
Abstract
The purpose of this study was to develop folic acid functionalized long-circulating co-encapsulated docetaxel (DTX) and curcumin (CRM) solid lipid nanoparticles (F-DC-SLN) to improve the pharmacokinetic and efficacy of DTX therapy. F-DC-SLN was prepared by hot melt-emulsification method and optimized by face centered-central composite design (FC-CCD). The SLN was characterized in terms of size and size distribution, drug entrapment efficiency and release profile. The cytotoxicity and cell uptake of the SLN formulations were evaluated in MCF-7 and MDA-MB-231 cell lines. The in vivo pharmacokinetic and biodistribution were studied in Wistar rats. F-DC-SLN exhibited 247.5 ± 3.40 nm particle size with 73.88 ± 1.08% entrapment efficiency and zeta potential of 14.53 ± 3.6 mV. Transmission electron microscopy (TEM) revealed spherical morphology of the SLN. Fluorescence microscopy confirmed the targeting efficacy of F-DC-SLN in MCF-7 cells. F-DC-SLN exhibited a significant increase in area under the curve (594.21 ± 64.34 versus 39.05 ± 7.41 μg/mL h) and mean residence time (31.14 ± 19.94 versus 7.24 ± 4.51 h) in comparison to Taxotere®. In addition, decreased DTX accumulation from F-DC-SLN in the heart and kidney in comparison to Taxotere may avoid to toxicity these vital organs. In conclusion, the F-DC-SLN improved the efficacy and pharmacokinetic profile of DTX exhibiting enhanced potential in optimizing breast cancer therapy.
Introduction
Breast cancer is the second leading cause of cancer deaths presently after lung cancer and is the most common cancer in women (Ferlay et al., Citation2007). World health organization predicts nearly 11.5 million deaths by the year 2030. The overall survival rate of around 15% with the existing chemotherapy underscores the need for more effective and novel therapeutic strategies (Kawalec et al., Citation2014) Treatment strategies available to combat cancers are surgery, radiation therapy, hormonal therapy, immunotherapy and chemotherapy. Chemotherapy plays a pivotal role in treatment of cancer; wherein cytotoxic agents are employed to destroy the cancerous cells. An array of antineoplastic agents is available, which are either used alone or in combination to form an effective treatment modality (Payam et al., Citation2011).
Docetaxel (DTX), a member of the taxoid family, is a preferred cytotoxic agent used in the treatment of breast, ovarian, non-small cell lung cancer and other human malignancies (Hu et al., Citation2015). Although, the taxoids are widely employed in cancer treatment, one of their limitations is the development of drug resistance mediated by P-glycoprotein and multidrug resistance efflux pumps. Further, the taxoids activate anti-apoptotic defense system mediated by B cell lymphoma (Bcl-2) protein. This induces the release of transcriptional factor NF-kB, which causes drug resistance in cancer cells (Jinturkar et al., Citation2012). Curcumin (CRM) is a natural polyphenol found in rhizomes of Curcuma longa (turmeric). CRM has been reported to exhibit that several pharmacological activities including anti-inflammatory, antineoplastic, chemopreventive activities have been reported for CRM and found to be safe even at high doses (Agrawal et al., Citation2014). Further, it inhibits the proliferation of several tumor types by blocking activity of NF-kB and anti-apoptotic proteins, including Bcl-2. In addition, it is a potent inhibitor of three major ATP-binding cassette (ABC) drug transporters, namely multidrug resistance protein 1 (MDR1), multidrug resistance associated protein (MRP1) and ATP-binding cassette sub-family G member 2 (ABCG2), that result in multidrug resistance to chemotherapy (Kunnumakkara et al., Citation2008, Ullah, Citation2008). Thus, DTX multidrug resistance can be overcome by the co-administration with CRM, resulting in dose reduction and enhanced efficacy.
The effectiveness of nanomedicine in cancer therapy stems from its ability to deliver selectively drugs to the tumor site by enhanced permeability and retention (EPR) effect (Cho et al., Citation2014). Nanotechnology enables increased delivery of poorly water soluble drugs, targeted delivery, co-delivery of two or more therapeutic agents, increase in half-life, controlled and sustained release of drug, reduced multidrug resistance, and multimodality treatment by co-delivery of chemotherapeutic, radiotherapeutic, thermotherapeutic and biotherapeutics agents. In addition, visualization of sites of drug delivery is possible by co-delivery of drug/s and imaging agents (Kutty & Fen, Citation2013). As a well-developed delivery system, solid lipid nanoparticle (SLN) has several advantages over polymeric nanoparticles, liposomes and microemulsion (Shegokar et al., Citation2011). SLN exhibits biocompatibility, physical stability, controlled release properties and ease of manufacture and scale-up. In addition, exclusion of organic solvents during its preparation, ability to incorporate both hydrophilic and hydrophobic drugs, potential of passive drug targeting is other advantages of SLN formulation (Tan & Billa, Citation2014). SLN is essentially composed of a biocompatible lipid matrix in which the drug is uniformly dispersed and stabilized with the aid of surfactants.
The folate receptor (FR) is one of the most widely evaluated receptors for active targeting of anticancer therapeutics. Folic acid has many advantages over antibody ligands such as small size, non-immunogenicity, non-toxicity, ease of handling, stability and low cost (Li & Szoka, Citation2007). Further, for increased effectiveness of active targeting modalities, there is a need for a longer circulation time. This increase in circulation time can be achieved by surfaced modification by incorporation of hydrophilic polymer, polyethylene glycol (PEG) by conjugation. PEG has many desirable attributes such as low immunogenicity and antigenicity, chemical inertness and primary hydroxyl groups for modification (Olivier et al., Citation2002). SLN systems represent a promising platform for conjugating PEG and for incorporation of active targeting ligand such as folic acid. Hence, in this study, the aim was to develop long circulating, functionalized SLN for co-delivery of DTX and CRM.
In present work, we developed folic acid functionalized co-encapsulated DTX and CRM SLN (F-DC-SLN). DTX was combined with CRM to enhance its therapeutic efficacy on the basis of preliminary cytotoxicity study in highly resistant and metastatic breast cancer cells (MDA-MB-231). DTX and CRM SLN (DC-SLN) formulation was developed systematically and optimized by face centered-central composite design (FC-CCD) to obtain the optimum formulation and process parameters. Folic acid-stearic acid (FA–SA) conjugate was then synthesized by classical 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC) chemistry and incorporated in the DC-SLN. In addition, PEG–stearic acid (PEG–SA) was incorporated to obtain F-DC-SLN. F-DC-SLN was characterized for its size and size distribution, surface morphology, charge, chemistry and drug release profile. Further, in vitro cell uptake of the SLN was evaluated both qualitatively and quantitatively in MCF-7 cells. F-DC-SLN pharmacokinetic and biodistribution studies were conducted on Wistar rats after intravenous administration. Two-month stability study was conducted at accelerated and refrigerated conditions.
Materials and methods
Materials
Docetaxel trihydrate was generous gift sample from Panacea Biotec, and Naprod Life Sciences (Bombay, India). CRM (>99.9% pure) was procured from Sami Labs Ltd. (Bangalore, India). Compritol 888 ATO® was a gift sample from Gattefosse (St-Priest, France). Glyceryl monostearate (GMS) was obtained from Johnson Matthey Company (Gurgaon, India). Poloxamer 188 was procured from Signet Chemical Corporation (Bombay, India). Folic acid, polyoxyethylene (40) stearate (PEG–SA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), Dimethyl sulfoxide (DMSO), Coumarin-6 were purchased from Sigma Aldrich (Bombay, India). Stearic acid, EDAC, Mannose, Dulbecco’s modified eagle medium (DMEM) (high glucose) were purchased from HiMedia (Bombay, India). Fetal bovine serum and penicillin–streptomycin solution (100×) were purchased from Invitrogen (CA). All other chemicals used were of analytical reagent grade. Human breast cancer cell line, MCF-7 and MDA-MB-231 were purchased from National Center for Cell Sciences (NCCS), Pune, India.
