Abstract
Context: Quassinoids are biologically active secondary metabolites found exclusively in the Simaroubaceae family of plants. These compounds generally present important biological properties, including cytotoxic and antitumor properties.
Objective: In the present study, the cytotoxic effects of neosergeolide, a quassinoid isolated from Picrolemma sprucei Hook. f., were evaluated in human promyelocytic leukemia cells (HL-60).
Materials and methods: Cytotoxicity and antiproliferative effects were evaluated by the MTT assay, May-Grünwald-Giemsa’s staining, BrdU incorporation test, and flow cytometry procedures. The comet assay and micronuclei analysis were applied to determine the genotoxic and mutagenic potential of neosergeolide.
Results: After 24 h exposure, neosergeolide strongly inhibited cancer cell proliferation (IC50 0.1 µM), and its activity seemed to be selective to tumor cells because it had no antiproliferative effect on human peripheral blood mononuclear cells (PBMC) at tested concentrations. Apoptosis was induced at submicromolar concentrations (0.05, 0.1, and 0.2 µM) as evidenced by morphological changes, mitochondrial depolarization, phosphatidylserine externalization, caspases activation, and internucleosomal DNA fragmentation. Additionally, neosergeolide effects were prevented by cyclosporine A (CsA), an inhibitor of the mitochondrial permeability transition (MPT) pore, which reinforced the participation of intrinsic pathways in the apoptotic process induced by this natural quassinoid. Direct DNA damage was further confirmed by comet assay and cytokinesis-block micronucleus test.
Discussion and conclusion: The present study provided experimental evidence to support the underlying mechanism of action involved in the neosergeolide-mediated apoptosis. In addition, no antiproliferative effect or DNA damage effect of neosergeolide was evident in PBMC, highlighting its therapeutic potential.
Keywords::
Introduction
Quassinoids are degraded triterpenes frequently highly oxygenated (CitationPolonsky et al., 1980; CitationJiwajinda et al., 2002), found exclusively in plants belonging to the Simaroubaceae family. These compounds generally presented important biological properties (CitationOkano et al., 1990; CitationGuo et al., 2005a), including antimalarial (CitationGuo et al., 2005b; CitationBertani et al., 2006; CitationSilva et al., 2009), anti-HIV (CitationOkano et al., 1996), and cytotoxic and antitumor activities (CitationTischler et al., 1992; CitationItokawa et al., 1993; CitationMata-Greenwood et al., 2001; CitationMurakami et al., 2004; Citationvon Bueren et al., 2007; CitationSilva et al., 2009). Since the use of bruceantin (CitationKupchan et al., 1973) in phase II breast cancer and melanoma clinical trials and its subsequent withdrawal due to the concerns about toxicity (CitationWiseman et al., 1982; CitationArseneau et al., 1983), studies on the antitumor properties of quassinoids have increased steadily (CitationOkano et al., 1985; CitationMata-Greenwood et al., 2001; CitationGuo et al., 2005a).
Recently, it has been reported that neosergeolide (), quassinoid isolated from Picrolemma sprucei Hook. f. (Simaroubaceae), has significant in vitro antimalarial, larvicide, and cytotoxic properties (CitationSilva et al., 2009). Also, neosergeolide nonselectively inhibited cancer cell lines proliferation (SF295 (glioblastoma), MDA-MB435 (melanoma), HCT8 (colon), and HL-60 (leukemia)) as evidenced by quite similar IC50 values. In the present study, the underlying molecular mechanisms of neosergeolide’s antiproliferative activity were evaluated using human promyelocytic leukemia cells (HL-60). The HL-60 cells are extensively used in the examination of the effects of test drugs on cell proliferation, cell cycle, cell differentiation, and apoptosis events (CitationCollins, 1987; CitationMilitão et al., 2006). For comparison, the effect of neosergeolide on the growth of normal cells was evaluated using peripheral blood mononuclear cells (PBMC).
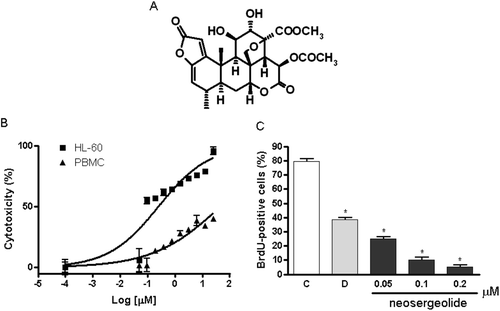
Figure 1. Chemical structure of neosergeolide (A), concentration-response curve of neosergeolide cytotoxicity (%) after 24 h exposure of HL-60 and PBMC (B), and its effects on 5-bromo-2-deoxyuridine (BrdU) incorporation (C) by HL-60 cells. *p < 0.001 compared to control by ANOVA followed by Student Newman–Keuls test. Data are presented as means ± S.E.M. for three independent experiments in triplicate.
Materials and methods
Drugs and reagents
The procedure used for the isolation of neosergeolide from the roots and stems of P. sprucei was described in a previous publication (CitationAndrade-Neto et al., 2007). Neosergeolide used in this study had purity greater than 98% based on HPLC (DAD/ESI-MS) and NMR analyses. Fetal calf serum and phytohemagglutinin were purchased from Cutilab (Campinas, SP, Brazil). RPMI 1640 medium, trypsin-EDTA, penicillin, and streptomycin were purchased from GIBCO® (Invitrogen, Carlsbad, CA, USA). Cytochalasin-B (Cyt-B), cyclosporine A (CsA), sulfanilamide, rhodamine 123 (Rho-123), and MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). Doxorubicin (Doxolem®) was purchased from Zodiac Produtos Farmacêuticos S. A. (São Paulo, SP, Brazil). All other chemicals and reagents were of analytical grade.
