Abstract
Context: Mitotic kinase enzymes regulate critical stages of mitosis and are amenable to pharmacological inhibition. Since natural products have been a rich source of antimitotic inhibitors, we postulated that natural products would also provide effective inhibitors of mitotic kinases.
Objective: To explore unique marine and terrestrial natural product sources for new anticancer drug leads, we screened our natural product extract library for polo-like kinase-1 (Plk1) kinase inhibitors.
Materials and methods: Extracts of the lichen Parmotrema sp. (Parmeliaceae) exhibited in vitro inhibitory activity. Bioassay-guided fractionation of the Parmotrema sp. extract led to the isolation of depside inhibitors.
Results: A new depside 1 has been isolated from the Sri Lankan lichen Parmotrema sp. along with the known metabolites 2 (β-collatolic acid) and 3 (β-alectoronic acid). The structure of depside 1 was elucidated by spectroscopic analysis. The three depsides 1–3 exhibited moderate inhibition of purified recombinant Plk1 kinase with IC50 of 2.8, 0.7, and 1.7 µM, respectively, at 1 µM ATP. Inhibitory activity was also observed at high concentrations of ATP, suggesting the potential for activity in a cellular environment. The depsides were also tested against a panel of 23 other recombinant kinases and were found to possess up to 30-fold selectivity toward Plk1.
Discussion and conclusion: These data suggest that the depsides 1–3 may serve as core structures that can be further explored as potential inhibitors of Plk1 and other kinases.
Introduction
Antimitotic drugs are commonly used in cancer therapy (CitationJiang et al., 2006). These agents primarily affect tubulin dynamics and interfere with cellular structures such as the mitotic spindle, which regulates the faithful segregation of chromosomes during cell division. An alternative approach to attenuate cell growth is to target other mediators of the cell division process, including mitotic kinases (CitationBlagden & de Bono, 2005). These enzymes regulate critical stages of mitosis and are amenable to pharmacological inhibition. Natural products, including taxanes, vincas, and other agents, have been a rich source of antimitotic inhibitors. Hence, we postulated that natural products might also be effective inhibitors of mitotic kinases.
Polo-like kinase-1 (Plk1) is a serine/threonine kinase that has several critical functions in cell division, including initiation of mitosis, centrosome maturation, spindle assembly, anaphase regulation, and cytokinesis (CitationBarr et al., 2004). These normal functions are compromised when the Plk1 pathway is deregulated. Plk1 overexpression can induce tumor growth in mouse models (CitationSmith et al., 1997). In patients, Plk1 protein or mRNA is reportedly overexpressed in many solid tumors, including those derived from colon (CitationTakahashi et al., 2003), pancreas (CitationGray et al., 2004), prostate (CitationWeichert et al., 2004), ovary (CitationTakai et al., 2001), head and neck (CitationKnecht et al., 1999), non-Hodgkin’s lymphoma (CitationMito et al., 2005), and others. For several of these tumor types, a positive correlation is observed between higher Plk1 expression, increased invasive potential, and reduced patient survival. Experimental reduction of Plk1 protein markedly impairs tumor cell growth in vitro and in vivo. Plk1 depletion using small interfering RNA (siRNA), antisense oligonucleotides, or neutralizing antibodies results in mitotic arrest and apoptosis of cancer cell lines (CitationLiu & Erikson, 2003; CitationSpänkuch et al., 2004). Hence, these depletion studies supported the hypothesis that inhibition of Plk1 may be useful for the treatment of cancers.
Multiple reports of Plk1 inhibitors have been emerging (CitationMcInnes et al., 2006; CitationStrebhardt & Ullrich, 2006). Wortmannin, a well-documented inhibitor of phosphoinositol-3-kinases (PI3K), inhibits Plk1 kinase activity (CitationLiu et al., 2007). Significantly, several highly potent and selective small molecule inhibitors of Plk1 were recently reported (CitationLansing et al., 2007; CitationSteegmaier et al., 2007). As molecular tools, these agents provide insight into Plk1 function in mitosis and other processes. Moreover, these inhibitors provide proof-of-concept that attenuation of Plk1 kinase activity effectively drives cells into mitotic arrest resulting in cell death. These data suggest that Plk1 kinase inhibitors may be useful in the treatment of cancers that depend upon Plk1 for cell division.
