Abstract
PEGylation is one of the most promising and extensively studied strategies for improving the pharmacological properties of proteins as well as their physical and thermal stability. Purified lysozyme obtained from hen egg white by batch mode was modified by PEGylation with methoxypolyethyleneglycol succinimidyl succinato (mPEG-SS, MW 5000). The conjugates produced retained full enzyme activity with the substrate glycol chitosan, independent of degree of enzyme modification, although lysozyme activity with the substrate Micrococcus lysodeikticus was altered according to the degree of modification. The conjugate with a low degree of modification by mPEG-SS retained 67% of its enzyme activity with the M. lysodeikticus substrate. The mPEG-SS was also shown to be a highly reactive polymer. The effects of pH and temperature on PEGylated lysozymes indicated that the conjugate was active over a wide pH range and was stable up to 50°C. This conjugate also showed resistance to proteolytic degradation, remained stable in human serum, and displayed greater antimicrobial activity than native lysozyme against Gram-negative bacteria.
Introduction
Lysozyme (mucopeptide N-acetylmuramoylhydrolase, EC 3.2.1.17) is widely found in nature and is used in a growing number of applications. Countries such as France, Germany, Japan and United Kingdom have acknowledged its safety and have approved the use of lysozyme (LZ) in several foods and in pharmacological and therapeutic applications (CitationMine et al., 2004). LZ belongs to a class of enzymes that lyse the cell wall of various Gram-positive bacteria by splitting β-(1-4) linkages between N-acetylmuramic acid and N-acetylglucosamine of peptidoglycan. However, LZ is largely ineffective against Gram-negative bacteria. The increasing development of bacterial resistance to traditional antibiotics has reached alarming levels, necessitating the development of new antimicrobial agents (CitationIbrahim et al., 2002). Microorganisms develop resistance to antimicrobial agents mainly by modifying cell targets and thus rendering themselves insensitive to antibiotics, and/or by inactivating those agents with microbial enzymes. Self-medication and improper use of antibiotics in developing countries can also lead to the development of microbial resistance (CitationRahman et al., 2008).
Various strategies have been tested for making lysozyme active against Gram-negative bacteria, including binding to dextran (CitationNakamura et al., 1991), lipophilization of LZ with fatty acid chains of different lengths (CitationLiu et al., 2000; CitationIbrahim et al., 1993, Citation1991), and adding hydrophobic peptides to the C-terminal of LZ through genetic modification (CitationIbrahim et al., 2001; CitationXu et al., 2004; CitationArima et al., 1997). There is some evidence that the catalytic properties of lysozyme can be enhanced through protein modification with methoxypolyethylene glycol (mPEG) (CitationNodake et al., 2002), and that these alterations promote the antimicrobial effects of LZ against Gram-negative bacteria while retaining full or partial activity against Gram-positive bacteria.
Lysozyme has become increasingly important in industrial processing due to its natural antibacterial properties, and simple and efficient methods for lysozyme production are greatly needed. While LZ can be obtained from various sources, hen egg white is preferred. Various steps are required to obtain high LZ purity, and several lysozyme-isolating methods have been investigated (CitationGhosh & Cui, 2000). Hen egg white is a commercially available mixture of proteins that can yield purified lysozyme using a combination of conventional processes. A rapid and simple method for batch purification of LZ was devised in the present study that overcomes some of the problems associated with packed-bed chromatography (such as consolidation of the packing material, formation of channels, and column blocking by particles in the feed solution).
Recent publications (CitationPasut et al., 2008; CitationZappe et al., 2008; CitationZalipsky et al., 2007) have shown PEGylation to be a procedure with significant potential for enhancing the therapeutic and biotechnological use of proteins by attaching an inert, non-toxic, and water-soluble PEG to a strategic protein (CitationTreetharnmathurot et al., 2008; CitationHarris & Veronese, 2003). PEGylated forms of adenosine deaminase and asparaginase (CitationVeronese & Pasut, 2005; CitationZhang et al., 2004; CitationSoares et al., 2002) are now approved for human use by the US Food and Drug Administration, and other PEG-modified proteins are being developed for possible therapeutic use, including PEG epidermal growth factor and PEGylated single-chain Fv proteins as well as PEGylated drugs for the treatment of hepatitis C, acromegaly, rheumatoid arthritis and various cancers (CitationVeronese & Pasut, 2005). When PEG is properly linked to a polypeptide it modifies many of its features, while maintaining its main biological function (such as enzymatic activity or receptor recognition). PEG conjugation results in a modified protein with enhanced solubility, temperature stability, serum half-life, and resistance to enzymatic degradation, as well as decreased renal clearance and lower immunogenicity, thereby enhancing its biological effectiveness (CitationRoberts et al., 2002; CitationHarris et al., 2001; CitationVeronese, 2001). The present work examined the modification of lysozyme by PEGylation with methoxypolyethylene glycol succinimidyl succinate (mPEG-SS), MW 5000, and determined some of the properties of the resulting conjugate, such as its degree of modification and reaction kinetics, as well as its stability and potential use as a new antimicrobial agent.