Cytotoxicity of DTX and CRM in MDA-MB-231 cells
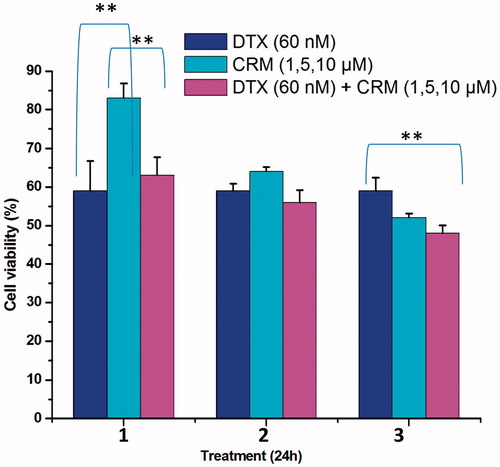
The cytotoxicity of DTX and CRM combination was evaluated in MDA-MB-231 cells, hormone receptor-negative advanced metastatic breast cancer cell line (Finn et al., Citation2007). In a media containing combinations of DTX and CRM, below their individual IC50 values, the cells were incubated for 24 h and cell viability measured by MTT assay. DTX at 60 nM was combined with 1, 5 and 10 µM of CRM for the evaluation study.
HPLC analysis
We developed a simultaneous method for estimation of DTX and CRM by HPLC (Shimadzu, MD, USA with SCL-10 A VP controller) equipped with class VP software. We selected Phenomenex C18 (250 mm × 4.5 mm, 5 μm) column and operated at 35 °C for analysis. A gradient (50:50, 50:50, 60:40, 50:50) of acetonitrile and water (pH of 4.0, adjusted with acetic acid) was optimized and formed the mobile phase maintained at flow rate of 1 mL/min. The samples were analyzed at 230 and 420 nm for DTX and CRM, respectively.
Solid lipid screening
The solubility of the drugs in solid lipids was estimated by visual inspection. Incrementally added (1% w/w) DTX, CRM and DTX/CRM (1:1) ratio into melted lipid at 80 °C and agitated at 100 rpm in an Equitron water shaker bath (Medica Mfg. Co., Mumbai, India) for 24 h. The addition of drug continued until the drug crystals remained un-dissolved in the melted lipid under the conditions of the study.
Synthesis of FA–SA conjugate
To a solution of stearic acid (400 mg) in dimethylformamide (DMF) (5 mL), added 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) (400 mg) and stirred thoroughly for 1 h at RT. To this mixture, added the folic acid (600 mg) solution in pyridine (3 mL) and stirred overnight at RT. Then, added water into the reaction mixture to precipitate the conjugate. The un-reacted folic acid, EDC and the water-soluble by products were removed by dialysis (MWCO-10 kDa, Sigma Aldrich) for 24 h in distilled water, filtered through a 0.45-μm Millipore filter and lyophilized to obtain FA–SA (Yuan et al., Citation2008). presents the schematic diagram of the preparation of FA–SA conjugate.
Figure 1. Schematic diagram of FA–SA conjugate synthesis.
The 1H NMR spectrum was recorded in 5 wt% (CD3)2SO solution employing ZH079807 Bruker 400 UltraShield™ spectrophotometer (MD, USA) (with B-ACS120 Autosampler and Topspin 2.7 software, MA, USA) at 298 K. The molecular mass was confirmed by LCQ (3D ion trap, ESI+ mode), Finnigan MAT spectrometer (Washington DC) with Xcaliber software (MA, USA). Infrared spectrum was obtained in FTIR (Perkin Elmer Synthesis Monitoring System) equipped with Spectrum-1 software (MA, USA).
Preparation of SLN
DC-SLN was prepared by method previously described by Mulik et al. (Citation2010) with slight modifications. First, Compritol and GMS (1:1 weight ratio) were heated to 80 °C. Into the melted lipid, added DTX and CRM, 5 mg each and stirred to dissolve. A solution of Poloxamer 188 (0.5% w/v) was heated to 80 °C. Both, the lipid and aqueous phases were mixed rapidly at 80 °C and homogenized for 10 min at 20 000 rpm (T 25 digital ULTRA-TURRAX®, IKA, NC, USA). The resultant hot nano-emulsion was cooled to 2–8 °C to obtain SLN. The obtained dispersion was centrifuged (Himac CR22-GII, Hitachi, Tokyo, Japan) at 22 000 rpm at 4 °C for 30 min. The residue obtained was re-suspended in distilled water and lyophilized with 10% w/v mannose as a cryoprotectant.
Blank SLN was prepared by above method excluding incorporation of the drugs. In order to formulate fluorescent SLN, the drugs were replaced with coumarin-6 (10 mg). Similarly, F-DC-SLN was formulated by replacing GMS and Compritol in DC-SLN with a mixture of PEG–SA (6 mg) and FA–SA (6 mg). For comparative evaluation study, DTX SLN (D-SLN) was formulated by incorporating 10 mg of DTX alone.
Optimization of SLN
Preliminary studies were conducted to determine the critical SLN formulation and process factors. The homogenization time, speed, stabilizer concentration and lipid amount affected SLN particle size, zeta potential, polydispersity index (PDI) and entrapment efficiency (EE). The compositions of the stabilizer, lipids and aqueous phase volume were optimized during the preliminary studies. FC-CCD by Design-Expert® software (Stat-Ease, Inc., Minnesota, USA) was adopted to optimize the factors at three levels (+1, 0 and −1) (Chauhan et al., Citation2012). displays the details of the factors, their levels and responses adopted in the optimization. The design generated 30 trials; including 16 factorial points, 8 axial points and 6 replicates as the center point for estimation of the pure error. The experimental runs were conducted in random in order to increase the predictability of the model.
Table 1. FC-CCD design with factors with corresponding levels and responses.
Characterization of SLN
Particle size, PDI and zeta potential
The SLN formulation was diluted in distilled water for determination of particle size, PDI and zeta potential in a Zetasizer (Nano ZS, Malvern Instruments, Malvern, UK). An average of 12 measurements was determined.
Entrapment efficiency
An indirect method was adopted for estimating EE of the SLN. Briefly, the SLN dispersion was centrifuged at 8000 rpm for 10 min to sediment the unentrapped drugs. The supernatant, containing the nanoparticles, was subjected to centrifugation at 22 000 rpm at 4 °C for 30 min. The supernatant was diluted with methanol and amount of DTX and CRM quantified by HPLC. The EE was calculated by the formula:
(1)
Shape and surface morphology
The SLN shape and morphology were studied by transmission electron microscopy (TEM) (Technai F20, FEI Netherland, Eindhoven, Netherland). The sample was stained with phosphotungstic acid (1.5%) solution and placed on 400 mesh carbon-coated grid and held for 90 s at RT and examined at an accelerating voltage of 100–200 kV.
Freeze drying
The SLN formulation(s) were freeze-dried (Vir Tis, Wizard 2.0, New York) after optimization of the freeze drying process (42 h). The condenser temperature was maintained at −60 °C at pressure of 200 Torr. The SLN formulation was centrifuged at 20 000 rpm at 4 °C for 30 min. The pellet obtained was dispersed in 5 mL of distilled water and freeze-dried in 5 mL vials with mannose (10% w/v) as a cryoprotectant. The product was evaluated for its physical appearance, particle size and EE.
