Abstract
Context: Liquid–liquid extraction (LLE) shows high efficiency in the plasma sample preparation. However, this extraction method is not optimal for the biological samples containing complex organic interferences, such as liver and brain tissues. Some plant secondary metabolites can be converted between water-insoluble and water-soluble forms by pH adjustment.
Objective: A two-step pH-dependent LLE method was introduced in this study to eliminate both water-soluble and lipidic interferences using the properties of pH-dependent interconvertible forms of analytes during sample preparation. A sensitive and reliable method using a reverse-phase HPLC coupled with a fluorescence detector was developed and validated.
Materials and methods: 10-Hydroxycamptothecin (HCPT) with internal standard camptothecin and liver tissues were used as model compounds and biological samples. The lactone form of HCPT was converted to the water-soluble carboxylate form under moderate alkaline conditions, and the water-insoluble interferences were extracted with a nonpolar solvent. Afterward, the water-insoluble lactone form of HCPT was regenerated by acidification and then extracted using an organic solvent in a second LLE step.
Results: The calibration curve was linear (r2 > 0.999) for HCPT concentrations ranging from 2.5 to 160 ng/mL. The mean recoveries of HCPT were 114.94 ± 3.98, 104.30 ± 2.44 and 95.90 ± 1.40% (n = 6) at concentrations of 2.5, 10 and 80 ng/mL, respectively. The stability determination data showed that no significant degradation occurred under the experimental conditions. This method was successfully applied to liver tissue distribution study of HCPT in mice.
Discussion and conclusion: This two-step LLE can be applied to distribution studies of compounds with pH-dependent interconvertible forms in other biological matrices.
Introduction
Sample preparation impacts nearly all the later analytical steps and is crucial for the accuracy, precision and robustness of the qualitative and quantitive bioanalysis. An efficient sample preparation also prolongs the life-time of instruments (e.g., HPLC instruments and chromatography columns) and in turn lowers the cost of assays (CitationChen et al., 2008). Numerous methods have been applied for sample preparation including protein precipitation (PP), Liquid–liquid extraction (LLE) and solid phase extraction (SPE) (CitationNováková & Vlčková, 2009). LLE is based on the distinct distribution of analytes between two immiscible solvents (CitationAlbaseer et al., 2010). Despite the disadvantages of large volumes of organic solvents and the poor potential for automation (CitationBjoërklund et al., 2002; CitationPalit et al., 2005), LLE was widely used for the extraction of aqueous samples (CitationNováková & Vlčková, 2009; CitationWu et al., 2010) because of its fast and effective separation of two phases, and especially high efficiency for the preparation of plasma sample (CitationAymard et al., 2000; CitationGauvin et al., 2000; CitationLiu et al., 2008; CitationSparreboom et al., 1998). Nevertheless, some shortcomings such as emulsion and its unsuitability for the removal of hydrophilic interfering substances restricted its application (CitationNováková & Vlčková, 2009). Another drawback of the conventional LLE may be its inapplicability for the preparation of biological matrices that contain large amounts of lipidic tissues, because lots of hydrophobic interferences would be co-extracted with the analytes (CitationPosyniak et al., 2001).
Plant secondary metabolites and many naturally occurring chemical compounds are widely used for medicinal and pharmacological purposes, such as paclitaxel, berberine, Vinca alkaloids, ephedrine and phenolic acids (CitationNardini & Ghiselli, 2004; CitationSingla et al., 2002; CitationTang et al., 2009). Some of these natural compounds are amphiphilic which have acidic or basic properties that allow them to be converted to water-soluble salts by pH adjustment (CitationAymard et al., 2000; CitationHertzberg et al., 1989; CitationHsiang et al., 1989; CitationJaxel et al., 1989; CitationNardini & Ghiselli, 2004). The pH adjustment in bioanalytical sample preparation is one of the most common sample treatments. Adjustment of the pH to less extreme values could be favorable for unstable compounds under basic or acidic conditions or in order to achieve different extraction selectivity. Proper pH adjustment of the sample to be extracted can be a powerful tool to achieve a different selectivity in the LLE (CitationHendriks et al., 2007, Citation2008). When proper pH adjustment introduced to these amphiphilic compounds, a two-step pH-dependent LLE can be used for sample preparation to remove water-soluble and lipidic interferences from the biological matrices.
