
Abstract
Context: Proniosomes offer a versatile vesicle drug delivery concept with potential for delivery of drugs via transdermal route.
Objectives: To develop proniosomal gel using cremophor RH 40 as non-ionic surfactant containing the antihypertensive drug lacidipine for transdermal delivery so as to avoid its extensive first pass metabolism and to improve its permeation through the skin.
Materials and methods: Proniosomes containing 1% lacidipine were prepared by the coacervation phase separation method, characterized, and optimized using a 23 full factorial design to define the optimum conditions to produce proniosomes with high entrapment efficiency, minimal vesicle size, and high-percentage release efficiency. The amount of cholesterol (X1), the amount of soya lecithin (X2), and the amount of cremophor RH 40 (X3) were selected as three independent variables.
Results and discussion: The system F4 was found to fulfill the maximum requisite of an optimum system because it had minimum vesicle size, maximum EE, maximum release efficiency, and maximum desirability. The optimized system (F4) was then converted to proniosomal gel using carbopol 940 (1% w/w). In vitro permeation through excised rabbit skin study revealed higher flux (6.48 ± 0.45) for lacidipine from the optimized proniosomal gel when compared with the corresponding emulgel (3.04 ± 0.13) mg/cm2/h. The optimized formulation was evaluated for its bioavailability compared with commercial product. Statistical analysis revealed significant increase in AUC (0 − α) 464.17 ± 113.15 ng h/ml compared with 209.02 ± 47.35 ng h/ml for commercial tablet. Skin irritancy and histopathological investigation of rat skin revealed its safety.
Conclusions: Cremophor RH 40 proniosomal gel could be considered as very promising nanocarriers for transdermal delivery of lacidipine.
Introduction
Proniosomes are non-ionic-based surfactant vesicles, which are converted to niosomes upon hydration or by absorbing water from the skin (Yadav et al., Citation2010). Physical stability problems of niosomes such as aggregation, fusion, and leaking and additional convenience in transportation, distribution, storage, and dosing can be minimized with proniosomes (Yasam et al., Citation2014). Proniosomes offer a versatile vesicle drug delivery concept with potential for delivery of drugs via transdermal route (Asija et al., Citation2014).
Cremophor RH 40 is Polyoxyl 40-hydrogenated castor oil non-ionic surfactant with HLB value 14–16 and melting point 30 °C (Rowe et al., Citation2006). Cremophor RH 40 is non-toxic, tasteless, colorless, and transparent, stable over a wide temperature range and shows little tendency to foaming. It is highly effective solubilizing for hydrophobic drugs, fat soluble vitamins, and essential oils (Grove & Müllertz, Citation2007). Yet, and to the best of our knowledge, none of the published work used cremophor RH 40 as non-ionic surfactant in the preparation of vesicular systems. In this study, a novel approach was developed that utilizes cremophor RH 40 as non-ionic surfactant in formulating proniosomal systems for transdermal delivery.
Lacidipine is a calcium channel blocker developed for oral administration and used in the treatment of hypertension and atherosclerosis. It also possesses an antioxidant effect (Lee & Bryson, Citation1994; Mc Cormack & Wagstaff, Citation2003). Lacidipine is a highly lipophilic drug of poor water solubility and undergoes extensive first-pass hepatic metabolism with a mean absolute bioavailability of about 10% (range 3–59%). This very low oral bioavailability restricts its use. So, lacidipine could be an excellent candidate for transdermal delivery. There is only one report till now to enhance the bioavailability of lacidipine through microemulsion based transdermal gels (Gannu et al., Citation2010).
Therefore, the aim of the present work was to develop a novel proniosomal gel using cremophor RH 40 as non-ionic surfactant for transdermal delivery of lacidipine. Also, the present study focused on investigating the effect of cholesterol amount, soya lecithin amount, and non-ionic surfactant (cremophor RH 40) amount in the 23 full factorial design on the vesicle size, the percentage of entrapment efficiency (%EE), and the percentage release efficiency (%RE).
Materials and methods
Materials
Lacidipine (kindly supplied by EgyPharm Company, Cairo, Egypt), Soya Lecithin, (Sigma-Aldrich, St. Louis, MO); Cholesterol (Acros Organics, Morris Plains, NJ), Cremophor® RH 40 (Polyoxyl 40 hydrogenated castor oil) (BASF, Ludwigshafen, Germany), deionized water for injection (Otsuka Pharmaceutical Company, Cairo, Egypt), acetonitril HPLC grade and methanol HPLC grade (Fisher Co., Leicestershire, UK). All other chemicals were of analytical grade and used without further purification.
Experimental design
A 23 full factorial design was used to define the optimum conditions regarding the selected factors. The design involved three factors at two levels as shown in . The amount of cholesterol (X1), the amount of soya lecithin (X2), and the amount of non-ionic surfactant (cremophor RH 40) (X3) were selected as three independent variables. The vesicle size, the %EE, and the %RE after 24 h through semi-permeable membrane (cellulose nitrate with a pore size of 0.45 μm) were selected as dependent variables. All data were statistically subjected to analysis of variance (ANOVA) using Design-Expert® software (version 7; Stat-Ease, Inc., Minneapolis, MN) and suitable regression models were driven to enable navigation of the experimental space. Significance level was set at p < 0.05 (Abdel Malak, Citation2012).
Table 1. The experimental plan of the factorial design 23 for the preparation of lacidipine proniosomes for transdermal delivery.
Preparation of lacidipine proniosomes
Proniosomes were prepared by a coacervation phase separation method (Gadekar et al., Citation2013; Ramkanth et al., Citation2014; Rajabalaya et al., Citation2015). Precisely weighed amounts of surfactant, soya lecithin, cholesterol, and drug were taken in a clean and dry wide mouthed glass vial of 5.0 ml capacity and absolute ethanol (0.25 ml) was added to it. All the ingredients were mixed well with a glass rod; the open end of the glass bottle was covered with a lid to prevent the loss of solvent from it and warmed over water bath at 60–70 °C for about 5 min until the surfactant mixture was dissolved completely. Then 0.1 ml of distilled water at 60 °C was added and warmed on a water bath till a clear solution was formed. Then, the mixture should be allowed to cool down at room temperature for characterization. The composition of different proniosome formulations is given in .
Table 2. The composition of lacidipine proniosomes formulations and their characterization results.
Evaluation of lacidipine proniosome formulations
Optical microscopic examination
In a glass vial, 0.1 g of lacidipine proniosome formulation (F4) was hydrated with 10 ml distilled water. The aqueous suspension was sonicated in a bath sonicator (Crest Ultrasound, Imola, Italy) for 30 min, then a few drops of the formed niosomal dispersion were spread on a glass slide and fixed over by drying at room temperature (Kumar et al., Citation2012). The dry thin film of the hydrated proniosomal formulation was examined for the formation of niosomes using optical microscope (Olympus, Tokyo, Japan), with a magnification power of 20×. Photomicrographs were taken using a digital camera (Olympus, C-4040ZOOM, Tokyo, Japan).
Zeta potential (surface charge) (ζ) determination
The zeta potential of different formulations was measured by Laser Doppler velocimetry using Malvern Zetasizer (Malvern Instruments, Malvern, UK). The instrument is a laser-based multiple angle particle electrophoresis analyzer. The instrument measures the electrophoretic mobility and surface charge (El-Nabarawi et al., Citation2013; Waddad et al., Citation2013). An aliquot of 0.1 ml of the prepared niosomes derived from proniosomal formulation was added to the zeta cell using an automatic pipette. The zeta potential was measured in triplicate and the resultant value was presented as the mean (± standard deviation (± SD)).