Solid state characterization
Differential scanning calorimetry
Differential scanning calorimetry (DSC) thermograms of DTX, CRM, GMS, Compritol, blank SLN, DC-SLN and F-DC-SLN were recorded (Mettler Toledo, DSC, 821 e, Greifensee, Switzerland). The DSC system was previously calibrated with indium. A heating rate of 20 °C/min was employed over a temperature range of 0–200 °C in nitrogen atmosphere (80 mL/min). Samples (2–8 mg) were weighed into aluminum pan and analyzed with empty pan employed as a reference.
Powder X-ray diffraction
The X-ray diffraction patterns of DTX, CRM, blank SLN, DC-SLN and F-DC-SLN were acquired from X-ray diffractometer (Bruker D8 Advance, Germany). Measurements were performed at a voltage of 40 kV and 25 mA. The scanned angle was set from 3° < 2θ > 40° and scanned at rate of 1 min−1.
Cross polarized light microscopy
The reconstituted DC-SLN and F-DC-SLN formulations were examined under cross polarized light microscope (Leica Microsystems, Wetzlar, Germany) to determine the presence of free drug crystals. The SLN was diluted with distilled water (1:10) before the study. The images were captured by a digital camera (Leica DC 300) present in the microscope and analyzed by software (Leica IM 50, version 1.20).
Characterization of folic acid on surface of F-DC-SLN
The FTIR spectra of folic acid, blank SLN, DC-SLN and F-DC-SLN were obtained in transmission mode in 400–4000 cm−1 region (infrared spectrometer, Perkin Elmer, MA, USA). Potassium bromide pellets were prepared by gently mixing 1 mg of the sample with 100 mg of KBr and the spectra obtained was analyzed by Spectrum V5.02 software (CIC Photonics, Inc., NM, USA).
In vitro drug release
The in vitro release profile of DTX and CRM from F-DC-SLN was evaluated by dialysis bag method in medium composed of phosphate buffer saline (pH 7.4, 0.01 M, 50% v/v ethanol). Accurate quantity (3 mL) of the SLN suspension was placed in sealed dialysis bag (MWCO 12 kDa, Sigma Aldrich) and suspended in a vial containing 20 mL of the medium, stirred constantly in a shaker water bath at 100 rpm at 37 °C. Aliquots of 1 mL of sample were withdrawn at different time intervals, and estimated the drug concentration by a validated HPLC method. To analyze the release kinetics and its mechanism, fitted the data to zero-order, first-order, Hixson Crowell, Weibull, Higuchi’s, Baker–Lonsdale and Korsemeyer–Peppas release models.
In vitro cell uptake
The cell uptake potential of F-DC-SLN was evaluated in MCF-7 cells. The cells were seeded into 96-well black plates (Costar, IL) at 5 × 103 cells/well (0.1 mL). After the cells reached 80% confluence, replaced the medium with a suspension of fluorescent SLN (0.250 mg/mL) and incubated for 2 h. Then, aspirated the SLN suspension, and washed the wells with 0.1 mL phosphate-buffered saline (PBS) thrice to remove the traces of SLN present on the surface. The cells were lyzed with 50 μL of 0.5% Triton X-100 in 0.2 N NaOH, and measured the fluorescence by the microplate reader (Molecular Devices, CA, USA) at an excitation wavelength of 430 nm and emission wavelength of 485 nm. The cellular uptake efficiency was expressed as the percentage of fluorescence in testing wells over that of positive control wells.
The qualitative uptake was evaluated by fluorescent microscopy. In an eight-well cover glass chamber MCF-7 cells were seeded and incubated until 70% confluence. Free coumarin, fluorescent SLN (0.250 mg/mL), dispersed in the cell culture medium, was added to the wells and incubated for 2 h. Then, cells were washed thrice with PBS to remove surface adsorbed SLN from the cells and observed under the microscope (Nikon Eclipse, Te2000-S, Tokyo, Japan).
In vitro cell cytotoxicity
For cytotoxicity measurement, MCF-7 cells were incubated in 96-well transparent plates (Costar) at 1 × 105 cells/well (0.2 mL) in DMEM medium. After 24 h, the medium was removed and the cells were incubated for 48 h in the media containing D-SLN, DC-SLN and F-DC-SLN at DTX concentrations of 0.25, 2.5, 10, 15 μg/mL. MTT assay was used to measure the cell viability. The absorbance of the wells was measured by the microplate reader at 550 nm and reference wavelength at 630 nm. Cell viability is defined as the percentage of absorbance of wells containing cells incubated with the SLN over that of cells only.
In vitro hemolysis
The hemolysis test was conducted as described in the literature with slight modifications. Fresh rat blood was collected in heparin tubes (Labtech Disposables, Gujarat, India) for the study. In brief, the blood sample was washed thrice with PBS and the red blood cells (RBCs) collected by centrifugation at 2800 rpm for 5 min and dispersed in normal saline. The washing step was repeated to remove debris and serum proteins. Taxotere, DC-SLN, F-DC-SLN were evaluated. Typically, 100 μL of the erythrocyte suspension was added to 900 μL of the test formulations. The samples were incubated at 37 °C for 60 min, with intermittent shaking and centrifuged at 3000 rpm for 60 min. The released hemoglobin in the supernatant was quantified in UV–visible spectrophotometer (Hitachi High Technologies Inc., Tokyo, Japan) at 540 nm. Erythrocytes incubated with saline and distilled water served as negative (0% hemolysis) and positive controls (100% hemolysis), respectively, and all hemolysis data points are presented as the percentage of the complete hemolysis. The % hemolysis rate (HR) was calculated from the following equation:
(2)
where DTest, DNegative and DPositive are the absorbance of test sample, negative control and positive control, respectively (Yueying et al., Citation2010).
Pharmacokinetic study
Animals and dosing
Female Wistar rats of 100–120 g and 3–4 weeks old were obtained from the central animal facility, National Institute of Pharmaceutical Education & Research (NIPER), India. The animal study protocols were duly approved by the Institutional Animal Ethics Committee, NIPER. The animals were acclimatized at temperature of 25 ± 2 °C and relative humidity of 50–60% under natural light/dark conditions for 1 week before the experimentation. The animals were randomly distributed into three groups each containing five animals. At DTX dose of 4 mg/kg, Group I received Taxotere as a reference, Group II received DC-SLN and Group III received F-DC-SLN, administered through the tail vein. The blood samples (∼0.2 mL) were collected from the retro-orbital plexus under the mild anesthesia into the microcentrifuge tubes containing heparin (40 IU/mL blood). Plasma was separated by centrifugation at 5000 rpm for 5 min at 4 °C. To 100 µL of plasma added 150 µL of acetonitrile to precipitate proteins and 10 µL of paclitaxel (20 µg/mL) as an internal standard; vortexed and centrifuged at 14 000 rpm at 4 °C for 10 min. The DTX amount in the supernatant was quantified by HPLC at 230 nm.
Bioanalysis of DTX in plasma
Calibration curves were used for the conversion of the DTX/paclitaxel chromatographic area to the DTX concentration. Standard and quality control samples were prepared by adding appropriate volumes of standard DTX solution in acetonitrile to drug free plasma. Calibration curve was designed over the range of 50–5000 ng/mL (R2 0.9766). Briefly, mixed an aliquot (100 µL) of plasma sample with 10 µL of internal standard solution (paclitaxel 20 µg/mL) and 50 µL of drug solution. After vortexing for 30 s, added acetonitrile (150 µL) as a protein precipitating agent and vortexed for 5 min. The mixture was centrifuged for 10 min at 14 000 rpm at 4 °C. After centrifugation, transferred the supernatant to autosampler vials, capped and placed in the HPLC (Waters 717plus autosampler, Tokyo, Japan). An 80 -µL aliquot of each sample was injected onto the HPLC column maintained at 40 °C. The mobile phase of acetonitrile and water (pH 4 adjusted with acetic acid, 52:48) at flow rate of 1.5 mL/min was employed for analysis.