Camptothecin (CPT, ) is a naturally occurring pentacyclic indole alkaloid first isolated from extracts of the Chinese tree Camptotheca acuminata Decne. (Nyssaceae) by CitationWall et al. (1966). CPT was the first compound that was reported to stop cell division by directly blocking the DNA replication enzyme topoisomerase I (CitationHsiang et al., 1985; CitationMontoro et al., 2010). CPT shows high tissue selectivity because, the level of topoisomerase I is higher in the malignant cells than in the normal cells (CitationThomas et al., 2004). CPT and its derivatives share the common feature of an E-ring lactone structure that could undergo a rapid, reversible and non-enzymatic hydrolysis to the water-soluble carboxylate form at basic pH and readily reconvert to the lactone form at acidic pH () (CitationHertzberg et al., 1989; CitationHsiang et al., 1989; CitationJaxel et al., 1989). The pharmacokinetics of CPTs in plasma samples by HPLC with fluorescence detection has been well established (CitationGuo et al., 2011a; CitationLiu et al., 2008; CitationLoh & Ahmed, 1990; CitationSun et al., 2004). The biological samples were prepared using relatively simple approaches such as protein precipitate with acetonitrile (ACN)/methanol (CitationFraier et al., 2000; CitationGuo et al., 2003), SPE (CitationD’Esposito et al., 2008; CitationFraier et al., 2000) or extraction with organic solvents using a conventional LLE (CitationKoshkina et al., 1999; CitationLiu et al., 2008; CitationSun et al., 2004; CitationZhao et al., 2006). However, the same sample preparation methods for plasma samples were used for tissue samples in most cases (CitationKoshkina et al., 1999; CitationWu & Zhang, 2001). Due to the different physiological profiles, liver and other tissues contain high hydrophobic compositions. A large quantity of interferences could be co-extracted with the analytes using the simple PP and conventional LLE process (CitationWu et al., 2010). Thus, a modification of the traditional sample preparation method is needed to determine the amount of CPTs in tissue samples.
Figure 1. Chemical equilibrium reaction between the lactone (a) and carboxylate (b) forms of CPTs (R=H, CPT; R=OH, HCPT).
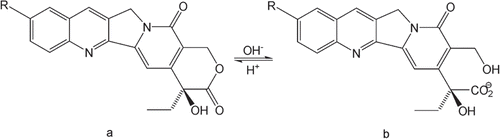
In the present study, we investigated the quantification of 10-hydroxycamptothecin (HCPT, ), a natural derivative of CPT with the same chemical property of pH-dependent lactone/carboxylate conversion. HCPT has been shown to be more biologically active and less toxic than CPT in animal experiments and human clinical evaluations (CitationWang et al., 2009; CitationZhang et al., 1998; CitationZunino & Pratesi, 2004). Liver tissue was chosen as a representative animal tissue with a high quantity of interferences. To separate HCPT from the hydrophobic interferences, the lactone form of the molecule was converted to the water-soluble carboxylate form under moderate alkaline conditions, and the hydrophobic interferences were removed by extracting with dichloromethane. Afterward, the water-insoluble lactone form was reformed by acidification and a second LLE was used to extract HCPT into diethyl ether. This sample preparation method separated HCPT from both hydrophilic and hydrophobic interferences and was suitable for tissues with highly hydrophobic compositions as well as plasma samples. Hence, a HPLC method with a two-step pH-dependent LLE to determine the HCPT concentration in liver tissues was developed and fully validated according to the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use guidelines set forth for bioanalytical method validation (CitationLiu et al., 2008). CPT was used as an internal standard (I.S.) for measuring HCPT concentrations by HPLC in this study.
Materials and methods
Chemicals and reagents
CPT and HCPT with purities of 99% were purchased from Harbin Foran High-Tech Development Ltd. (Harbin, Heilongjiang, China).
HPLC-grade ACN was obtained from Sigma-Aldrich (St. Louis, MO, USA). HPLC-grade water (resistivity 18.2 MΩ cm) was produced by purification of distilled water with a Milli-Q gradient system (Billerica, MA, USA). Other chemicals were of analytical reagent grade and were purchased from Sinopharm Chemical Reagent Co., Ltd (Shanghai, China).