Materials and methods
Materials
Fresh hen eggs were purchased from a local market, while Amberlite® IRC-50 and IRP-88 resins, methoxypolyethylene glycol-succinimidyl succinate 5000 Da (mPEG-SS 5000) and glycol chitosan were purchased from Sigma (St. Louis, MO), Gelcolde® Blue Stain P Gelcolde® Blue Stain Reagent from Pierce (Rockford, USA) and Sephadex G-50 from Amersham Pharmacia Biotech AB (Uppsala, Sweden). Micrococcus lysodeikticus was obtained from ICN Pharmaceuticals (Costa Mesa, CA-USA), strain E. coli ATCC 29255, Proteomix® (trypsin and chymotrypsin) from Biobrás (Montes Claros, MG, Brazil), and Protex 6L® (bacterial protease) and fungal protease from Genencor International, INC (New York-USA). Micrococcus lysodeikticus was obtained from ICN Pharmaceuticals, Protemix® (trypsin and chymotrypsin) from Biobrás, and Protex 6L® (bacterial protease) and fungal protease from Genencor International. All other reagents used were of analytical grade.
Enzyme assays
Lytic activity with Micrococcus lysodeikticus
The enzymatic activities of lysozyme and its conjugate were determined turbidimetrically by measuring the decrease in absorbance at 450 nm of a cell suspension (0.5 mg mL−1) of M. lysodeikticus in 100 mM phosphate buffer at pH 7 as previously reported by CitationShugar (1952). A decrease of 0.001 in absorbance was defined as 1 unit of lysozyme activity.
Enzyme activity with glycol chitosan
The enzyme activities of lysozyme and modified lysozymes were also assayed using the glycol chitosan substrate method of CitationImoto and Yagishita (1971). A mixture of 1 mL of 0.05% (w/v) glycol chitosan in 100 mM acetate buffer, pH 5.5, plus 100 µL of lysozyme or a modified lysozyme solution (2 mg mL−1) was incubated at 40°C for 30 min. After reacting, 2 mL of the color-developing reagent (0.5 g of potassium ferricyanide in 1 L of 0.5 M sodium carbonate) was added and the mixture immediately heated to boiling and allowed to boil for 15 min. The control solution contained no enzyme. Absorbance was read at 420 nm after cooling.
Protein content determination
Protein concentration was determined using the Bradford method (CitationBradford, 1976) with Bio-Rad protein assay dye reagent at 595 nm, using a standard curve of lysozyme.
SDS-polyacrylamide gel electrophoresis (SDS-PAGE)
Electrophoresis under denaturing conditions was performed using polyacrylamide gel concentrations of 10% and 12% (CitationLaemmli, 1970); staining was performed with Gelcolde® blue stain reagent.
Batch mode lysozyme purification on a cation exchanger
IRC-50 Resin: 45 mL of resin was placed in a 250 mL graduated beaker and activated with 60 mL of 0.5 N HCl for 30 min while stirring. After successive washes with distilled water until attaining pH neutrality, 100 mL of 0.5 M Tris-HCl buffer, pH 9, was added and stirred for 30 min at 8°C. An aliquot of 90 mL of egg white that had been previously passed through a sieve was added to the resin, at a resin to egg white ratio of 1:2 (v/v), and stirred for 1 h at 8°C. The egg white supernatant was then removed and the resin washed with 50 mM Tris-HCl buffer, pH 9. Elution was carried out by adding 90 mL of 1 M ammonium sulfate in 50 mM Tris-HCl buffer, pH 7, while stirring for 30 min at 8°C. The supernatant was then recovered and submitted to ultrafiltration through a 50,000 nominal molecular weight limit (NMWL) membrane (Ultrafree® MC centrifugal, Amicon Bioseparations Millipore) at 4000 g and 5°C for 20 min, yielding the permeate. IRP-88 Resin: 40 mL of the resin was placed in a 250 mL graduated beaker and activated with 80 mL of 0.5 N KCl with stirring for 30 min. Afterward, 100 mL of 0.5 M Tris-HCl buffer, pH 9, was then added and stirred for 30 min at 8°C. After successive washes of resin with 50 mM Tris-HCl buffer, pH 9, 80 mL of egg white that had been previously passed through a sieve, was added to the resin, at a resin to egg white ratio of 1:2 (v/v), and stirred for 1 h at 8°C. The egg white supernatant was then removed and the resin washed with 50 mM Tris-HCl buffer, pH 9. Elution was carried out by adding 90 mL of 1 M ammonium sulfate in 50 mM Tris-HCl buffer, pH 7, and stirring for 30 min at 8°C. The supernatant was then removed and submitted to ultrafiltration through a 50,000 NMWL membrane (Ultrafree®) at 4000 g and 5°C for 20 min, yielding the permeate.
Reaction kinetics of PEGylation of lysozyme
The reaction kinetics of lysozyme PEGylation with mPEG-SS (5000 MW) was followed for differing time periods. An aliquot of 500 µL of the LZ solution at a concentration of 4 mg mL−1 was added to 200 µL of the mPEG-SS 5000 MW solution at 10 mg mL−1 in Hepes buffer, pH 7.5, in a reaction tube. The reaction was allowed to proceed at 30°C for 2 h. Aliquots (20 µg) were withdrawn after 1, 3, 5, 7, 10, 20, 60 and 120 min and added to 30 µL of sample buffer. The aliquots were separated on 10% SDS-PAGE gels and immediately boiled to halt the reaction.
Chemical modification of lysozyme with mPEG-SS 5000
Lysozyme at 20 mg mL−1 in 100 mM sodium phosphate buffer (pH 7.3) was added to a stoichiometric quantity of mPEG-SS 5000 at different molar ratios of mPEG-SS/NH2 (), and incubated at 30°C for 2 h. The lytic activities of each sample (at different molar ratios of mPEG-SS 5000/NH2) with the substrates M. lysodeikticus and glycol chitosan were measured. The assays were conducted in three independent experiments.