Pharmacokinetic data analysis
The pharmacokinetic analysis of plasma concentration time data was analyzed by Kinetica 5.0 software (Thermo Scientific, MA, USA). Required pharmacokinetics parameters like total area under the curve (AUC), elimination half-life (t1/2), peak plasma concentration (Cmax), time to reach the maximum plasma concentration (Tmax) and mean residence time (MRT) were determined.
Biodistribution study
The biodistribution of SLN formulations were studied in Wistar rats. For the study, 18 female Wistar rats were fasted overnight and randomly divided into three groups of six animals each. At DTX dose of 4 mg/kg, Group I received Taxotere® as a reference, Group II received DC-SLN, while Group III received F-DC-SLN, administered through the tail vein. Three animals served as control without treatment and their tissues used as blank controls and for preparation of controlled spiked samples. Animals were decapitated in group of three at 7 and 120 min and the heart, liver, spleen, lungs, kidneys and brain removed, washed of residual blood, air dried and weighed (Li et al., Citation2009). Tissue samples were homogenized with saline to obtain 0.2 mg/mL, then added 40 μL paclitaxel solution (20 μg/mL) as an internal standard. The samples were vortexed for 3 min and centrifuged at 14 000 rpm for 10 min. The supernatants were collected and analyzed by HPLC.
Stastical analysis
The results of in vivo experiments are reported as mean ± SD (n = 5). The difference between the groups was tested by two-way analysis of variance (ANOVA), followed by Bonferroni post-tests using Graph Pad Prism 5.0 software (San Diego, USA). The difference was considered to be statistically significant when p < 0.05.
Stability study
Stability study was conducted at refrigerator (2–8 °C) and accelerated (25 °C/65% RH). Freeze-dried SLNs were transferred to 5 mL glass vials, sealed with aluminum caps for the study. Samples were taken at 1 and 2 months and examined for changes in their particle size, PDI and EE, in addition to its physical appearance and ease of reconstitution.
Results
Cytotoxicity of DTX and CRM in MDA-MB-231 cells
Anticancer potential of DTX and CRM combination was evaluated in MDA-MB-231 cells. presents the cell viability data of DTX and CRM combination after 24 h of incubation. Combination of DTX (60 nM) and CRM (5 and 10 µM) displayed higher cytotoxicity as indicated by decreased cell viability. Several studies have reported the synergistic effect of DTX and CRM in cancer cells (Sarkar & Li, Citation2006; Barthomeuf et al., Citation2010). MDA-MB-231 cells are human breast cancer cells obtained from the metastatic site, and exhibit advanced cancer manifestations (Wong & Chiu, Citation2011). Since DTX is used clinically to treat advanced breast cancer, MDA-MB-231 cells were selected in this study.
Figure 2. Cytotoxicity of DTX and CRM in MDA-MB-231 cells. Results are mean ± SEM (n = 3). p value significant at ***0.01.
Selection of solid lipids
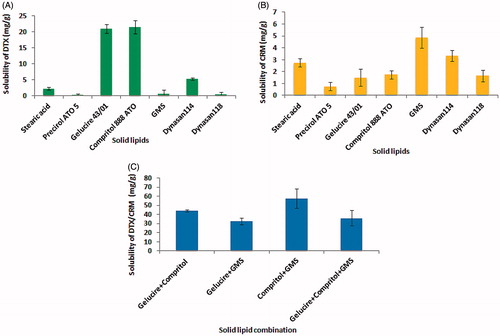
presents the results of the solubility study. DTX exhibited maximum solubility in Compritol, while CRM was most soluble in GMS. The mixture of DTX and CRM at 1:1 (w/w) proportion displayed increased solubility in a combination of Compritol and GMS.
Figure 3. Solubility study of DTX (A), CRM (B) and 1:1 mixture of DTX and CRM (C) in solid lipids. Values are mean ± SD (n = 3).
Optimization of SLN
FC-CCD design was adopted to optimize the SLN formulation. presents the data of the experimental runs. The quadratic model was statistically significant for particle size, zeta potential and PDI (p = 0.0001, 0.0287, 0.0003, respectively). Two factor-interaction model was statistically significant (p = 0.0120) for EE. The following polynomial equations were obtained by software:
(3)
(4)
(5)
(6)
Table 2. Results of FC-CCD experiment trials.
ANOVA was applied to determine significance and magnitude of the effects of critical factors and their interaction. The regression model obtained was applied to generate contour plots. The ANOVA confirmed the adequacy of quadratic and two factor-interaction models (model Prob > F is less than 0.05). It also identifies the significant factors that affect particle size (Y1), zeta potential (Y2), PDI (Y3) and EE (Y4). Particle size was significantly affected by homogenization time (X1), homogenization speed (X2) and stabilizer concentration (X3). PDI was affected by homogenization time, while zeta potential and EE remained unaffected under the evaluated conditions.
At homogenization time of 10 min, speed of 20 000 rpm, stabilizer concentration of 0.5% w/v and lipid concentration of 1% w/v resulted in DC-SLN with particle size of 188.5 nm, PDI of 0.353, zeta potential of −27.5 mV and EE of 72.35%. Reliability of optimization was confirmed by preparing three SLN batches and comparing the observed results with the experimental data. The desired limits for obtaining the optimized formulation were particle size in the range of 180–250 nm, PDI (0.2–0.3), negative zeta potential (22–30 mV), EE (70–80%). There was good correlation between obtained and predicted values.
Shape and surface morphology
TEM image exhibited distinct spherical particles (). Although narrow size distribution was observed with ZetaSizer, some particles lower than 100 nm were observed in TEM. This broader size distribution could be attributed to differential shear force in the different regions of formulation vessels.
Figure 4. TEM image of F-DC-SLN.

Freeze drying
The SLN formulations were freeze-dried by adopting an optimized stepwise freeze drying cycle developed in our lab. Mannose (10% w/v) was added as a cryoprotectant to 5 mL of SLN suspension before freeze drying. The obtained cake was re-dispersed in 5 mL of distilled water and determined the particle size, PDI and EE. Further, we studied the size ratio (Sf/Si, before and after freeze drying) and the data obtained is presented in . Incorporation of 10% mannose as a cryoprotectant resulted in intact fluffy cake, which was easily re-dispersed. No significant changes were observed in PDI and EE in the reconstituted SLN formulation. In addition, Sf/Si ratio (ratio of particle size after and before freeze drying) was 1.38 for DC SLN and 1.4 with F-DC-SLN, ratio near to unity demonstrate the efficiency of freeze-drying. However, in the absence of mannose, the freeze-dried blank SLN particle size increased significantly (p < 0.05) with unacceptable PDI and Sf/Si.
Table 3. Particle size, PDI and EE of SLN before and after freeze drying.
Solid state characterization
Differential scanning calorimetry (DSC)
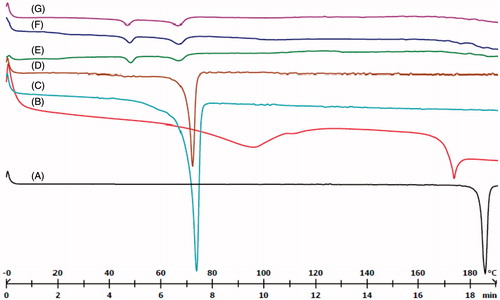
presents an overview of the melting process of DTX, CRM, GMS, Compritol, blank SLN, DC-SLN and F-DC-SLN. Bulk Compritol and GMS exhibited their characteristics melting endotherm near 71 °C, indicating their crystalline nature. The SLN formulations did not exhibit the characteristic melting endotherm of DTX and CRM at 172.3 °C and 185.1 °C, respectively, indicating amorphous nature of both drugs in the SLN matrix.
Figure 5. DSC characterization of SLN. (A) CRM, (B) DTX, (C) GMS, (D) Compritol, (E) blank SLN, (F) DC-SLN, (G) F-DC-SLN.