Instruments and analysis conditions
A Waters HPLC system equipped with a 1525 binary pump, a 717 plus autosampler, and a 2475 fluorescence detector was used (Milford, MA, USA). Reverse-phase chromatography was performed on a Hypersil GOLD C18 column (5 µm, 250 × 4.6 mm, Thermo Fisher Scientific Inc., Waltham, MA, USA). The optimized method used a binary gradient mobile phase with water as mobile phase A and ACN as mobile phase B. A flow rate of 1.2 mL/min was used with 50 µL of injection volumes. The time program of the gradient was as follows: Phase B linearly increased from 20 to 60% in the first 12 min and from 60 to 90% within the next 3 min, then phase B decreased from 90 to 20% in 4 min and kept stably at 20% for 6 min. The total running time was 25 min including a 9 min equilibration time for each sample analysis. An injection of ACN with 10% acetic acid was used to wash the column every 3–5 injections. The eluent was monitored at the excitation and emission wavelengths of 383 and 553 nm (CitationGuo et al., 2011a,Citationb), respectively. Waters Empower software was used for system control and data acquisition.
Preparation of mouse liver homogenate
After collected blood via a cardiac puncture, the mouse was dissected and its liver tissues were excised. The liver tissue samples were carefully washed with an isotonic sodium chloride solution. The accurately-weighted tissue was homogenized in 1% aqueous sodium phosphate solution (tissue/solution 1/2, w/v) using a crux homogenizer and stored at −80°C until analysis.
Preparation of standard and quality control samples
A stock solution of HCPT (40 µg/mL) was prepared by dissolving 10 mg of HCPT in a 250 mL volumetric flask with ACN and further diluted to a series of working solutions with ACN. Calibration curves were prepared by spiking 100 µL of the appropriate standard solution with 100 µL of mouse liver tissue homogenate. The effective concentrations of HCPT in liver tissue homogenate samples were 2.5, 5, 10, 20, 40, 80 and 160 ng/mL. A stock solution of I.S. (CPT) was prepared in ACN at a concentration of 40 µg/mL using the same method as for HCPT (above), and a 20-fold dilution of the stock solution with ACN gave the working solution with a concentration at 200 ng/mL. Quality control (QC) samples were prepared in pool as a single batch for each concentration at concentrations of 2.5 (low), 10 (medium) and 80 ng/mL (high), then aliquoted and stored in the freezer at −80°C until analysis. The spiked liver tissue homogenate samples (standards and quality controls) were treated following the same sample processing procedure as that for unknown samples.
Sample processing
One-hundred microliters of liver tissue homogenate and 100 µL of I.S. working solution (CPT, 200 ng/mL) were mixed in a 2 mL test tube. Two-hundred microliters of 1% trisodium phosphate solution was added to the mixture, and it was incubated for 30 min in an ultra-sonic bath. Following this, 500 µL of dichloromethane was added to the mixture, and then vortexed for 20 s. The mixture was incubated at room temperature for 30 min on a shaker and then centrifuged at 13,000g for 15 min. The upper layer was transferred to a clean test tube, mixed with 120 µL of 1% phosphoric acid solution and extracted with 700 µL of diethyl ether twice in a shaker at room temperature for 30 min. The organic layers were combined in a clean tube after centrifuging at 13,000g for 10 min, and dried in an MCFD5-3B freeze dryer (SIM International Group Co., Newark, DE, USA). The dried extract was reconstituted in 200 µL of ACN. After centrifuging at 13,000g for 10 min, a 50 µL aliquot was injected into the chromatographic system using a Waters 717 plus autosampler.
Method validation
Liver tissue homogenate samples were quantified using the peak area ratio of HCPT to that of the I.S. (CPT). A standard curve in the form of y = A x + B was used, where x represents HCPT concentration in the liver tissue homogenates and y represents the ratio of the HCPT peak area to that of the I.S. (CPT). A linear least-squares regression analysis was conducted to determine the slope, intercept and coefficient of determination (r2) to demonstrate the linearity of this method. To evaluate the linearity, liver tissue homogenate calibration curves were prepared and analysed in triplicate on 5 consecutive days.
The accuracy and precision were also determined by replicate analyses (n = 6) of QC samples at three concentration levels (2.5, 10 and 80 ng/mL) on different validation days. The accuracy was expressed by (mean observed concentration)/(spiked concentration) × 100% and the precision by relative standard deviation (RSD %). The concentration of each sample was determined by using the calibration curve and analysed on the same day.