Table 1. Different molar ratio mPEG-SS 5000/NH2.
Preparation and purification of modified lysozyme
To determine the enzymatic activity and stability of the modified lysozyme conjugate; was prepared with low degrees of modification were prepared and purified. Lysozyme was modified with mPEG-SS 5000 at a molar ratio of 0.11. Then 1 mL of lysozyme solution (20 mg mL−1) in 100 mM sodium phosphate buffer (pH 7.3) was added to 5 mg of mPEG-SS 5000. The reaction was carried out at 30°C for 2 h and incubated at 8°C for 16 h. Excess free mPEG-SS and native lysozyme were removed by size exclusion chromatography using Sephadex G-50. The column was pre-equilibrated with 100 mM phosphate buffer, pH 7, and eluted with the same buffer. Each eluted 1 mL fraction was analyzed by determining its enzyme activity and by 12% SDS-PAGE electrophoresis. Fractions that did not contain the conjugate were discarded. The enzymatic activity and stability of the conjugate was evaluated as follows below.
Evaluation of the enzymatic activity and the stability of native and modified lysozyme
Enzymatic activity was determined in buffers varying in pH from 4 to 12. Suspensions of Micrococcus lysodeikticus (0.5 mg mL−1) in different buffers were prepared (pH 4-5, 50 mM acetate; pH 6-8, 50 mM Hepes; and pH 9-12, 50 mM Tris-HCl) and enzymatic activity at 25°C was immediately measured by measuring absorbance at 450 nm. The pH at which the samples showed the highest activity was considered to represent 100% activity. Stability was determined after incubation for 1 h at 50°C in the same buffers (pH 4-5, 50 mM acetate; pH 6-8, 50 mM Hepes; and pH 9-12, 50 mM Tris-HCl). At the end of this time, enzymatic activity was determined with the substrate M. lysodeikticus as described above. All the assays were done in triplicate.
Resistance to proteolysis
The resistance of native LZ and its conjugates to proteolysis was examined using Proteomix® (trypsin and chymotrypsin), Protex 6L® (bacterial protease), and fungal protease. Protease (20 U) (was added to 100 µL of native LZ (600 U) or to 100 µL of LZ-mPEG-SS (450 U) in a total final volume of 1 mL of 100 mM phosphate buffer, pH 7. The reaction mixture was incubated at 37°C, and aliquots were withdrawn at 0, 15, 30, 60, and 120 min for immediate determination of enzymatic activity. The assays were conducted in triplicate.
Enzyme stability of native and modified lysozymes in human serum
The stability of the native lysozyme and its conjugates (LZ-mPEG-SS) was determined in human serum. An aliquot of 500 µL LZ-mPEG-SS (2250 U) was mixed with 1.5 mL human serum and the tubes incubated at 37°C. Aliquots of 50 µL were withdrawn at 0, 15, 30, 60, and 120 min and enzymatic activity against the substrate M. lysodeikticus was determined as described above.
Antibacterial assays
Gram-negative bacteria (Escherichia coli ATCC 29255) were grown for 18 h in Luria-Bertani (LB) medium at 37°C, at 150 rpm. The culture was then centrifuged at 2000 g for 10 min. The sedimented bacteria were washed three times with 50 mM phosphate buffer, pH 7, and the cells were collected and resuspended in buffer to 1 × 105 cells/mL. Then 500 µL of lysozyme (3000 U) or LZ-mPEG-SS (2250 U) were added to 150 µL of the cell suspension. The mixture was incubated at 37°C for 2 h and serially diluted. Each dilution (100 µL) was spread over an LB agar (2.5%) plate. Colony-forming units (CFU) were obtained after incubation at 37°C for 16 h. The antimicrobial assay with lysozyme was also performed in the presence of free mPEG-SS. The assays were conducted in three independent experiments. Antimicrobial activity was expressed as the percentage of colonies observed after treatment, where the number of colonies obtained after incubation with buffer alone was taken to be 100%.
Results and discussion
Extraction and purification of lysozyme
Lysozyme was extracted from hen egg white obtained from a local market. The extraction was performed by a batch method using a simple and direct procedure that did not require any pre-treatment of the egg white beyond its adsorption onto cation exchange resin, followed by washing of the resin and eluting the enzyme. According to the isoelectric point (pI) of lysozyme (pI = 11), five different Amberlite cation exchange resins (IRP-64, IRP-88, IRC-50, CG-50, and Amberlite 15 Merck®) were tested. Preliminary results indicated that Amberlite IRC-50 and IRP-88 had greater retention. These resins are weakly acidic, with wet mesh sizes of 16-50 and 100-500 respectively.
According to the results shown in , the yields obtained using the IRC-50 and IRP-88 resins for lysozyme extraction and purification were comparable. Lysozyme activity was highest in the eluate after 10 min of stirring (results not shown), indicating that the elution of lysozyme occurred quickly with either resin. This is a very interesting aspect of batch mode purification as compared to ion exchange column chromatography (in which protein elution can take hours). The permeate obtained using IRC-50 gave yields of 61%, with a 58-fold increase in specific activity as compared to the crude extract as indicated by fold purification ().The permeate obtained using IRP-88 showed a comparable yield of 62%, but a lower increase in specific activity (36x) as assayed by fold purification.
Table 2. Purification of hen egg white lysozyme.