Powder X-ray diffraction
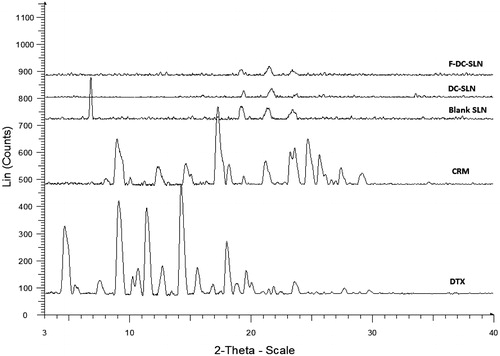
presents the characteristic XRD patterns of DTX, CRM, blank SLN, DC-SLN and F-DC-SLN. DTX and CRM characteristics peaks were absent in the SLN formulations; which further confirms the amorphous nature of both the drugs in the SLN.
Figure 6. PXRD characterization of SLN.
Cross polarized light microscopy
The cross polarized light microscopy was employed to determine presence of unencapsulated drug crystals in the reconstituted freeze-dried DC-SLN and F-DC-SLN formulations (). No birefringence was observed in the dispersions indicating absence of the free drug.
Figure 7. Polarized light microscopy image shows absence of birefringence in SLN dispersion.

Characterization of folic acid on surface of F-DC-SLN
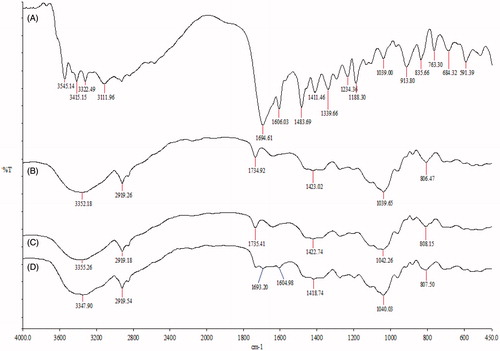
presents the FTIR spectra of folic acid, blank SLN, DC-SLN and F-DC-SLN. Folic acid exhibited characteristics stretching vibrational frequency peak of UC = O at 1694.61 cm−1 and bending mode of UN–H vibration at 1606.03 cm−1. Both these peaks are visible in F-DC-SLN and absent in DC-SLN. However, the visible peaks in F-DC-SLN were not very sharp, which can be attributed to decreased number of folic acid moieties (lesser ligand density) on the surface of F-DC-SLN.
Figure 8. FTIR characterization of folic acid on surface of SLN. (A) Folic acid, (B) blank SLN, (C) DC-SLN, (D) F-DC-SLN.
In vitro release study
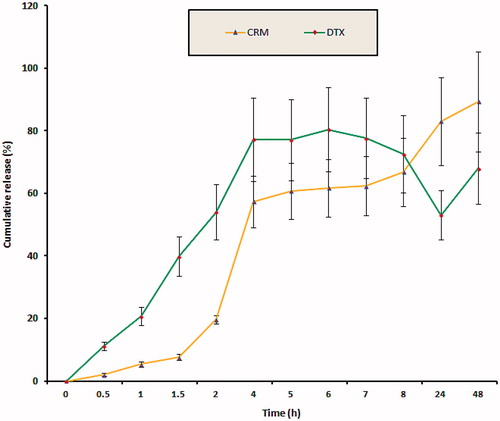
presents the drug release profile of optimized F-DC-SLN. The formulation exhibited a slow-release of DTX of 67.94% and CRM of 89.27% at the end of 48 h, while almost 80% of DTX was released in 20 h from Taxotere (in a release medium similar to SLN formulation except 3% w/v Tween 80 instead of 50% v/v ethanol). Weibull model best fitted the release kinetic data indicating SLN as a diffusion matrix. Model correlation coefficient of 0.9188 and 0.9975 was observed for DTX and CRM, respectively.
Figure 9. In vitro release of F-DC-SLN in PBS (0.01 M, pH 7.4, 50% v/v ethanol). Values are mean ± SD (n = 3).
In vitro cell uptake
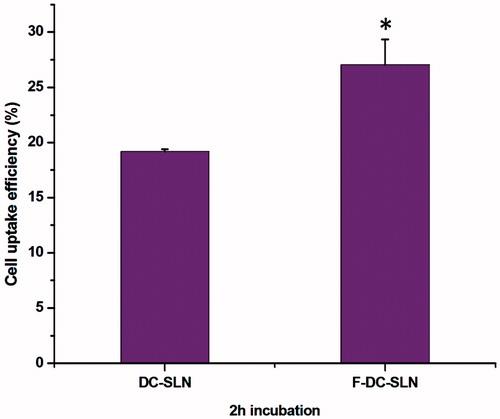
The cell uptake study was conducted quantitatively and qualitatively to evaluate the penetration potential of DC-SLN and F-DC-SLN. The quantitative uptake of SLN in MCF-7 cells was studied by measuring the fluorescence intensity after incubation for 2 h. Folic acid conjugation significantly (by 36.84%) enhanced the uptake (27.02 ± 3.98% versus 19.17 ± 0.34%) in comparison to DC-SLN (, two-tailed Student’s t test, p < 0.05).
Figure 10. Cell uptake efficiency of SLN formulations. Values are mean ± SEM (n = 3). p value significant at *0.05.
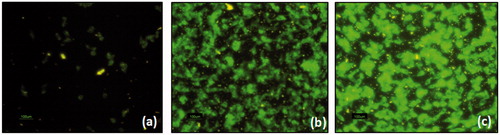
The concentrations of the SLN formulations remained constant as in the quantitative study (0.250 µg/mL). The internalization of the SLNs was visualized by the fluorescence microscope after incubation for 2 h. presents the comparative cellular uptake of free coumarin, DC-SLN and F-DC-SLN. Folic acid conjugation exhibited a considerable increase in fluorescence indicating enhanced cell uptake from F-DC-SLN.
Figure 11. Fluoroscence microscope images shows internalization of free coumarin (a), DC-SLN (b) and F-DC-SLN (c) in MCF-7 cells.
In vitro cell cytotoxicity
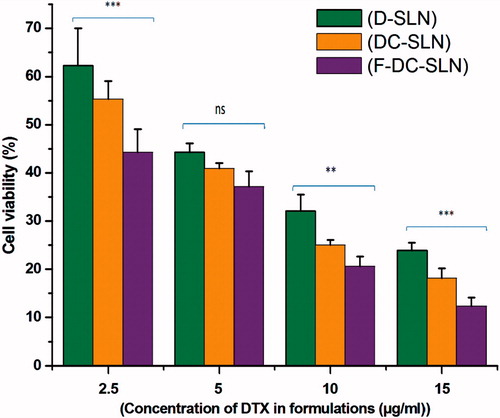
The cytotoxicity of SLN formulations in cancer cells was studied in MCF-7 cells. illustrates the cell viability data of MCF-7 cells treated with D-SLN, DC-SLN, F-DC-SLN each containing DTX at 15, 10, 5, and 2.5 μg/mL concentrations and incubated for 48 h. As expected, cell viability decreased with an increase in DTX concentration in all the SLN formulations. F-DC-SLN decreased the viability to the maximum extent followed by DC-SLN and D-SLN. Thus, functionalization of DC-SLN increased the cell uptake, which in turn resulted in increased cytotoxicity.
Figure 12. In vitro cytotoxicity of SLN in MCF-7 cells. Values are mean ± SEM (n = 3). p value significant at **0.01; ***0.001.