Vesicle size determination
Vesicle size analysis was determined using Malvern Zetasizer (Malvern Instruments, Malvern, UK) which employed photon correlation spectroscopy (PCS) for the prepared formulations (Gupta et al., Citation2007). Exactly 0.1 g of lacidipine proniosomes was hydrated with 10 ml distilled water. The aqueous suspension was sonicated in a sonicator bath for 30 min. Size analysis was conducted by adding niosomes derived from proniosomal formulation (1 ml) into a Malvern’s disposable vial, also the span or polydispersity index (PDI) value which indicate the homogeneity of the size distribution of the preparations was determined.
Determination of percentage entrapment efficiency (%EE)
About 0.1 g of lacidipine proniosomes were reconstituted with 10 ml distilled water and sonicated in a bath sonicator for 30 min. Lacidipine niosomes derived from proniosomal formulation were separated from the untrapped drug by filtrating 5 ml of the formed suspension using Whatman filter paper (Grade no. 1, 11 μm). For its very low water solubility, the unentrapped lacidipine (forms large crystals) was sedimented and retained over the filter paper, while lacidipine vesicles (present within the niosome bilayers) passed through the filter paper pores into the filtrate (Xu et al., Citation2009, Aburahma & Abdelbary, Citation2012; Elhissi et al., Citation2013). Then, 1 ml of the filtrate was mixed with methanol and sonicated for 10 min to disrupt the lacidipine niosomes and form a solution. The concentration of lacidipine in the resulting solution was analyzed spectrophotometrically at a predetermined λmax of 283.5 nm and the percentage of drug entrapped was calculated using the following formula:
where %EE is the percentage entrapment efficiency, A is the concentration of lacidipine in niosomes after filtration and B before filtration.
In vitro release studies
The in vitro release of lacidipine from the prepared proniosome formulations was performed using a USP dissolution tester (apparatus II) (USP dissolution tester XXXI, Pharma Test, Type PTW, Hainburg, Germany) with a slight modification. Proniosomes (0.5 g), equivalent to 5 mg of lacidipine, were spread over the surface of a glass plate of 4.2 cm diameter and then covered with the semi-permeable membrane (cellulose nitrate with pore size 0.45 μm). The glass and the membrane were held together by blaster and equally spaced plastic clips. This assembly was placed at the bottom of the dissolution vessel containing 500 ml of 30% methanolic solution (v/v) with constant speed (100 rpm) at 37 ± 0.5°C. Methanol was added to maintain the sink conditions during the release study due to the extreme poor solubility of lacidipine in water (Fang et al., Citation2001; El-Laithy et al., Citation2011). At predetermined time intervals (0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, and 24 h), aliquots of 5 ml of the release medium were withdrawn and replaced with equal volume of fresh medium to maintain a constant volume (Soliman et al., Citation2011). The concentration of lacidipine of the collected samples was determined spectrophotometrically at the predetermined λmax of 286 nm. The mean percentage of lacidipine released across the membrane was plotted as a function of time. All experiments were run in triplicates and the results were expressed as the mean values ± SD. The in vitro release of a plain drug suspension, equivalent to 5 mg of lacidipine, was carried out also in a similar manner as control formulation to compare the results of the percentage of drug released from different proniosome formulations. Release efficiency (RE) was calculated from the area-under-the-release curve (using the trapezoidal rule). It is expressed as a percentage of the area of the rectangle corresponding to 100% release for the same total time (24 h) (Salama et al., Citation2012). The relative release rate was also determined by dividing the release efficiency of proniosome formulations with control formulation (plain drug suspension) (Janga et al., Citation2012).
Optimization of lacidipine proniosomes
Optimization was performed to find out the level of independent variables (X1, X2, and X3) that would yield a minimum value of vesicle size, a maximum value of %EE, and a maximum value of %RE by applying the point prediction method of the Design Expert software (Ahad et al., Citation2012).
Preparation of lacidipine proniosomal gel
The optimized formulation was formulated as proniosomal gel. Lacidipine proniosomes were prepared by a coacervation phase separation method as mentioned in the “Preparation of lacidipine proniosomes” section. Carbopol 940 (1% w/w) was added to the hot mixture and the mixture was converted into lacidipine proniosomal gel on cooling (Patil et al., Citation2012). The gels were kept at room temperature for further characterization.
Evaluation of the optimized lacidipine proniosomal gel formulation
In vitro drug permeation studies through excised rabbit skin
Permeation studies give an important insight into the drug behavior in vivo, since the amount of the drug permeated dictates the amount of drug available for absorption into systemic circulation (Vora et al., Citation1998).
Preparation of full excised abdominal rabbit skin barrier membrane
Rabbits were sacrificed and the full thickness of rabbit skin was excised from the abdominal region and hair was removed with an electric clipper. The subcutaneous tissue was removed surgically and the dermis side was wiped with isopropyl alcohol to remove adhering fat. The cleaned skin was washed with distilled water and stored in the deep freezer until further use. The skin was brought to room temperature and cut into 2 cm diameter circular patches when used (Kantarci et al., Citation2005; Soliman et al., Citation2010).
In vitro permeation
In vitro skin permeation study was performed in a Franz diffusion cell with a diffusion area of 1.7 cm2 and a receptor volume of 14 ml. The receptor compartment was filled with 30% methanolic solution (v/v) with a constant speed (100 rpm) at 37 ± 0.5 °C. The excised rabbit skin was then mounted between the donor and receptor chambers of the Franz diffusion cell with the dermal side in contact with the receptor medium and the stratum corneum side facing upwards into the donor compartment. Then, the optimized lacidipine proniosomal gel formulation (0.5 g), equivalent to 5 mg of lacidipine was spread evenly on the skin surface in the donor compartment. At predetermined time intervals (0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, and 24 h), aliquots of 2 ml were withdrawn from the receptor compartment and replaced with an equal volume of fresh medium to maintain a constant volume. The concentration of lacidipine of the collected samples was determined using HPLC (Gannu et al., Citation2009). All experiments were run in triplicates and the results were expressed as the mean values ± SD. Emulgel containing 1% w/w lacidipine was used as control formulation to compare drug permeation through rabbit skin from optimized proniosomal gel formulation with that from the emulgel.
Permeation data analysis
The average cumulative amount of lacidipine permeated through the skin per unit surface area (μg/cm2) was calculated and plotted as a function of time. The drug flux (permeation rate) at steady state (JSS) was calculated from the slope of the straight line. The permeability coefficient (KP) was calculated using the following equations:
where Co is the initial concentration of the drug (μg/ml). Enhancement ratio (Er) was calculated by dividing JSS of the optimized proniosomal gel formulation by JSS of the control formulation (Ustundag et al., Citation2011):
Transmission electron microscope (TEM)
The morphology of vesicles derived from hydrating the optimized lacidipine proniosomal gel formulation was examined using transmission electron microscope. One drop of the vesicular dispersion was deposited in a form of a thin film on a carbon-coated copper grid, and then stained with one drop of 2% aqueous solution of phosphotungstic acid (PTA) and allowed to dry and any excess fluid was removed with filter paper before examination using a JEM-100 CX electron microscope (JEOL, Tokyo, Japan) (Aburahma & Abdelbary, Citation2012).