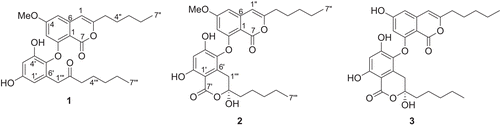
As part of a collaborative program designed to explore unique marine and terrestrial natural product sources for new anticancer drug leads, we screened our natural product extract library for Plk1 kinase inhibitors and found that extracts of the lichen Parmotrema sp. (Parmeliaceae), collected in Sri Lanka exhibited in vitro inhibitory activity. Bioassay-guided fractionation of the Parmotrema sp. extract led to the isolation of the inhibitors 1, β-collatolic acid (2) (CitationRangaswami & Rao, 1955), and β-alectoronic acid (3) (CitationKrivoshchekova et al., 1983) (). The structure of the new metabolite 1, along with the biological activities of despides 1–3, is described below.
Figure 1. The structures of depsides 1–3.
Materials and methods
The 1H and 13C NMR spectra were recorded on a Bruker AV-600 spectrometer (East Milton, ON, Canada) with a 5 mm CPTCI cryoprobe. 1H chemical shifts are referenced to the residual acetone-d6 signal (δ 2.05 ppm) and 13C chemical shifts are referenced to the acetone-d6 solvent peak (δ 29.92 ppm). Low- and high-resolution ESI-QIT-MS were recorded on a Bruker-Hewlett Packard 1100 Esquire-LC system mass spectrometer.
Merck Type 5554 silica gel plates and Whatman MKC18F plates were used for analytical thin-layer chromatography. Reversed-phase HPLC purifications were performed on a Waters 600E System Controller liquid chromatograph attached to a Waters 996 photodiode array detector. All solvents used for HPLC were of Fisher HPLC grade.
For the Plk1 enzyme assay, recombinant human Plk1 was expressed in Escherichia coli cells and purified by sequential chromatography on NiNTA, HQ/CM, and size-exclusion columns to >95% purity. Plk1 enzyme activity was assessed in a DELFIA format assay as follows. Plk1 enzyme was diluted in PKB buffer (20 mM HEPES, 10 mM MgCl2, 2 mM l-cysteine) and added to a 384-well streptavidin plate. Compounds to be evaluated were added to the reaction as 50× DMSO stocks in 0.5 µL. After 5-min preincubation of compound with enzyme, 5 µL of 5× peptide/ATP mixture was added to each well. Peptide substrate was derived from the C-terminal domain of Myt1 protein, including the T495 phosphorylation site (Btn-SFPSFEPRNLLSLFEDTLDPT). Reactions were incubated for 90 min at room temperature. Final reaction conditions were: 5 ng (2.9 nM or 200 ng/mL) Plk1, 0.5 µM MYT1 peptide, 10 µM ATP, 20 mM HEPES, 10 mM MgCl2, 2 mM l-cysteine, 2% DMSO. In ATP-dependent experiments, final ATP concentrations were 1, 10, 100, or 1000 μM. Plates were washed twice with 75 µL of Tris-buffered saline + 0.05% Tween-20 (TBST) in an automated plate washer. A mixture of primary and secondary antibodies was added (anti-phosphothreonine, Cell Signaling Technologies (Danvers, MA), diluted to 1:4000, plus anti-rabbit-IgG-Eu, Perkin Elmer (Waltham, MA), diluted to 1:5000 in DELFIA assay buffer). Antibodies were incubated on the plate for 60 min at room temperature. Plates were washed twice with TBST. DELFIA enhancement solution was added, incubated for 30 min, and then plates were read in a PerkinElmer Victor5 microplate reader. Signal-to-noise ratio was calculated from the ratio of +ATP/–ATP wells. The inhibitory effect of compounds on Plk1 kinase activity was determined by calculating the decrease in signal between DMSO and control versus compound-treated wells. IC50 values were calculated using the Excel add-in module XLfit, model 203 (IDBS, Guildford, UK). Statistical analyses were conducted with a two-tailed t-test to determine differences between IC50 values. All compounds were resuspended as 10 mM stocks in 100% DMSO and serially diluted into DMSO for assays. BI3536 was provided by Wyeth Chemical Sciences (Pearl River, NY). Wortmannin (catalog no. 681675) and staurosporine (catalog no. 569397) were purchased from Calbiochem/EMD Chemicals (Gibbstown, NJ).