In vitro hemolysis
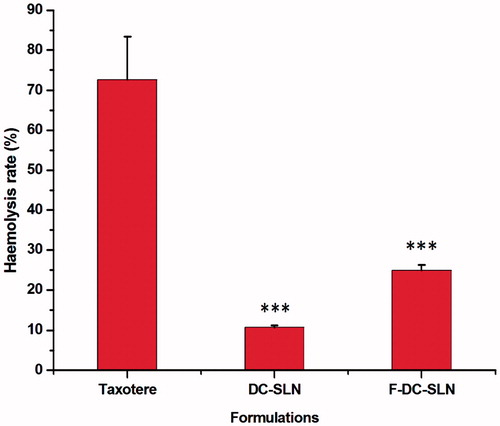
It is known that surfactants have an ability to interact with the lipid bilayer of RBC (Italia et al., Citation2009). Since Poloxamer 188 was incorporated in the SLN formulations, we investigated their toxicity on RBC. From , it is evident that the hemolytic activity of DC-SLN and F-DC-SLN is significantly (p < 0.001) less than Taxotere at 50 µg/mL DTX concentration.
Figure 13. In vitro hemolysis test of SLN formulations. Values are mean ± SD (n = 3). p value significant at ***0.001.
Pharmacokinetic study
presents the plasma concentration–time profiles of DTX following single dose i.v. administration of Taxotere, DC-SLN and F-DC-SLN. Taxotere was rapidly cleared from the circulation within 4 h, whereas F-DC-SLN exhibited a prolonged plasma circulation time. presents the corresponding pharmacokinetic parameters. F-DC-SLN exhibited maximum AUC followed by DC-SLN and Taxotere. F-DC-SLN exhibited a 15-fold increase (594.21 ± 64.34 versus 39.05 ± 7.41 μg/mL h) in comparison to Taxotere, while DC-SLN exhibited 3.5-fold increase (138.26 ± 40.19 versus 39.05 ± 7.41 μg/mL h) in AUC. Similarly, F-DC-SLN exhibited 4-fold increase in MRT (31.14 ± 19.94 versus 7.24 ± 4.51 h) in comparison to Taxotere, while DC-SLN displayed 2-fold increase (13.19 ± 6.80 versus 7.24 ± 4.51 h).
Figure 14. Plasma concentrations versus time profile of DTX after i.v. administration of DTX formulations. Values are mean ± SD (n = 5).
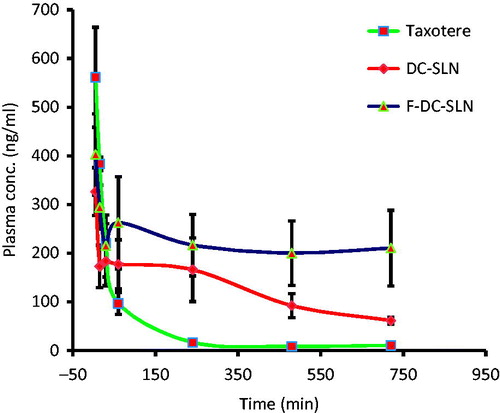
Table 4. Pharmacokinetic parameters of DTX after i.v. administration of Taxotere, DC-SLN and F-DC-SLN in rats.
Biodistribution study
presents the biodistribution data of Taxotere, DC-SLN and F-DC-SLN at 7 and 120 min of i.v. administration. At 7 min, Taxotere accumulated in the heart, liver, lung, kidney, with a negligible amount in the brain and spleen. But at the same time interval, lesser distribution in the heart, liver, lung, kidney and spleen was observed from F-DC-SLN compared to Taxotere. Further, higher accumulation of DC-SLN was observed in the liver, lung, spleen and brain compared to Taxotere and F-DC-SLN; whereas lesser distribution was observed in the heart and kidney compared to Taxotere.
Figure 15. Biodistribution of DTX after i.v. administration of DTX formulations. Stastitical diffrence between Taxotere 7 min and F-DC-SLN 7 min is shown that values are mean ± SD (n = 6). p value significant at *0.05, **0.01 and ***0.001.
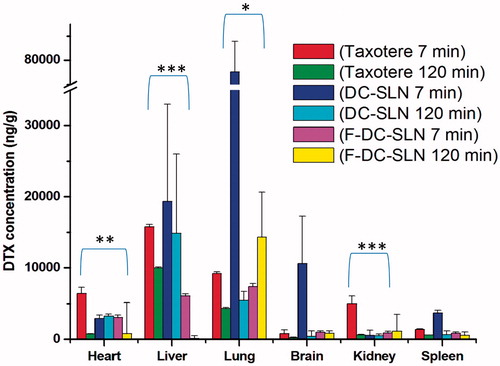
At 120 min, Taxotere exhibited decreased concentration in all the organs evaluated in study. Similarly, decreased concentration of DC-SLN in liver, lung, brain and spleen was observed at 120 min; but remained constant in the heart and kidney. F-DC-SLN concentration decreased in the heart and liver, whereas increased in lung after 120 min. It remained constant in the brain, kidney and spleen. To conclude, higher accumulation of DC-SLN was observed in the liver, lung, spleen and brains, which are organs of reticuloendothelial system (RES) (Li et al., Citation2009). F-DC-SLN exhibited reduced distribution in all the organs, except in the lungs, in comparison with Taxotere and DC-SLN.
Stability study
DC-SLN and F-DC-SLN formulations were found to be stable at refrigerated condition for 2 months. Both the formulations showed sign of instability at accelerated condition (25 °C/65% RH).
Discussion
Numerous SLN-based delivery systems have been attempted for the delivery of single anticancer to treat cancer. However, the complex molecular mechanisms in cancer render single drug therapy ineffective. Therefore, co-delivery of DTX and CRM in SLN was adopted to achieve improve pharmacokinetic and enhanced anticancer efficacy through their synergistic effect. DTX is a chemotherapeutic drug, which stabilizes microtubules and retains cell cycle in the late G2/M phase, thereby mediating cancer cell death (Lopes et al., Citation1993). CRM can suppress many survival signal pathways and promotes apoptosis in cancer cells (Reuter et al., Citation2008). In addition, several reports demonstrated the synergistic anticancer activity of DTX and CRM, which was further confirmed by preliminary cytotoxicity study in MDA-MB-231 cells. The aim of the present work was to enhance efficacy of DTX by improving its pharmacokinetic and biodistribution profiles. The SLN-based drug delivery is a promising approach for co-delivery of anticancer agents. It also offers advantage of ligand binding due to presence of surface groups of the solid lipids. To achieve the aforementioned objectives, folic acid functionalized long-circulating co-encapsulated DTX and CRM solid lipid nanoparticles were developed and evaluated biologically.
Based on the solubility, 1:1 mixture of GMS and Compritol was selected as the lipids to be incorporated in SLN formulation. Solubility of drug in the lipid matrix is a key factor in achieving high EE of SLN. As the melted lipids solidify, drug molecules get entrapped in their crystal lattice. Very ordered lattice is formed from single type of lipid with few void spaces for entrapment of the drug molecules resulting in lower EE (Vivek et al., Citation2007). This challenge was of poor EE, which was addressed by selecting a mixture of Compritol and GMS as the SLN matrix. SLN has the ability to achieve increased drug loading in comparison to liposomes along with its biocompatibility. Hot melt-emulsification method was adopted because it is simple, economical and avoids the use of organic solvents.
FC-CCD was employed to optimize the SLN process and formulation based on particle size, zeta potential, PDI and EE of the SLN. Particle size was significantly affected by homogenization time, speed and stabilizer concentration; an increase in homogenization time and speed resulted in decreased particle size. Further, increased homogenization speed in combination with a low level of stabilizer concentration resulted in smaller particle size. PDI was significantly affected by homogenization conditions. At higher level of homogenization time and speed, PDI increased, while it decreased at lower lipid amount and stabilizer concentration. Zeta potential decreased with increased stabilizer concentration. This phenomenon could be attributed to masking of lipid charge by Poloxamer 188, the nonionic surfactant incorporated in the SLN. Optimization resulted in DC-SLN with particle size of 188.5 nm. This size would be advantageous since particle size below 250 nm evade the macrophages, circulate for longer time in plasma exhibiting potential of passive targeting to cancer tissue (Beija et al., Citation2012).