The recovery of HCPT was evaluated by analysing QC samples at three different concentrations as described above and comparing the area ratio of peaks for HCPT and I.S. with those for neat solutions of standard compounds without extraction.
Stability
The stability of analytes was examined by keeping replicates of the liver tissue homogenate QC samples on bench at room temperature for 2 h, in the autosampler tray for 24 h, and in a freezer at −80°C for 30 days; the freeze-thaw stability was obtained over three freeze-thaw cycles, by thawing at room temperature for 2–3 h and then refreezing for 12–24 h. The stability of the standard solutions was tested after standing at room temperature for 4 h and after refrigeration at 4°C for 30 days. For each concentration and each storage condition, six replicates were analysed in one analytical batch. The concentration of HCPT after each storage period was related to the initial concentration, which was determined when the samples were originally prepared and processed.
Liver tissue distribution studies in mice
The concentrations of HCPT in mouse liver tissue homogenate samples were determined by using the HPLC method described above. Groups of 48 healthy male mice (20–25 g) were chosen for the tissue distribution study (approved by the Animal Ethical Committee in Northeast Forestry University). An oral formulation of HCPT (8 mg/mL) was prepared by mixing HCPT in cottonseed oil via constantly grinding with an agate mortar and pestle. After fasting for 12 h, each mouse was orally administrated the appropriate dosage of HCPT (80 mg/kg) by gavage. A series of liver samples were collected into tubes before and at 0.17, 0.33, 0.67, 1.0, 1.33, 1.67, 2.0, 4.0, 8.0, 12 and 24 h time points after administration. The liver tissue samples were washed with an isotonic sodium chloride solution, and homogenized in 1% aqueous sodium phosphate (tissue/solution 1/2, w/v) with a crux homogenizer and stored frozen at −80°C until analysis.
Statistical analysis
A noncompartmental pharmacokinetic analysis using the Kinetica™ software package (version 5.0, Thermo Fisher Scientific Inc., MA, USA) was performed to determine key parameters including the maximum plasma concentration (Cmax), the time-to-maximum concentration (Tmax), the elimination half-life (T1/2), the mean residence time (MRT), the area under the plasma concentration-time curve from zero to 24 h (AUC0–24), and the area under the plasma concentration-time curve from time zero to infinity (AUC0–∞).
Results and discussion
Sample preparation
Some of plant secondary metabolites with acidic or basic properties allow them to be converted into water-soluble salt when pH value was adjusted (CitationAymard et al., 2000; CitationHertzberg et al., 1989; CitationHsiang et al., 1989; CitationJaxel et al., 1989; CitationNardini & Ghiselli, 2004). For these compounds, a two-step pH-dependent LLE can be used for sample preparation in order to remove water-soluble and lipidic interferences from the biological matrices. Both CPT and its natural derivative HCPT have the chemical properties of pH-dependent conversion between lactone and carboxylate forms. HCPT (with CPT as an I.S.) and liver tissue were chosen as the model compounds and biological matrices, respectively.
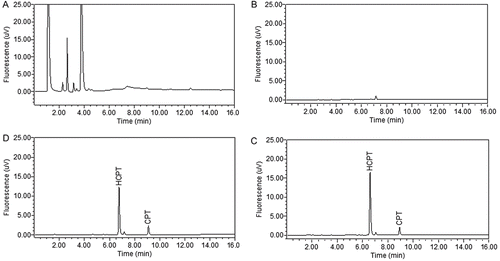
The two main steps for sample preparation in this study were a basic LLE followed by an acidic LLE. To separate HCPT from hydrophobic interferences, a basic aqueous solution was used to convert the lactone form of the analyte and I.S. into the water-soluble carboxylate form. Strong, moderate and weak bases, specifically sodium hydroxide, sodium phosphate, and sodium carbonate were tested at different concentrations (0.1–5%). It was proven that HCPT and CPT could be fully converted and hydrophobic interferences, such as lipid, were not affected by 1% sodium phosphate solution under ultrasonication. Dichloromethane was used as the organic phase in the first step of LLE to remove the hydrophobic interferences from HCPT and I.S. which remained in the water layer with their carboxylate forms. Next, 1% phosphoric acid was used to transform the carboxylate form of the analyte back into its lactone form. Finally, diethyl ether was added to extract HCPT and I.S. Ethyl acetate, diethyl ether and the mixtures of these two solvents in different ratios (20/80 and 50/50, v/v) were evaluated for extraction efficiency. Diethyl ether was found to be the optimal extraction solvent. Repetition of the second step of extraction was essential for a good recovery. To evaluate the necessity of the first step of LLE, extraction processes were also performed with and without this step. As shown in the chromatograms in , several interfering peaks were detected when the first step of LLE was omitted (, and the method was found to be lack of linearity and with lower sensitivity, whereas a much cleaner chromatogram was obtained when the first step of LLE was performed (. Although an endogenous peak was presented in blank liver tissue samples with a retention time close to that of HCPT, these two peaks were baseline resolved and there was no interference for the quantification of HCPT.