For the kinase selectivity profiling assay, all buffer salts and reagents were purchased from Sigma (St. Louis, MO), and were of the highest purity available. Kinases were purchased from Invitrogen (Carlsbad, CA) and were used without further purification. Fluorescently labeled peptide substrates were purchased from Anaspec (Freemont, CA). The peptide sequence was optimized for each kinase. The structural identity of the three depsidones was confirmed by mass spectrometry before sample testing. Compound plates were prepared by pipetting an 11-point dose–response titration in DMSO with final assay concentrations ranging from 10 μM to 5 nM. Assay plates were prepared with 150 nL addition of compound, followed by 7.5 μL of assay buffer (50 mM HEPES, 10 mM MgCl2, 0.005% Brij, 5 mM 2-mercaptoethanol) containing substrate and ATP, followed by 7.5 μL of buffer-containing enzyme. All reactions were performed at 1.5 μM peptide substrate ([S] << Km) and at the experimentally determined Km of ATP for each kinase. All final enzyme concentrations were below 50 nM, and in most cases were 10 nM or less. The reactions were incubated at room temperature for 1 to 2 h depending on kinase activity, but in all cases exhibited between 20% to 30% substrate phosphorylation. The reactions were quenched by the addition of 15 μL of stop buffer (100 mM HEPES, 20 mM EDTA, 0.005% Brij, and 0.2% Caliper coating reagent). Substrates and products were separated on the Caliper LC3000 using standard separation protocols. The percentage inhibition was calculated for each compound concentration and the IC50 was determined using Equation (1).
Specimens of Parmotrema sp. were found growing on a rock at 3500 ft, near Labukelle, Central Province, Sri Lanka. A herbarium specimen has been deposited in the Department of Chemistry, University of Peradeniya (LB-11).
Specimens of Parmotrema sp. (2.3 g) were repeatedly extracted with MeOH to give a MeOH extract (100 mg) that was fractionated via silica gel MPLC (eluent: 100% CH2Cl2 to 100% MeOH) to give a fraction (39.8 mg) that exhibited inhibitory activity along with the known and widely distributed depside atranorin (2.8 mg) (CitationJayaprakasha & Rao, 2000). The active fraction was further fractionated on a Sephadex LH-20 using MeOH as eluent to generate two active fractions, which after reversed-phase HPLC, using a CSC-Inertsil 150A/ODS2, 5 µm 25 × 0.94 cm column with 13:7 MeCN/(0.05% TFA/H2O) as eluent, gave pure samples of three depsides, the new compound 1 (0.8 mg) and the known metabolites 2 (2.9 mg) and 3 (1.1 mg).
Results
From the MeOH extracts of the lichen Parmotrema sp., one new depside, compound 1, along with the known compounds β-collatolic acid (2) (CitationRangaswami & Rao, 1955) and β-alectoronic acid (3) (CitationKrivoshchekova et al., 1983) were isolated ().