Mannose (10% w/v) proved to be suitable cryoprotectant for SLN stabilization during freeze drying. No significant (p > 0.05) difference in EE of DC-SLN and F-DC-SLN before and after freeze drying was observed (). However, particle size of the blank SLN changed significantly (p < 0.05) in the absence of mannose. Cryoprotectants form a glassy amorphous matrix around the NPs probably by hydrogen bonding with the polar groups of NPs, thereby preventing aggregation during freeze drying (Jain et al., Citation2011).
SLN formulations were characterized by DSC and powder X-ray diffraction (PXRD). Both DC-SLN and F-DC-SLN did not exhibit the melting endotherm of DTX and CRM, confirming the amorphous nature of both the drugs in the SLN formulation. However, GMS and Compritol exhibited melting endotherms at around 46 °C and 68 °C, respectively in the SLN. It has been reported that when the bulk lipid matrix turned into SLN, the melting point depressed regardless of the presence or absence of drug. The depressed melting point could further be attributed to the small size of SLN as explained by Thomson’s equation (Yamada et al., Citation2003). presents the XRD data of SLN formulations and pure drugs. No characteristic peaks of DTX and CRM were observed in DC-SLN and F-DC-SLN, which further supports the DSC results. These findings were further confirmed by cross polarized light microscopy, which did not exhibit birefringence of the free drug crystals in the reconstituted SLN suspensions (). The surface chemistry of F-DC-SLN studied by FTIR spectroscopy confirmed the presence of folic acid on the surface of SLN (). F-DC-SLN exhibited a positive zeta potential (14.53 ± 3.6 mV) in contrast to DC-SLN, confirming its functionalization with folic acid.
F-DC-SLN exhibited sustained DTX release for 48 h by diffusion as revealed from analysis of the release data by Weibull diffusion model. In general, drug release from SLN has been reported to follow Weibull and/or Higuchi’s mechanisms rather than by first-order release. Although drug release from SLN is by diffusion, several mechanisms could be involved including surface erosion, disintegration, diffusion and desorption (Venkateswarlu & Manjunath, Citation2004). Further, higher CRM release in comparison to DTX could be due to differences in the solubility of the two molecules in SLN matrix.
Active tumor targeting of SLN has been proposed by functionalization with folic acid. Binding of folic acid to the FR on the tumor cells results in internalization of the receptor via receptor-mediated endocytosis (RME) (Leamon & Low, Citation1991). Folic acid is an attractive target ligand owing to its high binding affinity (KD ∼10−10 M) to the FRs, which are overexpressed on human adenocarcinoma (MCF-7) cells. F-DC-SLN exhibited enhanced fluorescence in comparison to DC-SLN (), which can be attributed to the presence of the folic acid molecules on its surface internalized by probably by RME. This internalization has been reported to be not hampered even when folic acid is conjugated to a variety of nanocarriers (Liu et al., Citation2010). In this study, quantitative cellular uptake of F-DC-SLN was significantly higher (36.84%) in comparison to DC-SLN. The uptake of the DC-SLN could be explained by probable interaction between lipids and the cell membrane.
Cytotoxicity of the SLN was evaluated in MCF-7 cells by an indirect method. In the study, the SLN uptake by MCF-7 cells was studied. An increase in SLN uptake resulted in reduced cell viability indicating augmented cell toxicity. The order of cytotoxicity was in the order of F-DC-SLN > DC-SLN > D-SLN. Cell viability decreased with increased DTX concentration in all the SLN formulations. Thus, it can be concluded that the presence of folic acid on the F-DC-SLN resulted in enhanced uptake leading to decreased cell viability and increased cellular toxicity in comparison to DC-SLN and D-SLN. F-DC-SLN exhibited significantly (p < 0.05) lesser cell viability in comparison to D-SLN (). Furthermore, the SLN formulations has the potential to spare the normal tissue from the toxic effect of encapsulated chemotherapeutic drugs when administered intravenously and thus increasing the maximum tolerance dose (MTD). MTX (MTD 2700 ng/mL) dose of 15 μg/mL in the SLN formulation has the capacity to spare normal cells while causing toxicity to cancer cells by selective uptake. Thus, F-DC-SLN has the potential to enhance the therapeutic effect of DTX.
The toxicity of SLN formulations was compared with Taxotere by a simple in vitro hemolysis test. SLN formulation exhibited significantly lesser toxicity to RBC (). Polysorbate 80, added in Taxotere formulation as a solubilizer in high concentration, results in considerable toxicity to RBC (Ungaro et al., Citation2012). However, slight increase in hemolysis of F-DC-SLN in comparison to DC-SLN can be attributed to the presence of PEG–SA, which may exhibit amphiphilic properties.
In pharmacokinetic study, F-DC-SLN exhibited higher AUC in comparison with DC-SLN and Taxotere. This phenomenon could be due the slow release of DTX from the SLN matrix and evasion of SLN by RES uptake because of the presence of PEG on its surface (Li et al., Citation2009). This confirms the role PEGylation in improving the plasma half-life of the SLN. In one study in our lab, we observed enhanced anti-Xa activity by PEGylated enoxaparin administered subcutaneously indicating the potential of PEGylation in increasing plasma circulation time (Choubey et al., Citation2014). Increase in AUC of DC-SLN can be attributed to the slow release of DTX from the lipid matrix, resulting in reduced elimination rate and increase plasma concentration. Thus, SLN matrix presents an excellent diffusion barrier for sustained release of drugs and has the added advantage of protecting the loaded drug.
DTX concentration in different tissues was determined to assess its distribution upon i.v. administration of Taxotere, DC-SLN and F-DC-SLN in rats. Taxotere exhibited high concentration of DTX in heart and kidney, which could be responsible for its toxicity along with hypersensitivity reaction induced by Polysorbate 80. DC-SLN exhibited increased accumulation in liver, lung, spleen and brain at 7 min interval. This could be due to increased uptake by the RES resulting in increased DTX concentration liver, lung and spleen. The higher concentration of DTX found in liver, spleen, lung and brain in DC-SLN treated rats could be related to the slow drug release from SLN to the targeted organs. The higher accumulation of DC-SLN in lung could be attributed to the presence of mononuclear phagocytosis (MPS) in these organs. Further, the increased uptake of DC-SLN in the brain may be due to the interaction of between lipids in the SLN and membrane of the endothelial cells. The clearance and tissue distribution of particulate drug carriers administered intravenously have been reported to be greatly influenced by their size, surface features and opsonization. Opsonins are adsorbed on the surface of NPs and promote particle recognition by the RES organs (Moghimi et al., Citation2001). Various attempts have been made to achieve long circulation times by avoiding macrophage uptake, such as stealth SLN, for instance. F-DC-SLN exhibited considerably decreased concentration of DTX in heart and kidney, although a significantly higher concentration was observed in the lung. This may be due to the presence of a long hydrophilic chain of PEG in the F-DC-SLN, which results in increased circulation time in the plasma and probably leading to increased accumulation in the lungs where there is a rich blood flow because of high capillary permeability (Li et al., Citation2009). The uptake of F-DC-SLN by the brain had not improved probably because of increased hydrophilicity after functionalization.
Conclusions
In summary, folic acid functionalized PEGylated SLN co-encapsulating DTX and CRM were successfully developed. The SLN exhibited increased cell uptake, and cytotoxicity facilitated probably by the incorporation of folic acid. F-DC-SLN exhibited enhancement in AUC, MRT and t1/2, which can be attributed to PEGylation of the SLN formulation. Co-encapsulation of DTX with CRM resulted in enhanced DTX efficacy and synergistic activity of both the molecules. Thus, this study provides a platform to improve the efficiency of drug delivery to achieve optimization of anticancer treatment.