Figure 2. Representative chromatograms of: (A) a blank mouse liver tissue homogenate sample without the first LLE step; (B) a blank mouse liver tissue homogenate sample using the two-step LLE; (C) a blank mouse liver tissue homogenate spiked with HCPT (20 ng/mL) and I.S. (CPT 200 ng/mL); (D) a mouse liver tissue homogenate sample at 4 h after oral administration of HCPT at a dose of 80 mg/kg and spiked with I.S. (CPT 200 ng/mL).
Chromatography
Different chromatographic conditions including chromatographic columns have been evaluated. A good separation was obtained with a Hypersil GOLD C18 column using the binary gradient program described above. It was demonstrated that no endogenous substances interfered with the quantification of analytes in the liver tissue homogenates. Under this chromatographic condition, the monitoring time was 16 min followed by a 9 min equilibration time for each sample analysis. The retention times were 6.77 and 9.05 min for HCPT and CPT, respectively.
Method validation
Selectivity and specificity
Selectivity was assessed by evaluating the differences between six different batches of the spiked liver tissue homogenates sample and blank tissue homogenates. The representative chromatograms of a blank liver tissue homogenate, a blank liver tissue homogenate spiked with HCPT (20 ng/mL) and I.S., and a liver tissue homogenate sample after 4 h of oral administration of HCPT (80 mg/kg) are shown in . No interfering peak at the retention time of analytes, HCPT and I.S. was observed in all samples. The specificity was verified by comparing retention time of HCPT and CPT in QC samples (n = 10). The differences were less than 1%.
Linearity of calibration curves and lower limits of quantification
The calibration curve in this study was established by plotting the ratio of the peak areas of HCPT and I.S. against the expected concentration of calibration standards spiked in liver tissue homogenates. The linear-regression correlation coefficients were greater than 0.999 over the concentration range of 2.5–160 ng/mL in all validation runs. shows the classic linear calibration curve obtained for HCPT in liver tissue homogenate over the concentration range.
Figure 3. A representative calibration curve of HCPT in liver tissue homogenate samples.
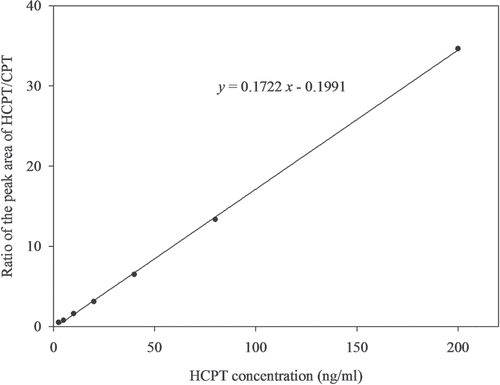
The lower limits of quantification (LLOQ) was defined as the lowest concentration on the calibration curve at which an acceptable accuracy of ±15% and a precision below 15% was obtained (CitationLiu et al., 2008). The present method provided a LLOQ of 2.5 ng/mL in 0.1 mL liver tissue homogenate samples. HCPT can be detected in liver tissue homogenate samples even 24 h after administration using this sensitive method. Thus, this method is sensitive enough to investigate the distribution of HCPT in liver tissue.
Precision and accuracy
The intra- and inter-day precision and accuracy for HCPT analysis were evaluated by assaying the QC samples (2.5, 10 and 80 ng/mL) in 5 consecutive days. The results are presented in . The inter-day precision was lower than 8.14%, and the accuracy was between 98.80 and 102.20%. The intra-day precision was lower than 10.02%, and the accuracy was between 96.80 and 99.98%. These data suggested that the method was accurate and precise for the quantitative analysis of HCPT in mouse liver tissue samples.