Accelerated stability study
The optimized formulation stored in glass vial covered with aluminum foil was kept in an oven at 40 ± 0.2°C for a period of 3 months. After 90 d, hydration step was carried out and the mean vesicle size as well as the percentage of entrapment efficiency (%EE) was determined and compared to the freshly prepared formulation.
In vivo bioavailability study in rabbit
Study design and sample collection
The animal study protocol was reviewed and approved by the Institutional Ethical Committee, Faculty of Pharmacy, Cairo University (S. no. PI 428). The bioavailability of lacidipine was determined from the optimized transdermal proniosomal gel formulation and was compared with the commercially oral available Lacipil® 4 mg tablet (GlaxoSimithKline, Philadelphia, PA). Six male New Zealand rabbits weighing 3.0 ± 0.3 kg were used for this study. The rabbits were randomly divided into two groups, each containing three rabbits. A single-dose, two phases with 1-week washout period randomized cross-over design was followed. The rabbits were fasted overnight before oral administration of Lacipil® tablet or transdermal application of the optimized formulation, but had free access to water. The dorsal side of rabbits used for the application of transdermal formulation was shaved carefully with the help of electrical shaver 1 d before application of the optimized formulation followed by cleaning with water. The transdermal formulation was applied on the dorsal side of rabbit for 24 h over a surface area of 4 cm2, then covered with a water impermeable back up membrane and further fixed with the help of adhesive membrane. Rabbits of Group I received transdermal application of optimized lacidipine proniosomal gel and rabbits of Group II received oral administration of Lacipil® tablet by suspending the crushed tablets in sufficient amount of water and administered orally by feeding tubing followed by rinsing with 10 ml of water. All groups received lacidipine equivalent to 4 mg (ElKasabgy et al., Citation2014). Blood samples (approximately 4 ml) were withdrawn from ear vein into evacuated heparinized glass tubes at the following times: 0.0, 0.5, 1.0, 2.0, 4.0, 6.0, 8.0, 12.0, 24.0, and 72.0 h following the transdermal applications and oral administration. The withdrawn blood samples were centrifuged at 4000 rpm for 10 min. Afterwards, plasma was separated and transferred directly into clean micro-centrifuge tubes and stored frozen at −70 °C pending for drug analysis. After a washing out period of 7 d, the study was repeated following the same procedures to complete the cross-over design.
Chromatographic condition
A sensitive, selective, and accurate liquid chromatography with double path mass spectroscopy method (LC-MS/MS) was developed and validated before the study for the determination of lacidipine in rabbit plasma samples. A Shimadzu Prominence (Shimadzu, Kyoto, Japan) series LC system equipped with degasser (DGU-20A3), solvent delivery unit (LC-20AB) with an autosampler (SIL-20 AC), was used in this study. The Guard column was Sunfire waters C18 (50 × 4.6 mm) (Waters Corporation, Milford, MA), 5 μm particle size. All analyses were carried out at room temperature. The isocratic mobile phase consisted of acetonitrile and (0.05 M) ammonium acetate buffer (8:2 v/v). After filtration and degassing of the mobile phase, it was delivered at a flow rate of 0.50 ml/min into the mass spectrometer’s electrospray ionization chamber. The volume of injection was 20 μl. The analytical data were processed by Analyst software version 1.6 (Applied Biosystems Inc., Foster city, CA).
Plasma sample preparation for LC-MS/MS injection
All frozen plasma samples were thawed at ambient temperature. Five hundred microliter of each rabbit plasma sample was transferred to a 10 ml glass test tube, and then 50 μl of internal standard (hydrochlorothiazide) working solution (100 ng/ml) was added and vortexed to mix for 1 min. After vortex mixing of the mixture for 1 min, 4 ml aliquot of tertiary butyl methyl ether was then added and the sample was vortex-mixed for 3 min. The tubes were then centrifuged for 10 min at 4000 rpm. The upper organic layer was transferred to clean glass tubes and evaporated to dryness using centrifugal vacuum concentrator Vacufuge 5301 (Eppendorf, Hamburg, Germany) at 40 °C. Dry residues were then dissolved in 200 μl of mobile phase and vortexed for 1 min to reconstitute residues, and 20 μl was injected using the autosampler to the LC–MS/MS system for analysis.
Pharmacokinetic parameters analysis
To assess the bioavailability of lacidipine, the pharmacokinetic parameters from plasma data following the two treatments were estimated and were analyzed for each rabbit by non-compartmental pharmacokinetic models (Shargel et al., Citation2005) using Kinetica® software (version 5, Thermo Fisher Scientific Inc., Waltham, MA). The relative bioavailability (F) for transdermal drug delivery relative to the oral commercial tablets lacipil® 4 mg was calculated by comparing the transdermal and oral AUCs.
Statistical analysis
One-way analysis of variance (ANOVA) followed by post hoc multiple comparisons using the least square difference (LSD) was performed to evaluate the difference between the pharmacokinetic parameters of lacidipine transdermal proniosomal gel formulation and lacipil® 4 mg tablet using statistical software of statistical package for social sciences (SPSS, Chicago, IL) version 14. Differences are considered to be significant at p < 0.05.
Skin irritation test
The skin irritancy test was carried out to determine any possible localized reaction of the optimized lacidipine proniosomal gel formulation on the skin of male albino rats (150–180 g). The hair on the dorsal side of albino rats was removed by clipping 1 d before the start of the experiment. The animals were divided into three groups (n = 6): the first group served as control (no treatment), the second group received 0.8% (v/v) aqueous formalin solution as a standard irritant, and the third group received the optimized formulation. A dose of 0.5 g of optimized formulation or 0.5 ml of formalin solution was applied on a 5 cm2 area of the shaved dorsal side of the rats. The development of erythema and edema was graded according to a visual scoring scale for 24 h (Azeem et al., Citation2009; Fouad et al., Citation2013). The mean erythemal or edema scores were recorded (ranging from 0 to 4) according to Draize (Draize et al., Citation1944), where 0 means no erythema or edema, 1 slight erythema or edema, 2 moderate erythema or edema, 3 moderate to severe erythema or edema, and 4 severe erythema or edema. The primary irritancy index (PII) was calculated by adding the edema and the erythema scores (Soliman et al., Citation2010,Citation2011).
Histopathological examination of skin specimens
To confirm previous irritation results, a histopathological study was carried. After 24 h, the rats were sacrificed and skin samples from treated and untreated (control) areas were taken. Each skin sample was stored in 10% (v/v) formalin saline solution. The skin samples were cut vertically in different sections. Each section was dehydrated using ethanol, embedded in paraffin for fixing, and stained with hematoxylin and eosin and then examined through the light electric microscope (Nikon, Tokyo, Japan) fitted with a Canon power shot G3 digital camera (Canon, Tokyo, Japan) and compared with control sample.
Results and discussion
Evaluation of lacidipine proniosome formulations
Optical microscopic examination
The photomicrograph of lacidipine proniosome formulation (F4) is shown in . It showed the presence of homogenous population of vesicles with spherical shape.
Figure 1. Optical photomicrograph of lacidipine proniosomes of F4.

Zeta potential (surface charge) (ζ) determination
The values of zeta potential for all proniosome formulations are illustrated in . It was found that the niosomes formed from lacidipine proniosome formulations had negative zeta potential values which were in the range of −18.85 ± 0.65 to −39.75 ± 0.85 mV and hence this was an indication that the prepared formulations were stable (Hernàndez & Goymann, Citation2005). The hydroxyl ions in the dispersion medium might localize at the surface of niosome membranes (Junyaprasert et al., Citation2008), thus being responsible for the negative surface charge of vesicles made from non-ionic surfactants.