The physical and spectroscopic data obtained for compound 1 are as follows:
Depsidone 1: Isolated as a purple powder; UV (MeOH) λmax (log ϵ) 206 (4.5), 244 (4.6), 279 (4.5), 326 (4.0) nm; 1H NMR, see ; 13C NMR, see ; positive ion HRESIMS [M+Na]+ m/z 505.2170 (calculated for C28H34O7Na, 505.2202).
Table 1. Comparison of the 1H and 13C NMR data for depsides 1 and 2 recorded at 600 MHz in acetone-d6.
The depsides 1–3 were assayed in a Plk1 enzyme assay at various ATP concentrations to evaluate the potency and ATP dependence of these inhibitors against Plk1. The Km(ATP) for Plk1 was determined to be ∼6 μM under the conditions of this assay (data not shown). Compounds were tested at 1, 10, 100, and 1000 μM ATP to span a range of ATP concentrations from near-Km to elevated levels that may mimic the high ATP concentrations present in cells where inhibitors must compete with ATP for binding to kinases. The IC50 values for the depsides at varied ATP concentrations are indicated in . At lower ATP concentrations (1 μM), the IC50 for depsides 1–3 were 2.8, 0.7, and 1.7 μM, respectively. Notably, minimal shift in IC50 was observed at high ATP concentrations for the three depsides. indicates the ratio of IC50 at each ATP concentration compared with the IC50 at 1 μM ATP. At 1000 μM ATP, the IC50 of depsides 1 and 2 increased by only 36-fold, while the IC50 of depside 3 changed minimally (1.4×). The ATP dependence of the depsides were compared with other compounds known to inhibit Plk1, including wortmannin, BI 2536, and staurosporine. Wortmannin is predicted to be an irreversible inhibitor of Plk1, as observed for PI3K kinases (CitationLiu et al., 2007), and hence would be expected to display minimal ATP dependence as a result of covalent binding. BI 2536 is reported to be an ATP competitive inhibitor; however, the compound is known to effectively inhibit Plk1 in cells and therefore is capable of action under high cellular ATP concentrations (CitationLansing et al., 2007; CitationSteegmaier et al., 2007). These two Plk1 inhibitors remained potent even at 1000 μM ATP ( and ). In contrast, staurosporine, a generic ATP competitive kinase inhibitor with submicromolar activity against Plk1 at low ATP concentrations, was clearly displaced under conditions of high ATP (IC50 ratio = 47× at 1000 μM ATP). These data suggest that the depsides either associate with Plk1 at a site other than the ATP-binding site (non-ATP competitive) or associate at the ATP-binding site (ATP competitive) but have a slow dissociation rate. Although these depsides are only moderately potent against purified Plk1 enzyme, they retain micromolar activity even at high ATP concentrations, suggesting that they could overcome elevated cellular levels of ATP.
Table 2. Inhibition of recombinant Plk1 kinase by depsides 1–3 and reference compounds.
Table 3. ATP dependence of depsides 1–3 and reference compounds.
The depsides 1–3 were also tested against a panel of 23 kinases to compare their selectivity profile among kinases (). These kinases are a small representative subset of different kinase classes spanning the kinome. The enzyme data suggest that the depsides possess differential activity between Plk1 and several other kinases, in many cases by more than an order of magnitude. While depsides 2 and 3 inhibited Plk1 with IC50 < 3 μM, they showed minimal activity against diverse kinases (IC50 > 10 μM against ∼14 of the 23 kinases tested). Moreover, for depside 3 the selectivity ratio between Plk1 and many other kinases was >35-fold. Depside 1 was the least potent against Plk1 (2–11 μM, at varied ATP concentrations) and showed the correspondingly lowest activity against the kinase panel.
Table 4. Kinase selectivity profile of depsides 1–3.
