brain-specific expression of an exogenous gene after i.v. administration. Proc Natl Acad Sci USA 98:12754–9
- Sweetman SC. (2007). Martindale, the complete drug reference. London: Pharmaceutical Press
- Tomoko N, Fumiyoshi I. (2005). Encapsulation efficiency of water-soluble and insoluble drugs in liposomes prepared by the microencapsulation vesicle method. Int J Pharm 298:198–205
- Türker S, Onur E, Özer Y. (2004). Nasal route and drug delivery systems. Pharm World Sci 26:137–42
- Ugwoke MI, Verbek N, Kinget R. (2001). The biopharmaceutical aspects of nasal mucoadhesive drug delivery. J Pharm Pharmacol 53:3–22
- Vincenzo P, Roberto M, Anna F, Sandra F. (2003). Quality control of commercial tablets containing the novel antipsychotic quetiapine. J Pharm Biomed Anal 32:1037–44
- Vyas SP, Goswami SK, Singh R. (1995). Liposomes based nasal delivery system of nifedipine: development and characterization. Int J Pharm 118:23–30
- Woensel M, Wauthoz N, Rosière R, et al. (2013). Formulations for intranasal delivery of pharmacological agents to combat brain disease: a new opportunity to tackle GBM? Cancers 5:1020–48
- Xie Y, Ye L, Zhang X, et al. (2005). Transport of nerve growth factor encapsulated into liposomes across the blood–brain barrier: in vitro and in vivo studies. J Control Release 105:106–19