One of the difficulties encountered in the purification procedures used for lysozyme is that egg white contains many other proteins with different isoelectric points (pI), such as ovalbumin (pI = 4.5), conalbumin (pI = 6.1) and ovomucin (pI = 4.7) (CitationGhosh & Cui, 2000) that electrostatically interact with lysozyme (pI = 11) and contribute to the viscous consistency of egg white.
shows a single band in 12% SDS-PAGE electrophoretic analysis, corresponding to lysozyme for the permeate obtained using IRC-50 (lane 3) having a molecular mass of approximately 14.4 kDa - which reflects the high purity achieved by this method. The permeate obtained with IRP-88, however, showed two bands (lane 5) - with a subtle band of approximately 46 kDa corresponding to ovalbumin, and a major band of lysozyme near 14.4 kDa, indicating only partial purification with IRP-88.
Figure 1. Polyacrylamide 12% SDS-PAGE electrophoresis of the products of the purification steps of LZ purification. P, molecular mass standard; 1, egg white (20 µg); 2, eluate of IRC-50 (20 µg); 3, permeate IRC-50 (20 µg); 4, eluate of IRP-88 (20 µg); 5, permeate IRR-88 (20 µg). The arrow indicates the LZ band around 14.4 kDa.
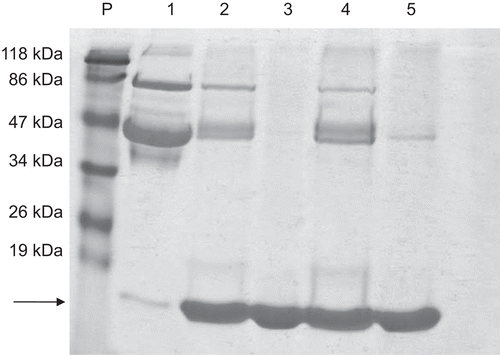
It can therefore be concluded that both resins were effective in batch purification of lysozyme. The yield, fold purification, and the specific activity (5,000-7,000 U/mg) obtained here were similar to those of previous workers (CitationRoy et al., 2003; CitationGhosh & Cui, 2000; CitationOwen & Chase, 1997) using other lysozyme extraction and purification procedures on egg white.
The main advantage of this batch method is the simplicity of the process, for the enzyme is obtained after only two purification steps without any pre-treatment of the egg white. This method is relatively fast and can be applied on an industrial scale and the protein-rich residue can be used for several other purposes (CitationTusge et al., 1996; CitationProctor & Cunningham, 1988).
The purified lysozyme obtained from hen egg white by batch mode was submitted to PEGylation with mPEG-SS (MW 5000). The specific effect of PEGylation on the physical, chemical, and biological properties of the protein is strictly determined by the nature of the proteins and polymers used, as well as by the PEGylation procedures adopted. Important parameters to be considered for the conjugated protein-polymer include: structure, molecular mass of protein and composition, purity of polymer, the number of polymer chains attached, in addition to the conjugation reaction chemistry (which can result in the loss of biological activity, depending on the extent of protein modification) (CitationCaliceti & Veronese, 2003).
To determine the influence of reaction time on the degree of lysozyme substitution, the reaction kinetics of LZ with mPEG-SS 5000 were followed over time, and the results presented in . Activated PEG typically links to proteins and polypeptides via lysine (K) and N-terminal amino acids groups, forming stable linkages such as amines, amides, or urethanes. A series of PEG-activated derivatives have been used as reagents in PEGylation technology, including PEG succinimidyl succinate as employed in the present work.
Figure 2. Electrophoresis in polyacrylamide SDS-PAGE 10%. Kinetics of the PEGylation reaction of LZ with mPEG-SS 5000 for 2 h at 30°C. P, standard of molecular mass; 1, 3, 5, 7, 10, 20, 30, 60 and 120 minutes, indicate the time interval in minutes of aliquots collected and submitted to electrophoresis.

The results obtained by PEGylation of LZ with mPEG-SS 5000 () demonstrated that the succinimidyl succinate functional group is highly reactive with lysine available in the lysozyme molecule and that the reaction was very fast–initiating after only 1 min.
The LZ-mPEG-SS 5000 conjugate displayed the same substitution profile and with the reaction time increased up to 2 h, the electrophoretic band near 30 kDa increased in intensity, demonstrating the high affinity of mPEG-SS 5000 by one of residues (K33, K97, K116), as suggested by CitationLee and Park (2003). However, as described by Swank & Munkers, (1971); Bailon & Berthold, (1998) may be an overestimation of the molecular mass of the conjugate by SDS-PAGE analysis. This phenomenon is related to the fact that the amount of SDS bound by LZ decreases when its amino groups are modified, leading to a decrease in its mobility on SDS-PAGE, as well as the fact the hydrodynamic volume of PEG is greater than that of the protein. As a result, SDS-PAGE analysis the band PEGylated shown in may have a molecular mass lower.
Our preliminary results indicated that the proportion of mPEG-SS/NH2 utilized for enzyme linkage should be more closely examined in order to better determine the effects of the degree of substitution on LZ enzymatic activity. shows the results for enzyme activity of native as well as modified LZ against Micrococcus lysodeikticus and with the substrate glycol chitosan.
Figure 3. Residual enzyme activity (%) of LZ-mPEG-SS 5000 in different molar ratio (mPEG-SS 5000/NH2) of 0.11, 0.21, 0.43, 0.86 toward substrate. (-•-) M. lysodeikticus; (-▪-) Glycol chitosan. Results are means ± SD, n = 3.