Table 1. Accuracy and precision for the analysis of HCPT in mouse liver tissue homogenates.
Recovery and stability
The recoveries of HCPT spiked into mouse liver tissue homogenates were determined at three QC sample concentrations (2.5, 10 and 80 ng/mL) and were 114.94 ± 3.98, 104.3 ± 2.44 and 95.9 ± 1.4% (n = 6), respectively.
The stability of HCPT was evaluated as described in the method section. The data are shown in , which suggested that no significant degradation occurred under the experimental conditions.
Table 2. Stability of HCPT in liver tissue homogenate samples (n = 6).
Application of the method to liver tissue distribution study
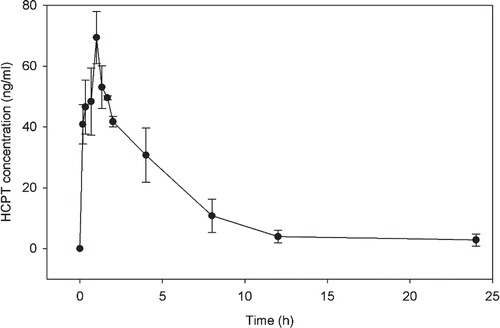
In this study, the mean concentration-time curve of HCPT in liver tissue homogenates was determined for four mice each receiving a single dose of 80 mg/kg. The curve and pharmacokinetic parameters of HCPT are shown in and , respectively. After oral administration of HCPT, the concentration of HCPT in the liver tissue homogenates reached the peak at Tmax of 1.0 h, which was later than that in plasma (0.17 h) base on the result of our related research (CitationGuo et al., 2011a,Citationb). This suggested that HCPT can quickly distribute to tissues after it was absorbed into the bloodstream. The concentration increased quickly to reach the peak concentration and then decreased slowly after 1 h. The Cmax values of HCPT were about 69.40 ± 8.59 ng/mL, i.e., there was about 138.8 ng HCPT in 1 g of live tissue. This value was four times higher than the mount of HCPT in mouse plasma samples (17.80 ± 7.14 ng/mL) in our previous study (CitationGuo et al., 2011a,Citationb), which suggested an accumulation of HCPT occurred in mouse liver tissues. The elimination half-time (T1/2, 4.73 ± 0.73 h) and the MRT (6.25 ± 1.17 h) of HCPT in liver tissue homogenates were both shorter than that in plasma samples (9.03 ± 2.58 h and 14.33 ± 4.56 h, respectively) (CitationGuo et al., 2011a,Citationb). The ratio (%) of the AUC0–24 value to the AUC0–∞ value was about 96.22% for HCPT in liver tissue. These results indicated that most HCPT was quickly eliminated from the liver tissue.
Table 3. The pharmacokinetic parameters of HCPT after oral administration of 80 mg/kg of HCPT.
Figure 4. Mean concentration-time profiles of HCPT in mouse liver tissue homogenate samples from four subjects each receiving a single dose of 80 mg/kg HCPT.
Conclusion
It is possible to interconvert some plant secondary metabolites between water-insoluble and water-soluble forms by pH adjustment. A sensitive, accurate and precise RP-HPLC with fluorescence detection was developed and validated using HCPT with I.S. (CPT) and mouse liver tissues as model compounds and biological matrices. A two-step pH-dependent LLE was employed sample preparation. This LLE process could separate HCPT from hydrophobic or hydrophilic interferences by dissolving HCPT in the basic aqueous solution and then the lactone form in an organic solvent at acid condition. This method was convenient for the quantification of HCPT in mouse liver tissue homogenate samples and fully validated. In addition, this method was also successfully applied to the determination of liver distribution profiles of HCPT in male mice with a LLOQ of 2.5 ng/mL. This modified LLE method can also be applied to distribution studies of other compounds with pH-dependent interconvertible forms in biological matrices, especially tissue samples.
Declaration of interest
This research is financially supported by the “948” Project of the State Forestry Administration (2011-4-16), the Scientific Research Foundation of Heilongjiang Province for the Returned Overseas Scholars (No. LC 201004) and the Cultivation Program of the Northeast Forestry University for Excellent Doctoral Dissertation.