Vesicle size determination
All formulations were found in nanosize range of 162.43 ± 0.77 nm to 547.30 ± 2.10 nm with adequate values of polydispersity index of 0.352 ± 0.038 (F4) to 0.676 ± 0.036 (F7). Low value of PDI means high uniformity between the vesicles. The mean vesicle sizes of proniosome formulations are shown in . Needless to say that small diameter is advantageous to decrease irritation and improve the penetration of the vesicles into skin (Rahman et al., Citation2014). Also, achievement of nanovesicles can help in passage across the anatomical constraints in the skin (Kumar et al., Citation2007).
Analysis of vesicle size data
The effect of different formulation variables, namely the amount of cholesterol (X1), the amount of soya lecithin (X2), and the amount of cremophor RH 40 (X3) on the mean vesicle size of lacidipine proniosomes, was assessed using the software Design-Expert® (version 7; Stat-Ease, Inc., Minneapolis, MN). The 3D response surface plots are shown in . The ANOVA analysis of the data of the vesicle size revealed that all the three variables and their interaction affected significantly the mean vesicle size (p < 0.05).
Figure 2. 3D response surface plots showing the effect of independent variables on the mean vesicle size of the prepared lacidipine proniosomes formulations (Y1).
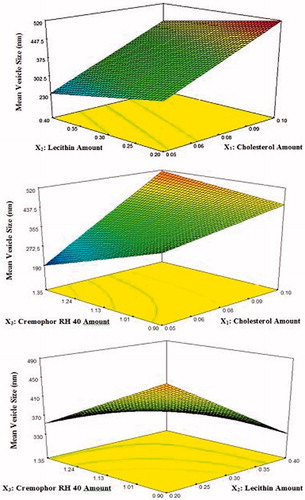
A polynomial equation of mean vesicle size was also obtained and shown below in the terms of coded factors:
where adjusted R2 is 0.9997, indicating good correlation between the independent variables (Xu et al., Citation2009). A positive sign of coefficient indicates that the output increases with an increase in parameter level, and a negative sign of coefficients indicates that the output increases with a decrease in parameter level. The larger coefficient means the independent variable has more potent influence on the mean vesicle size.
Effect of the amount of cholesterol on the vesicle size
The lacidipine proniosomes prepared at higher cholesterol amount had significantly higher mean vesicle size than those prepared at lower cholesterol amount. This may be attributed to formation of rigid bilayer structure (Hosny, Citation2010). Cholesterol is the main additive which affects the physical stability of the vesicles as it provides the rigidity to the bilayer membrane. It strengthens the bilayer and diminishes the bilayer fluidity by eliminating the phase transition temperature peak of the vesicles (Essa, Citation2010). Similar results were previously published that the vesicle size was increased with increase the amount of cholesterol (Negi et al., Citation2011; El-Nabarawi et al., Citation2013; Kamboj et al., Citation2014).
Effect of the amount of lecithin on the vesicle size
On increasing the amount of lecithin, a significant decrease in vesicle size was observed. Increasing the lecithin content may contribute an increase in the hydrophobicity, with subsequent reduction of vesicle size (Yadav et al., Citation2010; Chavan et al., Citation2012).
Effect of the amount of cremophor RH 40 on the vesicle size
It was found that the size of the vesicles significantly decreased with increasing the amount of cremophor RH 40. This could be explained by the easier formation of the vesicle and/or the better accommodation of the surfactant in the vesicle structure (Shatalebi et al., Citation2010). These results fit well with the results obtained by Negi et al. (Citation2011) when they studied the effect of the concentration of tween 80 on vesicle size of venlafaxine niosomes. Rahman et al. (Citation2014) also stated that increased surfactant concentration led to a decrease in vesicle size.
Determination of percentage entrapment efficiency (%EE)
The entrapment efficiency of niosomes is governed by the ability of formulation to retain drug molecules in the aqueous core or in the bilayer membrane of the vesicles (El-Nabarawi et al., Citation2013). The entrapment efficiency is one of the most important parameters from pharmaceutical viewpoint in the evaluation of niosomal formulations and formulation scientist’s effort are always directed towards achieving high entrapment efficiency (Balakrishnan et al., Citation2009). The length of alkyl chain and phase transition temperature of surfactants was affecting the entrapment efficiency. Surfactants of longer alkyl chains and higher phase transition temperature showed higher entrapment efficiency (Guinedi et al., Citation2005). The results of %EE of lacidipine in niosomes are illustrated in and was found to range from 44.06 ± 0.06% to 98.01 ± 0.683%. It was noticed that the formulation with smaller mean vesicles size possessed higher entrapment value. As the size of vesicle decreases, surface area increases and hence drug entrapment also increases (Ammar et al., Citation2011). A similar result was previously published that the entrapment efficiency was increased with a decrease in the vesicle size (Akhtar et al., 2014).
Analysis of %EE data
Vesicle entrapment efficiency relies on the stability of the vesicle which is highly dependent on amount of cholesterol (X1), the amount of soya lecithin (X2), and the amount of surfactant (cremophor RH 40) (X3) forming the bilayers. The 3D response surface plots are shown in . The ANOVA analysis of the data of the percentage entrapment efficiency revealed that all the three variables and their interaction affected significantly (p < 0.05) the percentage entrapment efficiency except X1 X2 interaction (p > 0.05). A polynomial equation of mean percentage entrapment efficiency were also obtained and shown below in the terms of coded factors:
where the adjusted R2 was 0.995.
Figure 3. 3D response surface plots showing effect of independent variables on the percentage entrapment efficiency (%EE) of lacidipine in the prepared proniosomes formulations (Y2).
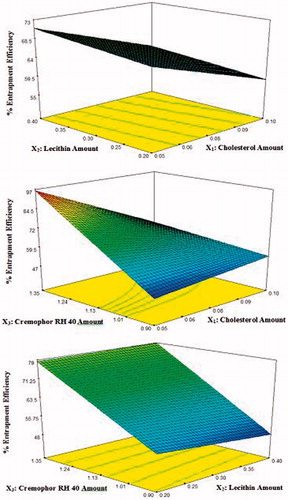
Effect of the amount of cholesterol on the %EE
The variation in the amount of cholesterol significantly affects the %EE. Increasing the cholesterol content was accompanied by significant decrease in %EE. This may be due to two reasons. First, addition of cholesterol might be disrupting the regular linear structure of vesicular membrane. Second, the cholesterol will compete with the drug for the space within the bilayer, hence remove the drug from the vesicles bilayer (Wang et al., Citation2010; Abdelbary, Citation2011). These results fit well with previously published results regarding the effect of cholesterol on %EE of niosomes (Aboelwafa et al., Citation2010; Thomas & Viswanad, Citation2012).
Effect of the amount of lecithin on the %EE
On increasing the amount of lecithin added, a significant decrease in %EE was observed. Hao & Li (Citation2011) found that the number of niosomes taking part in encapsulation decreases as increasing the lipid concentration, which might be ascribed to the aggregate of niosomes at high lipid level. Similar results were also obtained by Xu et al. (Citation2009) who found that if the amount of lecithin was too high, the entrapment efficiency would decrease.
Effect of the amount of cremophor RH 40 on the %EE
It was found that the %EE of lacidipine significantly increased with increasing the amount of cremophor RH 40 used. This can be explained as follows: cremophor RH 40 is the main component responsible for vesicle formation and hence, increasing its amount resulted in the increase in the number of formed niosomes and consequently the volume of the hydrophobic bilayer domain, the available housing for entrapment lacidipine hydrophobic drug (Hao et al., Citation2002). Similar results were previously reported (El-Laithy et al., Citation2011; Ahmed et al., Citation2014).