Discussion
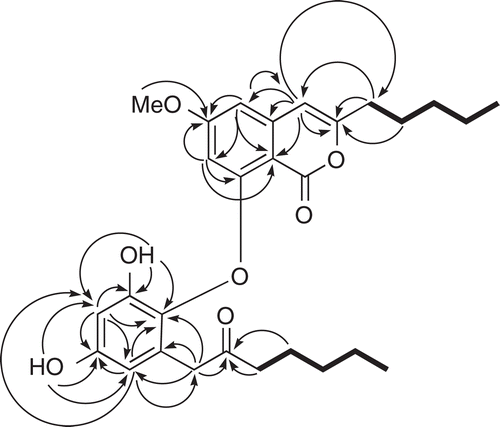
Depside 1 was obtained as a purple powder that gave a [M+Na]+ ion in the HRESIMS at m/z 505.217 that was appropriate for a molecular formula of C28H34O7 differing from that of the known compound 2 by loss of CO2. Comparison of the 13C NMR spectrum of depside 1 with that of depside 2 () indicated that decarboxylation had taken place at C-7′ in depside 1 since the C-7′ lactone carbonyl and the C-2″′ hemiketal resonances in depside 2 were no longer present but instead a ketone carbon was observed resonating at δ 207.2 ppm in depside 1. Careful analysis of the gCOSY, gHSQC, and gHMBC data confirmed the structure of depside 1 () as the decarboxylated adduct of β-collatolic acid (2) (CitationRangaswami & Rao, 1955).
Figure 2. gCOSY connectivities (in bold) and key gHMBC (arrow) correlations used to assign the structure of depside 1.
A new finding was the biological activity of these three depsides against Plk1 and a panel of diverse kinases. Depsides 1–3 were found to inhibit recombinant purified Plk1 kinase activity at micromolar levels. These compounds retain activity even at high ATP concentrations, consistent with ATP competitive inhibitors possessing a slow off-rate. In addition, the depsides possessed differential activity against a small panel of diverse kinases. Moreover, these data suggest some preferential activity of the depsides for Plk1 versus other kinases. For example, all three depsides inhibited the mitotic kinase aurora B at equivalent potency as Plk1. Of the kinases tested, aurora is the most closely related to Plk1. In addition, moderate activity was also observed against other mitotic kinases, including Cdk2 and Cdk1. Interestingly, depsides 2 and 3 also inhibited the non-receptor tyrosine kinases (NRTK) Hck, Fyn, and Abl with IC50 of 3–10 µM. In contrast, minimal activity was observed against other NRTKs such as Src and Lyn (IC50 > 50 µM). These kinases are structurally distant from Plk1 in the kinome, but these data suggest related binding sites for the depsides on the serine–threonine kinase Plk1 and some tyrosine kinases, and may allow the use of the depsides as useful probes to interrogate these unrelated kinases.
The inhibitory activity of the depsides 1–3 against Plk1 and other kinases suggests that these are likely to bind to the ATP pocket of kinases. The relative ATP independence of these entities also suggests tight binding that may be beneficial in a high ATP cellular environment. The depsides were tested for their ability to inhibit phosphorylation of the Plk1 substrate Myt1 in a cellular assay; however, no inhibition was observed up to 30 µM (data not shown). Hence, these depsides may either have low cell membrane permeability or lack sufficient enzyme potency to possess cell activity.
Conclusion
Our data suggest that depsides 2 and 3 have similar kinase inhibition profiles. The subtle replacement of a hydroxyl in depside 3 by a methoxy residue in depside 2 suggests that this site imparts minimal effect on kinase binding. In contrast, the structural differences between depside 1 compared with depsides 2 and 3 are sufficient to reduce potency by more than 5–10-fold against a panel of kinases. The depsides are less potent than other reported Plk1 inhibitors such as wortmannin and BI 2536, which inhibit Plk1 at low nanomolar concentrations; however, they represent a new structural class of Plk1 inhibitors. These data suggest that the depsides 1–3 may serve as core structures that can be further explored as potential inhibitors of Plk1 and other kinases.
Declaration of interest
The research was supported by National Cancer Institute-National Cooperative Drug Discovery Group grant CA 67786. The authors alone are responsible for the content and writing of the paper.