References
- Albaseer SS, Rao RN, Swamy YV, Mukkanti K. (2010). An overview of sample preparation and extraction of synthetic pyrethroids from water, sediment and soil. J Chromatogr A, 1217, 5537–5554.
- Aymard G, Labarthe B, Warot D, Berlin I, Diquet B. (2000). Sensitive determination of ephedrine and norephedrine in human plasma samples using derivatization with 9-fluorenylmethyl chloroformate and liquid chromatography. J Chromatogr B Biomed Sci Appl, 744, 25–31.
- Bjoërklund E, Holst CV, Anklam E. (2002). Fast extraction, clean-up and detection methods for the rapid analysis and screening of seven indicator PCBs in food matrices. Trends Analyt Chem, 21, 39–52.
- Chen Y, Guo Z, Wang X, Qiu C. (2008). Sample preparation. J Chromatogr A, 1184, 191–219.
- D’Esposito F, Tattam BN, Ramzan I, Murray M. (2008). A liquid chromatography/electrospray ionization mass spectrometry (LC-MS/MS) assay for the determination of irinotecan (CPT-11) and its two major metabolites in human liver microsomal incubations and human plasma samples. J Chromatogr B Analyt Technol Biomed Life Sci, 875, 522–530.
- Fraier D, Frigerio E, Brianceschi G, Casati M, Benecchi A, James C. (2000). Determination of MAG-camptothecin, a new polymer-bound camptothecin derivative, and free camptothecin in dog plasma by HPLC with fluorimetric detection. J Pharm Biomed Anal, 22, 505–514.
- Gauvin A, Pinguet F, Poujol S, Astre C, Bressolle F. (2000). High-performance liquid chromatographic determination of vinorelbine in human plasma and blood: Application to a pharmacokinetic study. J Chromatogr B Biomed Sci Appl, 748, 389–399.
- Guo H, Zheng J, Jing L, Li S, Yan X, Wang Y. (2011a). Determination of the plasma concentration of 10-hydroxycamptothecin in mice by RP-HPLC. China Pharm, 22, 13–15.
- Guo S, Zheng J, Dong J, Guo N, Jing L, Yue X, Yan X, Wang Y, Dai Z. (2011b). Iron/dextran sulfate multilayered microcapsules for controlled release of 10-hydroxycamptothecin. Int J Biol Macromol, 49, 409–415.
- Guo W, Ahmad A, Khan S, Dahhani F, Wang YF, Ahmad I. (2003). Determination by liquid chromatography with fluorescence detection of total 7-ethyl-10-hydroxy-camptothecin (SN-38) in beagle dog plasma after intravenous administration of liposome-based SN-38 (LE-SN38). J Chromatogr B Analyt Technol Biomed Life Sci, 791, 85–92.
- Hendriks G, Uges DR, Franke JP. (2007). Reconsideration of sample pH adjustment in bioanalytical liquid-liquid extraction of ionisable compounds. J Chromatogr B Analyt Technol Biomed Life Sci, 853, 234–241.
- Hendriks G, Uges DR, Franke JP. (2008). pH adjustment of human blood plasma prior to bioanalytical sample preparation. J Pharm Biomed Anal, 47, 126–133.
- Hertzberg RP, Caranfa MJ, Holden KG, Jakas DR, Gallagher G, Mattern MR, Mong SM, Bartus JO, Johnson RK, Kingsbury WD. (1989). Modification of the hydroxy lactone ring of camptothecin: Inhibition of mammalian topoisomerase I and biological activity. J Med Chem, 32, 715–720.
- Hsiang YH, Hertzberg R, Hecht S, Liu LF. (1985). Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J Biol Chem, 260, 14873–14878.
- Hsiang YH, Liu LF, Wall ME, Wani MC, Nicholas AW, Manikumar G, Kirschenbaum S, Silber R, Potmesil M. (1989). DNA topoisomerase I-mediated DNA cleavage and cytotoxicity of camptothecin analogues. Cancer Res, 49, 4385–4389.
- Jaxel C, Kohn KW, Wani MC, Wall ME, Pommier Y. (1989). Structure-activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: Evidence for a specific receptor site and a relation to antitumor activity. Cancer Res, 49, 1465–1469.