In vitro release studies
In vitro release studies are often performed to predict how a delivery system might work in an ideal situation as well as give some indications of its in vivo performance since drug release dictates the amount of drug available for absorption (Gupta et al., Citation2007; Rahman et al., Citation2014).
Results of lacidipine in vitro release from different proniosomal formulations and the plain drug suspension are shown graphically in . As observed in the in vitro release profile, the release of lacidipine from all proniosome formulations was apparently biphasic process where an initial rapid release phase was observed in the first hour where about 21.97 ± 1.24–73.56 ± 1.34% of the drug was released from various proniosome formulations followed by a slow release phase. The initial rapid phase was due to desorption of lacidipine from the surface of niosomes while the drug release in the slower phase was regulated by diffusion through the swollen niosomal bilayers (Pardakhty et al., Citation2007; Mokhtar et al., Citation2008). This profile could be advantageous if we considered the importance of epidermis saturation with initial fast drug released to achieve high-concentration gradient required for successful drug delivery to the blood (Csoka et al., Citation2007). This biphasic release profile agreed with the release profile of previously published studies (Aboelwafa et al., Citation2010; El-Laithy et al., Citation2011; Marwa et al., Citation2013).
Figure 4. Release pattern of lacidipine from proniosomes formulations and from the plain drug suspension.
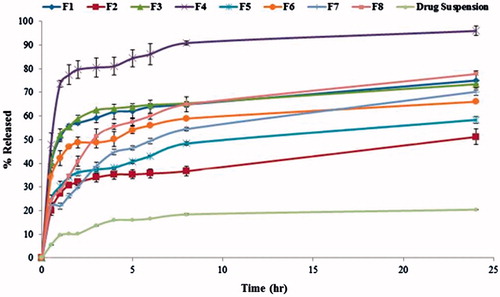
The %RE after 24 h and the relative release rate were calculated and shown in . It was found that the %RE of lacidipine from all proniosome formulations was higher than the plain drug suspension. The relative release rate greater than 1 indicates release enhancement and, in our case, it was noticed that all proniosome formulations achieved release enhancement more than 1. This might be due to the emulsification effect of the surfactant after the hydration of the proniosomes by the release medium followed by the disruption of vesicles structure and/or the change in the physical state of the drug from crystalline to amorphous during preparation (Hiremath et al., Citation2009; Pankaj et al., Citation2013). Interestingly, the proniosome formulation (F4) promoted higher release of lacidipine, higher release efficiency and relative release rate compared with other formulations.
Analysis of mean percentage release efficiency data
The 3D response surface plots are shown in . The ANOVA analysis of the data of the percentage release efficiency of lacidipine proniosomes revealed that all the three variables and their interaction affected significantly the percentage release efficiency (p < 0.05). A polynomial equation of mean percentage release efficiency was also obtained and shown below in the terms of coded factors:
where the adjusted R2 was 0.972.
Figure 5. 3D response surface plots showing effect of independent variables on the percentage release efficiency (%RE) of lacidipine from the prepared proniosomes formulations (Y3).
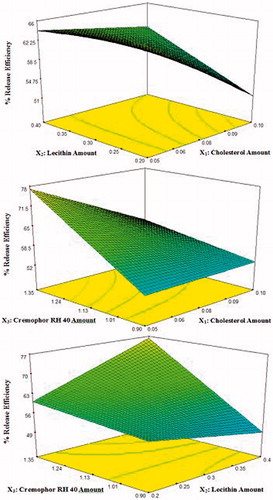
Effect of the amount of cholesterol on the %RE
It was found that the %RE of lacidipine significantly decreased generally with increasing the amount of cholesterol. This is probably due to the fact that the presence of cholesterol in niosomal formulations reduces the leakage or permeability of entrapped drug by decreasing membrane fluidity which led to lower drug elution from the vesicles (Abd-Elbary et al., Citation2008; Ali et al., Citation2010). These results agreed with previous studies (Samyuktha Rani & Vedha Hari, Citation2011; Marwa et al., Citation2013; Rahman et al., Citation2014).
Effect of the amount of lecithin on the %RE
Generally, the %RE of lacidipine significantly increased with increasing the amount of lecithin. This could be explained on the basis that the lecithin may act as a permeation enhancer in the preparation of proniosomes (Yadav et al., Citation2010; Singla et al., Citation2012).
Effect of the amount of cremophor RH 40 on the %RE
The release profile of the proniosome-derived niosomes prepared using cremophor RH 40 (HLB 14-16) revealed significant increase in the %RE of lacidipine with the increase in the amount of surfactant. This may be due to its high solubilizing power on the hydrophobic lacidipine in aqueous medium, since hydrophilic surfactants have higher solubilizing power on hydrophobic solutes in aqueous medium compared with hydrophobic surfactants (Szuts et al., Citation2008). Similar results were reported by El-Laithy et al. (Citation2011) who found that increasing the amount of the hydrophilic sucrose stearate L-1965 (SE L-1965) from 180 to 270 mg was associated with higher drug release.
Optimization of lacidipine proniosomes
The optimum values of the independent variables were obtained using the Design-Expert® software and based on the criterion of desirability. Upon trading of various response variables and comprehensive evaluation of feasibility and exhaustive grid search, the formulation F4 containing cholesterol (10 mg), soya lecithin (80 mg), cremophor RH 40 (270 mg), absolute ethanol (0.25 ml), and water (0.1 ml) was found to fulfill the maximum requisite of an optimum formulation because it had minimum mean vesicle size (162.43 nm), maximum EE (98.01%), maximum RE (88.33%) values, and maximum desirability (0.98). The optimized formulation (F4) was then formulated as proniosomal gel using carbopol 940 (1% w/w).
Evaluation of the optimized lacidipine proniosomal gel formulation
In vitro drug permeation studies through excised rabbit skin
The results of the cumulative amount permeated per unit area (1.7 cm2) are shown graphically in and permeation data analysis is represented in . As shown in , no lag phase was detected and lacidipine was detected in the receptor compartment in the first 0.5 h (minimum sampling time). This could indicate that all processes (water permeation from the receptor compartment to the skin, proniosomes conversion to niosomes and eventually the permeation of lacidipine across the skin) took place very rapidly. Similar observations were previously reported (Vora et al., Citation1998; Fang et al., Citation2001; Aboelwafa et al., Citation2010; El-Laithy et al., Citation2011).
Figure 6. Permeation profile of lacidipine from optimized proniosomal gel formulation and from the control emulgel through excised rabbit skin.
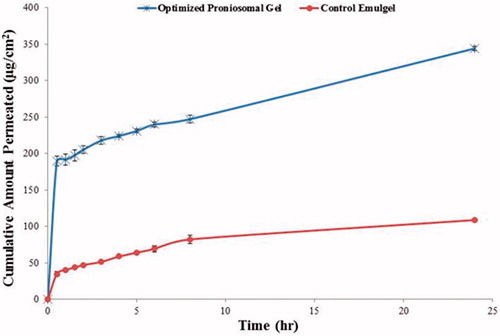
Table 3. Permeation data parameters.