References
- Barr FA, Silljé HH, Nigg EA. (2004). Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol 5:429–440.
- Blagden S, de Bono J. (2005). Drugging cell cycle kinases in cancer therapy. Curr Drug Targets 6:325–335.
- Gray PJ Jr, Bearss DJ, Han H, Nagle R, Tsao MS, Dean N, Von Hoff DD. (2004). Identification of human polo-like kinase 1 as a potential therapeutic target in pancreatic cancer. Mol Cancer Ther 3:641–646.
- Jayaprakasha GK, Rao LJ. (2000). Phenolic constituents from the lichen Parmotrema stuppeum (Nyl.) Hale and their antioxidant activity. Z Naturforsch, C, J Biosci 55:1018–1022.
- Jiang N, Wang X, Yang Y, Dai W. (2006). Advances in mitotic inhibitors for cancer treatment. Mini Rev Med Chem 6:885–895.
- Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, Strebhardt K. (1999). Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res 59:2794–2797.
- Krivoshchekova OE, Mishchenko NP, Stepanenko LS, Maksimov OB. (1983). Aromatic metabolites of Parmeliaceae lichens. I. Depsidones. Khimiya Prirodnykh Soedinenii 1:13–19.
- Lansing TJ, McConnell RT, Duckett DR, Spehar GM, Knick VB, Hassler DF, Noro N, Furuta M, Emmitte KA, Gilmer TM, Mook RA Jr, Cheung M. (2007). In vitro biological activity of a novel small-molecule inhibitor of polo-like kinase 1. Mol Cancer Ther 6:450–459.
- Liu X, Erikson RL. (2003). Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc Natl Acad Sci USA 100:5789–5794.
- Liu Y, Jiang N, Wu J, Dai W, Rosenblum JS. (2007). Polo-like kinases inhibited by wortmannin. Labeling site and downstream effects. J Biol Chem 282:2505–2511.
- McInnes C, Mazumdar A, Mezna M, Meades C, Midgley C, Scaerou F, Carpenter L, Mackenzie M, Taylor P, Walkinshaw M, Fischer PM, Glover D. (2006). Inhibitors of polo-like kinase reveal roles in spindle-pole maintenance. Nat Chem Biol 2:608–617.
- Mito K, Kashima K, Kikuchi H, Daa T, Nakayama I, Yokoyama S. (2005). Expression of polo-like kinase (PLK1) in non-Hodgkin’s lymphomas. Leuk Lymphoma 46:225–231.
- Rangaswami S, Rao SV. (1955). Chemical components of Parmelia species of India. Indian J Pharm 7:50–53.
- Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, Ferris DK. (1997). Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Commun 234:397–405.
- Spänkuch B, Matthess Y, Knecht R, Zimmer B, Kaufmann M, Strebhardt K. (2004). Cancer inhibition in nude mice after systemic application of U6 promoter-driven short hairpin RNAs against PLK1. J Natl Cancer Inst 96:862–872.
- Steegmaier M, Hoffmann M, Baum A, Lénárt P, Petronczki M, Krssák M, Gürtler U, Garin-Chesa P, Lieb S, Quant J, Grauert M, Adolf GR, Kraut N, Peters JM, Rettig WJ. (2007). BI 2536, a potent and selective inhibitor of polo-like kinase 1, inhibits tumor growth in vivo. Curr Biol 17:316–322.
- Strebhardt K, Ullrich A. (2006). Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 6:321–330.
- Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, Kimura M, Okano Y, Saji S. (2003). Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci 94:148–152.
- Takai N, Miyazaki T, Fujisawa K, Nasu K, Hamanaka R, Miyakawa I. (2001). Expression of polo-like kinase in ovarian cancer is associated with histological grade and clinical stage. Cancer Lett 164:41–49.
- Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, Loening S, Dietel M, Kristiansen G. (2004). Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 60:240–245.