- Koshkina NV, Gilbert BE, Waldrep JC, Seryshev A, Knight V. (1999). Distribution of camptothecin after delivery as a liposome aerosol or following intramuscular injection in mice. Cancer Chemother Pharmacol, 44, 187–192.
- Liu X, Wang Y, Vardeman D, Cao Z, Giovanella B. (2008). Development and validation of a reverse-phase HPLC with fluorescence detector method for simultaneous determination of CZ48 and its active metabolite camptothecin in mouse plasma. J Chromatogr B Analyt Technol Biomed Life Sci, 867, 84–89.
- Loh JP, Ahmed AE. (1990). Determination of camptothecin in biological fluids using reversed-phase high-performance liquid chromatography with fluorescence detection. J Chromatogr, 530, 367–376.
- Montoro P, Maldini M, Piacente S, Macchia M, Pizza C. (2010). Metabolite fingerprinting of Camptotheca acuminata and the HPLC-ESI-MS/MS analysis of camptothecin and related alkaloids. J Pharm Biomed Anal, 51, 405–415.
- Nardini M, Ghiselli A. (2004). Determination of free and bound phenolic acids in beer. Food Chem, 84, 137–143.
- Nováková L, Vlcková H. (2009). A review of current trends and advances in modern bio-analytical methods: Chromatography and sample preparation. Anal Chim Acta, 656, 8–35.
- Palit M, Pardasani D, Gupta AK, Dubey DK. (2005). Application of single drop microextraction for analysis of chemical warfare agents and related compounds in water by gas chromatography/mass spectrometry. Anal Chem, 77, 711–717.
- Posyniak A, Zmudzki J, Semeniuk S. (2001). Effects of the matrix and sample preparation on the determination of fluoroquinolone residues in animal tissues. J Chromatogr A, 914, 89–94.
- Singla AK, Garg A, Aggarwal D. (2002). Paclitaxel and its formulations. Int J Pharm, 235, 179–192.
- Sparreboom A, de Bruijn P, Nooter K, Loos WJ, Stoter G, Verweij J. (1998). Determination of paclitaxel in human plasma using single solvent extraction prior to isocratic reversed-phase high-performance liquid chromatography with ultraviolet detection. J Chromatogr B Biomed Sci Appl, 705, 159–164.
- Sun J, Wang Y, Zhang Z. (2004). Determination of 10-hydroxycamptothecin in tissues of rats by HPLC-FD. J China Pharm Univ, 35, 540–544.
- Tang J, Feng Y, Tsao S, Wang N, Curtain R, Wang Y. (2009). Berberine and Coptidis rhizoma as novel antineoplastic agents: A review of traditional use and biomedical investigations. J Ethnopharmacol, 126, 5–17.
- Thomas CJ, Rahier NJ, Hecht SM. (2004). Camptothecin: Current perspectives. Bioorg Med Chem, 12, 1585–1604.
- Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AT, Sim GA. (1966). Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from Camptotheca acuminata. J Amer Chem Soc, 88, 3888–3890.
- Wang J, Wang R, Li LB. (2009). Preparation and properties of hydroxycamptothecin-loaded nanoparticles made of amphiphilic copolymer and normal polymer. J Colloid Interface Sci, 336, 808–813.
- Wu J, Lu J, Wilson C, Lin Y, Lu H. (2010). Effective liquid-liquid extraction method for analysis of pyrethroid and phenylpyrazole pesticides in emulsion-prone surface water samples. J Chromatogr A, 1217, 6327–6333.
- Wu Y, Zhang G. (2001). Pharmacokinetics and tissue distribute ion of HCPT capsules in mice. China Pharm, 12, 589–590.
- Zhang R, Li Y, Cai Q, Liu T, Sun H, Chambless B. (1998). Preclinical pharmacology of the natural product anticancer agent 10-hydroxycamptothecin, an inhibitor of topoisomerase I. Cancer Chemother Pharmacol, 41, 257–267.
- Zhao YX, Gao JQ, Qiao HL, Chen HL, Liang WQ. (2006). Development and validation of a sensitive reversed-phase HPLC method to determine intracellular accumulation of hydroxycamptothecin. J Pharm Biomed Anal, 41, 1007–1010.
- Zunino F, Pratesi G. (2004). Camptothecins in clinical development. Expert Opin Investig Drugs, 13, 269–284.