It is clear that the optimized proniosomal gel formulation exhibited higher skin permeation compared with the emulgel formulation containing equivalent amount of lacidipine. The observed penetration enhancement effect may be attributed to both the presence of no-nionic surfactant and the formation of niosomes on hydration of proniosomes. There are two possible mechanisms by which skin permeation is improved by surfactants. First, surfactants may increase fluidity, solubilize, and extract lipid component in the stratum corneum. Second, they may interact and bind with keratin filaments resulting in a disruption within the corneocyte (Park et al., Citation2000; Aboelwafa et al., Citation2010). Niosomes may enhance the permeability of drugs through structure modification of stratum corneum, previous researchers reported that the intercellular lipid barrier in the stratum corneum would be more permeable following treatment with niosomes (Aboelwafa et al., Citation2010). Additionally, adsorption and fusion of the formed niosomes onto the surface of skin leads to a high thermodynamic activity gradient of drug at the interface, which is thought to be the main driving force for the permeation of lipophilic species. This effect is signified by the presence of lecithin which is widely known to impart high affinity between the vesicle and skin surface layers (Fang et al., Citation2001).
Transmission electron microscope (TEM)
To confirm the formation of vesicle structures from the proniosomes, the morphology of the hydrated proniosomal gel in distilled water was investigated using TEM. Photograph of TEM depicted in reveals that the examined proniosomal gel appears as spherical nanovesicles.
Figure 7. TEM photograph of lacidipine proniosomal gel formulation.

Accelerated stability study
It was found that after 90 d of storage, the drug content, the mean vesicle size, and the percentage entrapment efficiency of the optimized lacidipine proniosomal gel formulation were not significantly different (p > 0.05) from the freshly prepared (data not shown). These results suggest that proniosomes offered a more stable system that could minimize the problems reported about conventionally prepared niosomes like degradation by hydrolysis or oxidation, sedimentation, aggregation, and fusion during storage. The same finding was previously observed, when they studied the effect of storage on the vesicle size and the percentage entrapment efficiency (Abd-Elbary et al., Citation2008; El-Laithy et al., Citation2011).
In vivo bioavailability study in rabbit
The LC–MS/MS assay has a good linearity from 0.0 5 to 15.00 ng/ml with acceptable within and between day reproducibility. The lower limit of lacidipine quantification in plasma was 0.05 ng/ml. The assay method showed acceptable precision with CV% < 20% where the calculated CV% values were 1.57–17.24% for the intra-day assay and 1.67–17.85% for the inter-day assay. In addition, the assay method showed acceptable accuracy with relative error < 20% where the values of the relative error ranged from −18.92% to 15.20% (data not shown).
The mean plasma concentration–time curves after oral administration of lacipil® 4 mg tablet and after transdermal application (equivalent to 4 mg lacidipine) of the optimized proniosomal gel formulation are shown in . Mean pharmacokinetic parameters are presented in . The mean values of Cpmax, Tmax, Kel, T1/2, AUC(0-72), and AUC(0–∞) after oral administration of lacipil® 4 mg tablet were found to be 44.05 ng/ml, 0.58 h, 0.121 h − 1, 5.76 h, 209.02 ng h/ml, and 209.02 ng h/ml, respectively, while after transdermal application of the optimized proniosomal gel formulation, they were found to be 101.38 ng/ml, 1.00 h, 0.053 h − 1, 13.37 h, 452.63 ng h/ml, and 464.17 ng h/ml, respectively.
Figure 8: Mean plasma concentration time curve of lacidipine after oral administration of lacipil® tablet and after transdermal application of the proniosomal gel formulation.
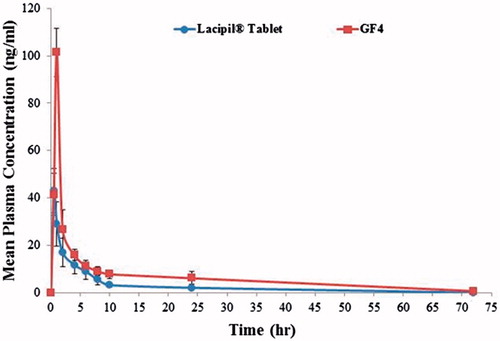
Table 4. Pharmacokinetic parameters of lacidipine following oral administration of lacipil® tablet and transdermal application of the optimized proniosomal gel formulation.
Overall, it is apparent from the previous results that the mean value of Cpmax after transdermal application of the optimized proniosomal gel formulation (2.22-folds increase) was significantly (p < 0.05) higher than that of the market product (Lacipil® tablets). Also, the mean value of AUC(0−∞) by the transdermal route was 2.22 times significantly (p < 0.05) higher than that of the market product (Lacipil® tablets). In addition, the relative bioavailability of the optimized proniosomal gel was 222.07% with respect to the market product.
The improved bioavailability of lacidipine after transdermal application may be due to two reasons. First, the ability of lacidipine to penetrate skin layers due to enhancement of its permeation through the skin via proniosomes. Second, this could be due to avoidance of first-pass hepatic metabolism by transdermal route. The reported oral bioavailability of lacidipine was 10% because of first-pass metabolism. Therefore, transdermal route of administration of lacidipine in the form of proniosomal gel could provide an effective treatment for the management of chronic hypertension.
Skin irritation test
Skin irritation is a common problem encountered with dermal and transdermal drug delivery, limiting its wide acceptance among patients in spite of its obvious benefits. For this reason, skin irritancy test was performed to confirm the safety of the optimized proniosomal gel formulation. Draize et al. (Citation1944) mentioned that a value of the primary irritancy index (PII) (erythema and edema) < 2 indicates that the applied formulation is not irritant to human skin. The mean value of PII of the optimized proniosomal gel formulation was found to be 0.33 ± 0.47. From this, it was concluded that the optimized lacidipine proniosomal gel formulation was safe for transdermal drug delivery.
Histopathological examination
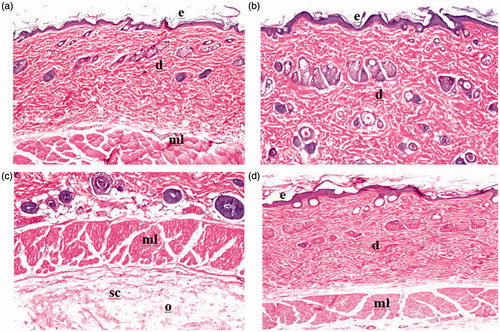
Histopathological examination of the skin rat specimens was performed using a Nikon light microscope (Nikon, Tokyo, Japan) to evaluate the cutaneous irritation potential of the optimized proniosomal gel formulation. The photomicrographs of control rat skin (untreated) group I showed normal skin with well-defined epidermal and dermal layers as shown in . While the photomicrographs of the second group (treated with formalin solution as standard irritant) showed inflammatory cell infiltration with edema in the subcutaneous connective tissue (). When the skin was treated with the optimized proniosomal gel formulation for 24 h, the epidermis and the dermis did not show any inflammatory cell infiltration. There was no histopathological alteration or apparent signs of skin irritation (erythema and edema) observed on skin specimens, indicating the absence of any skin irritation as a consequence of proniosomal gel treatment (). These results indicated that the developed lacidipine proniosomal gel is safe for transdermal delivery.
Figure 9. Light micrographs of rat skin untreated (a). Light micrographs of rat skin treated with standard irritant (b). Light micrographs of rat skin treated with standard irritant (c). Light micrographs of rat skin treated with optimized lacidipine proniosomal gel formulation (d).