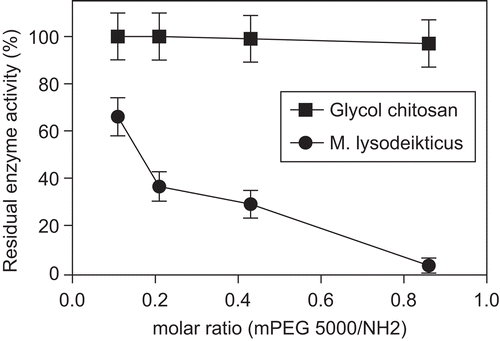
Previous studies indicated that the lysozyme is preferentially modified at lysines (K) 33, 97, and 116 (CitationYamada et al., 1986) and that the loss of enzymatic activity depended on the degree of substitution (CitationLee & Park, 2003; CitationLee et al., 2001; CitationNodake & Yamasaki, 2000; CitationRoberts & Harris, 1998). The degree of protein modification will vary depending on the quantity of activated mPEG available to react with the lysine residues of the protein–with a greater availability of mPEG resulting in a greater degree of protein modification. As such, multiple lysozyme lysine residues can be modified, depending on the proportions of mPEG/NH2 utilized in the reaction mixture. When multiple lysine residues are modified, a heterogeneous mixture is produced composed of populations of protein molecules with variable numbers of attached polyethylene glycol molecules, which can range from zero to the number of ϵ- and α-amine groups in the protein (CitationRoberts et al., 2002; CitationVeronese, 2001).
As can be observed in , the mixture with the highest proportion of mPEG-SS 5000/NH2 (0.86) resulted in excessive PEGylation of LZ and in the loss of 97% of its lytic enzyme activity through steric hindrance that prevents the formation of an enzyme-substrate complex. Steric hindrance is a particularly severe problem for proteins acting on macromolecular substrates (CitationNodake & Yamasaki, 2000; CitationZalipsky, 1995; CitationChiu et al., 1993; CitationCaliceti et al., 1990), and this effect was observed in the activity profile of PEGylated lysozyme with the cell wall substrate of M. lysodeikticus (). There have been a series of studies demonstrating the loss of enzyme activity as a consequence of PEGylation, and although this problem does not occur only with egg-white lysozyme, it has been reported for catalase (CitationAbuchowski et al., 1977), trypsin (CitationZhang et al., 1999) and l-asparaginase (CitationSoares et al., 2002; CitationZhang et al., 2004).
In the present work, decreasing the extent of LZ modification resulted in a greater retention of enzyme activity, and an mPEG-SS 5000/NH2 molar ratio of 0.11 resulted in a residual enzyme activity of 67%.
When a smaller and less complex substrate such as the polymer glycol chitosan was used, the modification of LZ with mPEG 5000 at different molar ratios () did not affect its catalytic activity. Similar results were reported for N-acetylated lysozyme (CitationYamasaki et al., 1968), linkage to hydrophobic peptides (CitationArima et al., 1997), and chemical modification (CitationNodake & Yamasaki, 2000). The results shown in indicate that steric hindrance caused by mPEG-SS 5000 modification of LZ interferes directly with enzyme activity with the cell wall of M. lysodeikticus, while results with a simple substrate (glycol chitosan) indicated that the conformational structure of the active site was not affected.
Enzyme activity stability of modified lysozyme
To determine the stability of LZ activity after mPEG-SS 5000 conjugation, the modified enzyme was isolated by gel filtration (Sephadex G-50) and any excess mPEG removed. The stability of the native and the modified LZ as based on enzymatic activity was determined in different buffers, varying in pH from 4 to 12. shows the effects of pH on enzymatic activity and indicates that the conjugation reaction caused striking alterations in the activity profile of LZ in terms of the cell wall substrate of Micrococcus lysodeikticus.
Figure 4. Evaluation of the enzymatic activity (A) and stability (B) of native LZ (-○-) and LZ-mPEG-SS 5000 (-▪-). The activity was determined in different buffers varying the pH from 4–12 (4–5, 50 mM acetate; 6–8, Hepes 50 mM and 9–12 Tris-HCl 50 mM) at 25°C. The stability was determined after incubation by 1 h in the same buffers at 50°C. At the end of this time the enzymatic activity was determined against substrate M. lysodeikticus as described in methods. Results are means ± SD, n = 3.
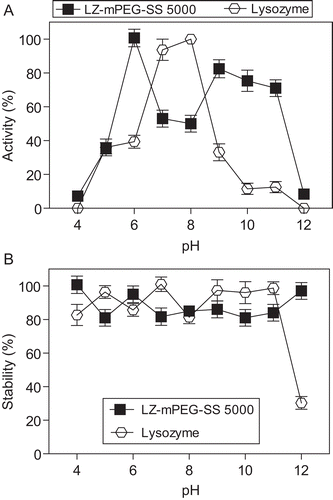
Native lysozyme activity showed optimum activity at pH between 7 and 8 under test assay conditions, but the optimum pH for LZ-mPEG-SS 5000 decreased to 6. At pH levels of between 9 and 11, native lysozyme showed a substantial decline in activity, while the derivative LZ-mPEG-SS 5000 retained between 70% and 80% of its activity. As noted by CitationHomchaudhuri et al. (2006), native LZ forms aggregates at pH 12.2 that can lead to the loss of enzymatic activity at 25°C.