We thank Mr. Pradeep Jadhavar for his valuable inputs in reaction chemistry. Further, we would like to thank Mr. Vinod Kumar (HRTEM facility, NIPER), Mr. Rajdeo Kumar, (CIL, NIPER), and Mr. Ashok Kumar Datusalia for their help and support.
Declaration of interest
The authors report no declarations of interest. Authors wish to thank Department of Biotechnology (DBT), New Delhi, India for providing the fellowship (DBT-JRF/2011-12/209) to Dr. Harish.
References
- Agrawal R, Sandhu SK, Sharma I, Kaur IP. (2014). Development and evaluation of curcumin-loaded elastic vesicles as an effective topical anti-inflammatory formulation. AAPS PharmSciTech 16:1–11
- Barthomeuf, C, Chollet, P, Bayet-Robert, M. (2010). Curcuminoids in combination docetaxel for the treatment of cancer and tumour metastasis. Google Patents No. US 20120022148 A1, 1-7
- Beija M, Salvayre R, Lauth-de Viguerie N, Marty JD. (2012). Colloidal systems for drug delivery: from design to therapy. Trends Biotechnol 30:485–96
- Chauhan MS, Kumar A, Pathak K. (2012). Osmotically regulated floating asymmetric membrane capsule for controlled site-specific delivery of ranitidine hydrochloride: optimization by central composite design. AAPS PharmSciTech 13:1492–501
- Cho H, Lai TC, Tomoda K, Kwon GS. (2014). Polymeric micelles for multi-drug delivery in cancer. AAPS PharmSciTech 16:111
- Choubey AK, Dora CP, Bhatt TD, et al. (2014). Development and evaluation of PEGylated Enoxaparin: a novel approach for enhanced anti-Xa activity. Bioorg Chem 54:1–6
- Ferlay J, Autier P, Boniol M, et al. (2007). Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol 18:581–92
- Finn RS, Dering J, Ginther C, et al. (2007). Dasatinib, an orally active small molecule inhibitor of both the src and abl kinases, selectively inhibits growth of basal-type/“triple-negative” breast cancer cell lines growing in vitro. Breast Cancer Res Treat 105:319–26
- Hu Q, Rijcken CJ, Bansal R, et al. (2015). Complete regression of breast tumour with a single dose of docetaxel-entrapped core-cross-linked polymeric micelles. Biomaterials 53:370–8
- Italia J, Yahya M, Singh D, Kumar MR. (2009). Biodegradable nanoparticles improve oral bioavailability of amphotericin B and show reduced nephrotoxicity compared to intravenous Fungizone®. Pharm Res 26:1324–31
- Jain AK, Swarnakar NK, Godugu C, et al. (2011). The effect of the oral administration of polymeric nanoparticles on the efficacy and toxicity of tamoxifen. Biomaterials 32:503–15
- Jinturkar KA, Anish C, Kumar MK, et al. (2012). Liposomal formulations of Etoposide and Docetaxel for p53 mediated enhanced cytotoxicity in lung cancer cell lines. Biomaterials 33:2492–507
- Kawalec P, Łopuch S, Mikrut A. (2014). Effectiveness of targeted therapy in patients with previously untreated metastatic breast cancer: a systematic review and meta-analysis. Clin Breast Cancer 15:90–100
- Kunnumakkara AB, Anand P, Aggarwal BB. (2008). Curcumin inhibits proliferation, invasion, angiogenesis and metastasis of different cancers through interaction with multiple cell signaling proteins. Cancer Lett 269:199–225
- Kutty RV, Fen SS. (2013). Cetuximab conjugated vitamin E TPGS micelles for targeted delivery of docetaxel for treatment of triple negative breast cancers. Biomaterials 34:10160–71
- Leamon CP, Low PS. (1991). Delivery of macromolecules into living cells: a method that exploits folate receptor endocytosis. Proc Natl Acad Sci USA 88:5572–6
- Li W Jr, Szoka FC. (2007). Lipid-based nanoparticles for nucleic acid delivery. Pharm Res 24:438–49
- Li X, Wang D, Zhang J, Pan W. (2009). Preparation and pharmacokinetics of docetaxel based on nanostructured lipid carriers. J Pharm Pharmacol 61:1485–92
- Liu Y, Li K, Pan J, et al. (2010). Folic acid conjugated nanoparticles of mixed lipid monolayer shell and biodegradable polymer core for targeted delivery of Docetaxel. Biomaterials 31:330–8
- Lopes NM, Adams EG, Pitts TW, Bhuyan BK. (1993). Cell kill kinetics and cell cycle effects of taxol on human and hamster ovarian cell lines. Cancer Chemother Pharm 32:235–42
- Moghimi SM, Hunter AC, Murray JC. (2001). Long circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev 53:283–318
- Mulik RS, Mönkkönen J, Juvonen RO, et al. (2010). Transferrin mediated solid lipid nanoparticles containing curcumin: enhanced in vitro anticancer activity by induction of apoptosis. Int J Pharm 398:190–203
- Olivier JC, Huertas R, Lee HJ, et al. (2002). Synthesis of pegylated immunonanoparticles. Pharm Res 19:1137–43
- Payam Z, Raquel D, Loan H, et al. (2011). Combination drug delivery strategy for the treatment of multidrug resistant ovarian cancer. Mol Pharm 8:260–9
- Reuter S, Eifes S, Dicato M, et al. (2008). Modulation of anti-apoptotic and survival pathways by curcumin as a strategy to induce apoptosis in cancer cells. Biochem Pharmacol 76:1340–51
- Sarkar FH, Li Y. (2006). Using chemopreventive agents to enhance the efficacy of cancer therapy. Cancer Res 66:3347–50
- Shegokar R, Singh K, Müller R. (2011). Production & stability of stavudine solid lipid nanoparticles – from lab to industrial scale. Int J Pharm 416:461–70
- Tan SW, Billa N. (2014). Lipid effects on expulsion rate of amphotericin B from solid lipid nanoparticles. AAPS PharmSciTech 15:287–95
- Ullah MF. (2008). Cancer multidrug resistance (MDR): a major impediment to effective chemotherapy. Asian Pac J Cancer Prev 9:1–6
- Ungaro F, Conte C, Ostacolo L, et al. (2012). Core-shell biodegradable nanoassemblies for the passive targeting of docetaxel: features, antiproliferative activity and in vivo toxicity. Nanomedicine 8:637–46
- Venkateswarlu V, Manjunath K. (2004). Preparation, characterization and in vitro release kinetics of clozapine solid lipid nanoparticles. J Control Release 95:627–38
- Vivek K, Reddy H, Murthy RS. (2007). Investigations of the effect of the lipid matrix on drug entrapment, in vitro release, and physical stability of olanzapine-loaded solid lipid nanoparticles. AAPS PharmSciTech 8:16–24
- Wong MY, Chiu GN. (2011). Liposome formulation of co-encapsulated vincristine and quercetin enhanced antitumor activity in a trastuzumab-insensitive breast tumor xenograft model. Nanomedicine 7:834–40
- Yamada K, Hikosaka M, Toda A, et al. (2003). Equilibrium melting temperature of isotactic polypropylene with high tacticity: 1. Determination by differential scanning calorimetry. Macromolecules 36:4790–801
- Yuan H, Miao J, Du YZ, et al. (2008). Cellular uptake of solid lipid nanoparticles and cytotoxicity of encapsulated paclitaxel in A549 cancer cells. Int J Pharm 348:137–45
- Yueying H, Yan Z, Chunhua G, et al. (2010). Micellar carrier based on methoxy poly(ethylene glycol)-block-poly(ɛ-caprolactone) block copolymers bearing ketone groups on the polyester block for doxorubicin delivery. J Mater Sci Mater Med 21:567–74