Conclusion
Different proniosome formulations containing 1% w/w lacidipine were prepared by a coacervation phase separation method, evaluated, and optimized using a 23 full factorial design to define the optimum amount of the cholesterol, soya lecithin, and non-ionic surfactant (cremophor RH 40) to produce proniosomes with minimum vesicle size, high EE, and high RE. The relative bioavailability of the optimized proniosomal gel was 222.2% with respect to the market product. Results of the skin irritation test and histopathological examinations indicated that developed lacidipine gel is safe for human use. From these results, it can be concluded that the developed cremophor RH 40 proniosomes could be considered as very promising carrier as an absorption and penetration enhancer for delivering of the antihypertensive lacidipine transdermally.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
Reference
- Abdel Malak NS. (2012). Formulation of coated polymer reinforced gellan gum beads of tizanidine HCl using fractional factorial design. Int J Pharm Pharm Sci 4:369–79
- Abd-Elbary A, El-Laithy HM, Tadros MI. (2008). Sucrose stearate-basedproniosome-derived niosomes for the nebulisable delivery of cromolyn sodium. Int J Pharm 357:189–98
- Abdelbary G. (2011). Ocular ciprofloxacin hydrochloride mucoadhesive chitosancoated liposomes. Pharm Dev Technol 16:44–56
- Aboelwafa AA, El-Setouhy DA, Elmeshad AN. (2010). Comparative study on the effects of some polyoxyethylene alkyl ether and sorbitan fatty acid ester surfactants on the performance of transdermal carvedilol proniosomal gel using experimental design. AAPS PharmSciTech 11:1591–602
- Aburahma MH, Abdelbary GA. (2012). Novel diphenyl dimethyl bicarboxylate provesicular powders with enhanced hepatocurative activity: preparation, optimization, in vitro/in vivo evaluation. Int J Pharm 422:139–50
- Ahad A, Aqil M, Kohli K, et al. (2012). Formulation and optimization of nanotransfersomes using experimental design technique for accentuated transdermal delivery of valsartan. Colloids Surf B: Biointerfaces 100:146–54
- Ahmed S, El-Setouhy DA, Badawi AA, El-Nabarawi MA. (2014). Provesicular granisetron hydrochloride buccal formulations: in vitro evaluation and preliminary investigation of in vivo performance. Eur J Pharm Sci 60:10–23
- Akhtara M, Imamb SS, Ahmada MA, et al. (2014). Neuroprotective study of Nigella sativa-loaded oral provesicular lipid formulation: in vitro and ex vivo study. Drug Deliv 21:487–94
- Ali MH, Kirby DJ, Mohammed AR, Perrie Y. (2010). Solubilisation of drugs within liposomal bilayers: alternatives to cholesterol as a membrane stabilising agent. J Pharm Pharmacol 62:1646–55
- Ammar HO, Ghorab M, El-Nahhas SA, Higazy IM. (2011). Proniosomes as a carrier system for transdermal delivery of tenoxicam. Int J Pharm 405:142–52
- Asija R, Sharma D, Nirmal H. (2014). Proniosomes: a review. Int J Curr Trends Pharm Res 2:337–41
- Azeem IA, Ahmad FJ, Khar RK, Talegaonkar S. (2009). Nanocarrier for the transdermal delivery of an antiparkinsonian drug. AAPS PharmSciTech 10:1093–103
- Balakrishnan P, Shanmugam S, Lee WS, et al. (2009). Formulation and in vitro assessment of minoxidil niosomes for enhanced skin delivery. Int J Pharm 377:1–8
- Biju SS. (2006). Vesicular systems: an overview. Ind J Pharm Sci 68:141–53
- Chavan P, Jain B, Jain P. (2012). Proniosomal gel: a novel approach for transdermal drug delivery: a review. Int J Pharm Res Dev 4:158–70
- Csoka G, Marton S, Zelko R, et al. (2007). Application of sucrose fatty acid esters in transdermal therapeutic systems. Eur J Pharm Biopharm 65:233–7
- Draize J, Woodward G, Calvery H. (1944). Methods for the study of irritation and toxicity of substances applied topically to the skin and mucous membranes. J Pharmacol Exp Ther 82:377–9
- Elhissi A, Hidayat K, David A, et al. (2013). Air-jet and vibrating-mesh nebulization of niosomes generated using a particulate-based proniosome technology. Int J Pharm 444:193–9
- ElKasabgy NA, Elsayed I, Elshafeey AH. (2014). Design of lipotomes as a novel dual functioning nanocarrier for bioavailability enhancement of lacidipine: in-vitro and in-vivo characterization. Int J Pharm 472:369–79
- El-laithy HM, Shoukry O, Mahran IG. (2011). Novel sugar esters proniosomes for transdermal delivery of vinpocetine: preclinical and clinical studies. Eur J Pharm Biopharm 77:43–55
- El-Nabarawi MA, Bendas ER, El Rehem RTA, Abary MYS. (2013). Transdermal drug delivery of paroxetine through lipid-vesicular formulation to augment its bioavailability. Int J Pharm 443:307–17
- Essa E. (2010). Effect of formulation and processing variables on the particle size of sorbitan monopalmitate niosomes. Asian J Pharm 4:227–33
- Fang JY, Yu SY, Wu PC, et al. (2001). In vitro skin permeation of estradiol from various proniosome formulations. Int J Pharm 215:91–9
- Fouad SA, Basalious EB, El-Nabarawi MA, Tayel SA. (2013). Microemulsion and poloxamer microemulsion-based gel for sustained transdermal delivery of diclofenac epolamine using in-skin drug depot: in vitro/in vivo evaluation. Int J Pharm 453:569–78
- Gadekar V, Bhowmick M, Pandey GK, et al. (2013). Formulation and evaluation of naproxen proniosomal gel for the treatment of inflammatory and degenerative disorders of the musculoskeletal system. J Drug Deliv Ther 3:36–41
- Gannu R, Palem CR, Yamsani VV, et al. (2010). Enhanced bioavailability of lacidipine via microemulsion based transdermal gels: formulation optimization, ex vivo and in vivo characterization. Int J Pharm 388:231–41
- Gannu R, Yamsani VV, Palem CR, et al. (2009). Development of high performance liquid chromatography method for lacidipine in rabbit serum: application to pharmacokinetic study. Anal Chim Acta 632:278–83
- Grove M, Müllertz A. (2007). Liquid self-microemulsifying drug delivery systems. In oral lipid-based formulations. In: Hauss DJ, ed. New York: Informa Inc., 107–27
- Guinedi AS, Mortada ND, Mansour S, Hathout RM. (2005). Preparation and evaluation of reverse-phase evaporation and multilamellar niosomes as ophthalmic carriers of acetazolamide. Int J Pharm 306:71–82
- Gupta A, Prajapati SK, Balamurugan M, et al. (2007). Design and development of a proniosomal transdermal drug delivery system for captopril. Trop J Pharm Res 6:687–93
- Hao Y, Zhao L, Li N, et al. (2002). Studies on a high encapsulation of colchicine by a niosome system. Int J Pharm 244:73–80
- Hao YM, Li K. (2011). Entrapment and release difference resulting from hydrogen bonding interactions in niosome. Int J Pharm 403:245–53
- Hernàndez JRV, Goymann CCM. (2005). Novel nanoparticulate carrier system based on carnauba wax and decyl oleate for the dispersion of inorganic sunscreens in aqueous media. Eur J Pharm Biopharm 60:113–22