The conjugate LZ-mPEG-SS 5000 also showed enhanced catalytic activity relative to native LZ between pH 9 and 11, indicating that PEGylation confers greater resistance to denaturation. CitationNodake et al. (2002) suggested that chemical modification by PEGylation does not involve the whole LZ molecule, but appears to create a hydrophobic region in a specific region that enhances its lytic activity at basic pH.
The biological stability of both the native lysozyme and the conjugate LZ-mPEG-SS 5000 at 50°C was determined in different buffers () over a wide pH range (4 to 12). The results indicated that the modified enzyme exhibited stability similar to that of native LZ, although greater stability was observed at pH 12. Therefore, the development of stable formulations of lysozyme could considerably extend the uses of this enzyme in terms of food preservation, with PEGylation protecting it against denaturation (likely as a result of increased hydrophobic interactions) caused by exposure to extreme temperatures and pH (CitationJohnson & Larson, 2005).
The resistance of modified lysozyme to proteolysis
Preliminary results (not shown) indicated that native lysozyme was relatively resistant to proteolytic degradation, retaining considerable enzymatic activity (98%) for 2 h at 37°C in the presence of 5 different proteases. This resistance to proteolytic degradation may be explained by the globular shape of LZ and its tightly packed and rigid molecular structure (CitationPolverino de Laureto et al., 1995). Previous studies have demonstrated lysozyme resistance to proteolytic degradation by papain, trypsin, chymotrypsin (CitationMine et al., 2004) as well as other proteases (CitationPolverino de Laureto et al., 1995). This proteolysis resistance has contributed to the analysis of the structure and dynamics of proteins (CitationPolverino de Laureto et al., 2002).
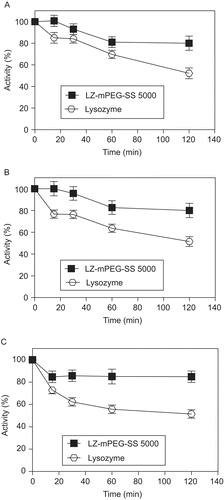
The degree of resistance of native and modified lysozyme to proteolytic degradation by different proteases as examined in the present study is shown in . The presence of excess protease (mass ratio of LZ/protease,1:25) resulted in a 50% decrease in native lysozyme enzyme activity after 2 h in the presence of 20 U of trypsin + chymotrypsin (A), bacterial protease (B), or fungal protease (C), while the enzymatic activity of the modified lysozyme remained at around 80% under the same conditions. This proteolytic resistance seems to be related to the fact that mPEG linkage causes an increase in molecular mass that increases steric hindrance and results in a decreased susceptibility to proteases (CitationLee & Park, 2003; CitationRoberts et al., 2002; CitationVeronese, 2001).These results are in accordance with CitationSo et al. (1996) who demonstrated increased resistance of modified LZ to the proteases trypsin, pronase P, and cathepsins B and D. Other enzymes modified by PEGylation, including ribonuclease, catalase, trypsin, and l-asparaginase (CitationSoares et al., 2002; CitationMonfardini et al., 1995), have also been shown to be more resistant to proteolytic degradation.
Figure 5. Resistance to proteolysis of native lysozyme (-○-) and LZ-mPEG-SS 5000 (-▪-), digestion as assessed by enzyme activity upon incubation with Proteomix® (A), Protex 6L® (B) and fungal protease (C). Results are means ± SD, n = 3.
When samples of native or modified lysozyme were submitted to in vitro incubation in human serum, the enzymes were found to remain stable, and demonstrated less than a 10% loss of enzymatic activity (results not shown), indicating that no alterations affecting the catalytic activity of either the native or modified lysozymes had occurred, such as binding by non-specific antibodies or enzymatic hydrolysis by serum proteases.
Antimicrobial activity of modified lysozyme
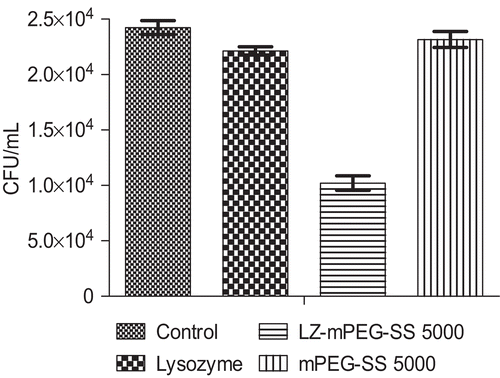
The LZ-mPEG-SS 5000 conjugate had higher antimicrobial activity than native lysozyme against E. coli (), producing a 58% greater decrease in the numbers of colonies, while free mPEG had no antimicrobial activity against E. coli. LZ-mPEG-SS 5000 showed greater antimicrobial activity against Gram-negative bacteria than did the native enzyme. As Gram-negative bacteria have a peptidoglycan layer composed mainly of lipopolysaccharides (LPS) and lipid bilayers that is located inside their outer membrane (CitationIbrahim et al., 2002), LZ-mPEG-SS 5000 apparently has a higher surface hydrophobicity (as compared to unmodified LZ) that allows better interaction with the lipid moiety of LPS, resulting in damage to the outer membrane and cell lysis (CitationVannini et al., 2004). Gram-negative bacteria are assumed to be lysed by lysozyme only after the permeability of the outer membrane has been altered, allowing enzyme access to the peptidoglycan layer (CitationIbrahim et al., 2002). LZ-mPEG-SS 5000 can therefore be considered a promising antimicrobial agent against E. coli.