- Hiremath PS, Soppimath KS, Betageri GV. (2009). Proliposomes of exemestane for improved oral delivery: formulation and in vitro evaluation using PAMPA, Caco-2 and rat intestine. Int J Pharm 380:96–104
- Hosny KM. (2010). Ciprofloxacin as ocular liposomal hydrogel. AAPS PharmSciTech 11:241–6
- Janga KY, Jukanti R, Velpula A, et al. (2012). Bioavailability enhancement of zaleplon via proliposomes: role of surface charge. Eur J Pharm Biopharm 80:347–57
- Junyaprasert VB, Teeranachaideekul V, Supaperm T. (2008). Effect of charged and non-ionic membrane additives on physicochemical properties and stability of niosomes. AAPS PharmSciTech 9:851–9
- Kamboj S, Saini V, Bala S. (2014). Formulation and characterization of drug loaded nonionic surfactant vesicles (niosomes) for oral bioavailability enhancement. Sci World J 2014:959741
- Kantarci G, Ozgungey I, Karasulu HY, et al. (2005). In vitro permeation of diclofenac sodium from novel microemulsion formulations through rabbit skin. Drug Dev Res 65:17–25
- Kumar R, Singh B, Bakshi G, Katare OP. (2007). Development of liposomal systems of finasteride for topical applications: design, characterization, and in vitro evaluation. Pharm Dev Technol 12:591–601
- Kumar S, Prajapati S, Sahu VK, Andprakash G. (2012). Proniosomal gel of flurbiprofen: formulation and evaluation. J Drug Deliv Ther 2:1–5
- Lee CR, Bryson HM. (1994). Lacidipine: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the treatment of hypertension. Drugs 48:274–96
- Mahale NB, Thakkar PD, Mali RG, et al. (2012). Niosomes: novel sustained release nonionic stable vesicular systems: an overview. Adv Colloid Interface Sci 183:46–54
- Marwa A, Omaima S, Hanaa E, Mohammed A. (2013). Preparation and in-vitro evaluation of diclofenac sodium niosomal formulations. Int J Pharm Sci Res 4:1757–65
- Mc Cormack PL, Wagstaff AJ. (2003). Lacidipine: a review of its use in the management of hypertension. Drugs 63:2327–56
- Mokhtar M, Sammour OA, Hammad MA, Megrab NA. (2008). Effect of some formulation parameters on flurbiprofen encapsulation and release rates of niosomes prepared from proniosomes. Int J Pharm 361:104–11
- Negi P, Ahmad FJ, Ahmad D, et al. (2011). Development of a novel formulation for transdermal delivery of an antidepressant drug. Int J Pharm Sci Res 2:1766–71
- Pankaj S, Rini T, Dandagi PM. (2013). Formulation and evaluation of proniosome based drug delivery system of the antifungal drug clotrimazole. Int J Pharm Sci Nanotechnol 6:1945–51
- Pardakhty A, Varshosaz J, Rouholamini A. (2007). In vitro study of polyoxyethylene alkyl ether niosomes for delivery of insulin. Int J Pharm 328:130–41
- Patil HN, Hardikar SR, Bhosale AV. (2012). Formulation development and evaluation of proniosomal gel of carvedilol. Int J Pharm Pharm Sci 4:191–7
- Park ES, Chang SY, Hahn M, Chi SC. (2000). Enhancing effect of polyoxyethylene alkyl ethers on the skin permeation of ibuprofen. Int J Pharm 209:109–19
- Rahman SA, Abdelmalak NS, Badawi A, et al. (2014). Formulation of tretinoin-loaded topical proniosomes for treatment of acne: in-vitro characterization, skin irritation test and comparative clinical study. Drug Deliv 1–9
- Rajabalaya R, David SR, Chellian J, et al. (2015). Transdermal delivery of oxybutynin chloride proniosomal gels for the treatment of overactive bladder. Drug Deliv 1–10
- Ramkanth S, Chetty CM, Sudhakar Y. (2014). Development and in vitro permeation studies of proniosomal based transdermal delivery system of Atenolol. Pak J Pharm Sci 27:115–20
- Rowe RC, Sheskey PJ, Owen SC. (2006). Handbook of pharmaceutical excipients. 5th ed. London: Pharmaceutical Press
- Salama HA, Mahmoud AA, Kamel AO, et al. (2012). Phospholipid based colloidal poloxamer-nanocubic vesicles for brain targeting via the nasal route. Colloids Surf B Biointerfaces 100:146–54
- Samyuktha Rani B, Vedha Hari BN. (2011). Niosomal formulation of orlistat: formulation and in-vitro evaluation. Int J Drug Dev Res 3:300–11
- Satturwar PM, Fulzele SV, Nande VS, Khandare JN. (2002). Formulation and evaluation of ketoconazole niosomes. Indian J Pharm 64:155–8
- Shargel L, Wu-Pong S, Yu ABC. (2005). Applied biopharmaceutics & pharmacokinetics. New York: Appleton & Lange Reviews/McGraw-Hill, Medical Pub, 457
- Shatalebi MA, Mostafavi SA, Moghaddas A. (2010). Niosome as a drug carrier for topical delivery of N-acetyl glucosamine. Res Pharm Sci 5:107–17
- Singla S, Hari Kumar SL, Aggarwal G. (2012). Proniosomes for penetration enhancement in transdermal system. Int J Drug Dev Res 4:1–13
- Soliman SMH, Abd El-Malak NS, El-Gazayerly ON, Abd El-Rehim AEA. (2010). Formulation of microemulsion gel systems for transdermal delivery of celecoxib: in-vitro permeation, anti-inflammatory activity and skin irritation tests. Drug Discov Ther 4:459–71
- Soliman SMH, Abd El-Malak NS, El-Gazayerly ON, Abd El-Rehim AEA. (2011). Preparation of celecoxib solid dispersions for dermal application: in vitro characterization and skin irritation test. J Drug Del Sci Tech 21:509–16
- Szuts A, Makai Z, Rajko R, Szabo-Revesz P. (2008). Study of the effects of drugs on the structures of sucrose esters and the effects of solid-state interactions on drug release. J Pharm Biomed Anal 48:1136–42
- Thomas L, Viswanad V. (2012). Formulation and optimization of clotrimazole-loaded proniosomal gel using 3(2) factorial design. J Sci Pharm 80:731–48
- Ustundag ON, Apaydin S, Karabay YNU, et al. (2011). Evaluation of skin permeation and anti-inflammatory and analgesic effects of new naproxen microemulsion formulations. Int J Pharm 416:136–44
- Vora B, Khopade AJ, Jain NK. (1998). Proniosome based transdermal delivery of levonorgestrel for effective contraception. J Control Release 54:149–65
- Waddad AY, Abbad S, Yu F, et al. (2013). Formulation, characterization and pharmacokinetics of Morin hydrate niosomes prepared from various non-ionic surfactants. Int J Pharm 456:446–58
- Wang Y, Tu S, Li R, et al. (2010). Cholesterol succinyl chitosan anchored liposomes: preparation, characterization, physical stability, and drug release behavior. Nanomed: Nanotechnol Biol Med 6:471–7
- Xu H, He L, Nie S, et al. (2009). Optimized preparation of vinpocetine proliposomes by a novel method and in vivo evaluation of its pharmacokinetics in New Zealand rabbits. J Control Release 140:61–8
- Yadav K, Yadav D, Saroha K, et al. (2010). Proniosomal gel: a provesicular approach for transdermal drug delivery. Der Pharm Lett 2:189–98
- Yasam VR, Jakki SL, Natarajan J, Kuppusamy G. (2014). A review on novel vesicular drug delivery: proniosomes. Drug Deliv 21:243–9