Figure 6. Antimicrobial activity of lysozyme, lysozyme-mPEG-SS 5000 and free mPEG-SS 5000 against E. coli. The survial of E. Coli was expressed as CFU/mL. Values are the means from three independent experiments.
In conclusion, a rapid and simple method for the batch purification of lysozyme from hen egg white was devised, allowing selective extraction of LZ with a yield of around 60% and a high degree of purification, without damaging the other egg-white proteins. The conjugate produced by lysozyme modification with mPEG succinimidyl succinate showed a fast binding reaction, with enzymatic activity being influenced by the degree of lysozyme modification. The conjugate retained biological activity over a wide pH range, showed resistance to proteolytic degradation, remained stable in human serum, and displayed greater antimicrobial activity against E. coli than the native lysozyme.
Declaration of interest
This work received financial support from the Brazilian Research Foundation- Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP)-05/50958-0.
References
- Abuchowski A, McCoy JR, Palczuk NC, Theo VE, Davis FF (1977): Effect of covalent attachment of poly(ethylene glycol) on immunogenicity and circulating life of bovine liver catalase. J Biol Chem 252: 3582–3586.
- Arima H, Ibrahim HR, Kinoshita T, Kato A (1997): Bacterial action of lysozyme attached with various sizes of hydrophobic peptides to the C-terminal using genetic modification. FEBS Letters 415: 114–118.
- Bradford MM (1976): A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72: 248–254.
- Caliceti P, Veronese FM (2003): Pharmacokinetic and biodistribution properties of poly(ethyleneglycol) protein conjugates. Adv Drug Deliv Rev 55: 1261–1277.
- Caliceti P, Schiavon O, Veronese FM, Chaiken IM (1990): Effects of monomethoxypoly(ethylene glycol) modification of ribonuclease on antibody recognition, substrate accessibility and conformational stability. J Mol Recognit 3: 89–93.
- Chiu HC, Zalipky S, Kopeckova P, Kopecek J (1993): Enzymatic activity of chymotrypsin and its poly(ethylene glycol) conjugates toward low and high molecular weight substrates. Bioconjug Chem 4: 290–295.
- Ghosh R, Cui ZF (2000): Purification of lysozyme using ultrafiltration. Biotechnol Bioeng 68: 191–203.
- Harris JM, Martin NE, Modi M (2001): Pegylation. A novel process for modifying pharmacokinetics. Clin Pharmacokinet 40: 539–551.
- Harris JM, Veronese FM (2003): Peptide and protein pegylation II-clinical evaluation. Adv Drug Deliv Rev 55: 1259–1260.
- Homchaudhuri L, Kumar S, Swaminathan R (2006): Slow aggregation of lysozyme in alkaline pH monitored in real time employing the fluorescence anisotropy of covalently labelled dansyl probe. FEBS Letters 580: 2097–2101.
- Ibrahim HR, Aoki T, Matsuzaki T (2001): Genetic evidence that antibacterial activity of lysozyme is independent of its catalitic function. FEBS Letters 506: 27–32.
- Ibrahim HR, Aoki T, Pellegrini A (2002): Strategies for new antimicrobial proteins and peptides: Lysozyme and aprotinin as model molecules. Curr Pharm Des 8: 671–693.
- Ibrahim HR, Kobayshi K, Kato A (1991): Antimicrobial effects of lysozyme against gram-negative bacteria due to covalent binding of palmitic acid. J Agric Food Chem 39: 2077–2083.
- Ibrahim HR, Kobayshi K, Kato A (1993): Length of hydrocarbon chain and antimicrobial action to Gram-negative bacteria of fatty acylated lysozyme. J Agric Food Chem 41: 1164–1168.
- Imoto T, Yagishita K (1971): A simple activity measurement of lysozyme. Agric Biol Chem 35: 1154–1156.
- Johnson EA, Larson AE (2005): Lysozyme, in: Davidson PM, Sofos JN, Branen AL, eds, Antimicrobials in Food, third edition. New York, Taylor & Francis Group, pp. 361–387.
- Laemmli UK (1970): Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685.
- Lee H, Park TG (2003): A novel method for identifying pegylation sites of protein using biotinylated peg derivatives. J Pharm Sci 92: 97–103.
- Lee S, Greenwald RB, Mcguire J, Yang K, Shi C (2001): Drug delivery systems employing 1,6-elimination: Releasable poly (ethylene glycol) conjugates of proteins. Bioconjug Chem 12: 163–169.
- Liu ST, Sugimoto T, Azakami H, Kato A (2000): Lipophilization of lysozyme by short and middle chain fatty acids. J Agric Food Chem 48: 265–269.
- Mine Y, Ma F, Lauriau S (2004): Antimicrobial peptides release by enzymatic hydrolysis of hen egg white lysozyme. J Agric Food Chem 52: 1088–1094.
- Monfardini C, Shiavon O, Caliceti P, Mopurgo M, Harris JM, Veronese FM (1995): A branched monomethoxypoly(ethylene glycol) for protein modification. Bioconjug Chem 6: 62–69.
- Nakamura S, Kato A, Kobayashi K (1991): New antimicrobial characteristics of lysozyme-dextran conjugate. J Agric Food Chem 39: 647–650.
- Nodake Y, Iwasaki K, Yamasaki N (2002): Interactions of a lysozyme monomethoxypolyethylene glycol conjugate with lipopolysaccharides and lipid bilayers and effects of conjugate on gram-negative bacteria. Biosci Biotechnol Biochem 66: 1848–1852.
- Nodake Y, Yamasaki N (2000): Some proprieties of a macromolecular conjugate of lysozyme prepared by modification with a monomethoxypolyethylene glycol derivative. Biosci Biotechnol Biochem 64: 767–774.
- Owen RO, Chase HA (1997): Direct purification of lysozyme using continuous counter current expanded bed adsorption. J Chromatogr A 757: 41–49.
- Pasut G, Mero A, Caboi F, Scaramuzza S, Sollai L, Veronese FM (2008): A new PEG-β-alanine active derivate for releasable protein conjugation. Bioconjug Chem 19: 2427–2431.
- Polverino de Laureto P, De filippis V, Scaramella E, Zambonin M, Fontana A (1995): Limited proteolysis of lysozyme in trifluoroethanol. Isolation and characterization of a partially active enzyme derivative. Eur J Biochem 230: 779–787.
- Polverino de Laureto P, Frare E, Gottardo R, Van dael H, Fontana A (2002): Partly folded states of members of the lysozyme/lactalbumin superfamily: A comparative study by dichroism spectroscopy and limited proteolysis. Protein Science 11: 2932–2946.
- Proctor VA, Cunningham FE (1988): The chemistry of lysozyme and its use as a food preservative and pharmaceutical. CRC Crit Rev Food Sci Nutr 26: 359–395.
- Rahman MM, Sarker SD, Gray AI, Khondkar P (2008): Antibacterial and antifungal activities of the constituents of Flemingia paniculata. Pharm Biol 46: 356- 359.
- Roberts MJ, Bentley MD, Harris JM (2002): Chemistry for peptide and protein PEGylation. Adv Drug Deliv Rev 54: 459–476.
- Roberts MJ, Harris JM (1998): Attachment of degradable poly(ethylene glycol) to proteins has the potential to increase therapeutic efficacy. J Pharm Sci 87: 1440–1445.
- Roy I, Rao MV, Gupta MN (2003): An integrated process for purification of lysozyme, ovalbumin, ovomucoid from hen egg white. Appl Biochem Biotechnol 111: 55–63.
- Shugar D (1952): The measurement of lysozyme activity and ultraviolet inactivation of lysozyme. Biochem Biophys Acta 8: 302–309.
- So T, Ito HO, Koga T, Ueda T, Imoto T (1996): Reduced immunogenicity of monomethoxypolyethyleneglycol-modified lysozyme for activation of T cells. Immunology Letters 49: 91–97.
- Soares LS, Guimaraes GM, Polakiewicz B, Pitombo RNM, Abrahao-Neto J (2002): Effects of polyethylene glycol attachment on physicochemical and biological stability of E. coli l-asparaginase. Inter J Pharm 237: 163–170.
- Treetharnmathurot B, Ovartlarnporn C, Wungsintaweekul J, Ducan R, Wiwattanapatapee R (2008): Effect of PEG molecular weight amd linking chemistry on the biological activity and thermal stability of PEGylated trypsin. Intl J Pharm 357:252–259.
- Tusge Y, Shimoyumada M, Watanabe K (1996): Binding of egg white proteins to viruses. Biosci Biotech Biochem 60: 1503–1504.
- Vannini L, Lanciotti R, Baldi D, Guerzoni ME (2004): Interactions between high pressure homogenization and antimicrobial activity of lysozyme and lactoperoxidase. Intl J Food Microbiol 94: 123–135.
- Veronese FM (2001): Peptide and protein PEGylation: a review of problems and solutions. Biomaterials 22: 405–417.
- Veronese FM, Pasut G (2005): Pegylation, successful approach to drug delivery. Drug Discov Today 10: 1451–1458.
- Xu X, Kashima O, Saito A, Azakami H, Kato A (2004): Structural and functional properties of chicken lysozyme fused serine-rich heptapeptides the C-terminus. Biosci Biotechnol Biochem 68: 1273–1278.
- Yamada H, Matsunaga N, Domoto H, IMOTO T (1986): Dinitrophenylation as a probe for the determination of environments of amino groups in protein. Reactivities of individual amino groups in lysozyme. J Biochem 100: 233–241.
- Yamasaki N, Hayasi K, Funatsu M (1968): Acetylation of lysozyme Part I. Preparation, fractionation and properties of acetylated lysozyme. Agric Biol Chem 32: 55–63.
- Yamasaki N, Hayasi K, Funatsu M (1968): Acetylation of lysozyme Part II. Mechanism of lysis by lysozyme. Agric Biol Chem 32: 64–68.
- Zalipsky S (1995): Chemistry of polyethylene glycol conjugates with biologically active molecules. Ad Drug Del Rev 16: 157–182.
- Zalipsky S, Mullah N, Engbers C, Hutchins MU, Kiwan R (2007): Thiolytically cleavable dithiobenzyl urethane-linkage polymer-protein conjugates as macromolecular products: reversible PEGylation of proteins. Bioconjug Chem 18:1869–1878.
- Zappe H, Snell ME, Bossard MJ (2008): PEGylation of cyanovirin-N, an entry inhibitor of HIV. Adv Drug Del Rev 60: 79–87.
- Zhang Z, He Z, Guan G (1999): Thermal stability and thermodynamic analysis of native and methoxypolyethylene glycol modified trypsin. Biotechnol Techn 13: 781–786.
- Zhang JF, Shi LY, Wei DZ (2004): Chemical modification of l-asparaginase from Escherichia coli with a modified polyethyleneglycol under substrate protection conditions. Biotechnol Letters 26: 753–756.