Cell cultures
The human promyelocytic leukemia cell line (HL-60) was donated by the National Cancer Institute (Bethesda, MD, USA). Cells were maintained in RPMI 1640 medium supplemented with 10% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin at 37°C with 5% CO2. Heparinized blood from healthy, non-smoker donors who had not taken any drug at least 15 days prior to sampling was collected and PBMC were isolated using density-gradient centrifugation over Histopaque-1077. The PBMC were washed and re-suspended in RPMI 1640 medium supplemented with 20% fetal bovine serum, 2 mM glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin at 37°C under a 5% CO2 atmosphere. Phytohemagglutinin (2%) was added at the start of cultures. After 24 h, cells were treated with the test compounds.
Neosergeolide toxicity to HL-60 cells and PBMC
The cytotoxicity of neosergeolide to HL-60 cells and PBMC was evaluated by the MTT assay (CitationMosmann, 1983). Briefly, cells were plated in 96-well plates (3 × 105 cells/mL for HL-60 and 4 × 105 cells/mL for PBMC). Neosergeolide was dissolved in 1% DMSO at concentrations of 0.006–4 μM and the resulting solutions were added to wells. After 24 h, the supernatant was replaced by fresh medium containing MTT (0.5 mg/mL). After 3 h, the MTT formazan product was dissolved in DMSO and absorbance was measured at 595 nm (Beckman Coulter® DTX-880 spectrometer). Doxorubicin (0.044–46 μM) was used as positive control.
Antiproliferative effect (inhibition of BrdU incorporation)
The HL-60 cells were plated in 24-well tissue culture plates (3 × 105 cells/mL) and treated with neosergeolide at different concentrations. After 21 h of exposure to sample, 20 μL of BrdU (10 mM) was added to each well and incubated for 3 h at 37°C. To determine the amount of BrdU incorporated into DNA (CitationPera et al., 1977), cells were harvested, transferred to cytospin slides and allowed to dry for 2 h at room temperature. Cells that had incorporated BrdU were labeled by direct peroxidase immunocytochemistry using the chromogen diaminobenzidine (DAB). Slides were counterstained with hematoxylin, mounted, and coverslipped. Determination of BrdU positivity was performed by light microscopy (Olympus, Tokyo, Japan). A total of 200 cells were counted per sample to determine the percentage of BrdU-positive cells. Doxorubicin (0.6 μM) was used as positive control.
Analysis of morphological changes
Untreated or neosergeolide-treated HL-60 cells were examined for morphological changes by light microscopy (Metrimpex Hungary/PZO-Labimex Modelo Studar Lab®). To evaluate morphology, cells were harvested, transferred to cytospin slides, fixed with ethanol for 1 min and stained with May-Grünwald-Giemsa. Doxorubicin (0.6 μM) was used as positive control.
Flow cytometric experiments
Cell membrane integrity
The HL-60 cell membrane integrity was evaluated by the exclusion of propidium iodide (PI) at 50 μg/mL. Aliquots were removed from cultures after 3, 6, 12, and 24 h of incubation. Cell fluorescence was then determined by flow cytometry in a Guava EasyCyte Mini (Guava Technologies, Inc., Hayward, CA, USA) using Guava Express Plus software. A total of 5,000 events were evaluated per experiment.
Internucleosomal DNA fragmentation
Aliquots were removed from HL-60 cell cultures after 3, 6, 12, and 24 h of incubation with neosergeolide. Then, the aliquots were incubated at 37°C for 30 min in the dark in a lysis solution containing 0.1% citrate, 0.1% Triton X-100, and 50 μg/mL PI. Cell fluorescence was then determined by flow cytometry in a Guava EasyCyte Mini (Guava Technologies, Inc., Hayward, CA, USA) using Guava Express Plus software. The percentage of degraded DNA was determined by the number of cells displaying subdiploid (sub-Go/G1) DNA divided by the total number of cells examined. A total of 5,000 events were evaluated per experiment.
Mitochondrial transmembrane potential (Δψm)
Mitochondrial depolarization was evaluated after 3, 6, 12, and 24 h of incubation with neosergeolide using the method of incorporation of Rho-123. Rho-123 is a cell-permeable, cationic, fluorescent dye that is readily sequestered by active mitochondria without inducing cytotoxic effects. Briefly, treated and untreated HL-60 cells were centrifuged at 2000 rpm for 5 min and the pellet was re-suspended in 200 μL of a 1 μg/mL solution of Rho-123 for 15 min in the dark. After incubation, cells were centrifuged at 2000 rpm for 5 min. The resulting pellet was re-suspended in 200 μL of phosphate-buffered saline (PBS) and incubated for 30 min in the dark. Fluorescence was measured and percentage of mitochondrial depolarization was determined (CitationCury-Boaventura et al., 2004).
Annexin V/PI and caspases (9, 3, and 7) detection
The Annexin V (AnnV) cytometry assay was used to detect cell population in viable, early and late apoptosis stage. After short exposure time (3 h) or pulse treatment (3 h of neosergeolide exposure following 21 h reincubation period without drug), HL-60 cells were stained with fluorescein isothiocyanate (FITC) conjugated AnnV (Guava Nexin kit, Guava Technologies, Inc., Hayward, CA, USA) and PI (necrotic-cell indicator), and then they were subjected to flow cytometry (Guava EasyCyte Mini). Cells undergoing early and late apoptosis were detected by the emission of the fluorescence from only FITC and, both FITC and PI, respectively. Also, the percentage of cells with active caspases (9, 3, and 7) were estimated by flow cytometry (Guava EasyCyte Mini) using a Guava Caspases 9 and 3/7 FAM Kit (Guava Technologies, Inc., Hayward, CA, USA).
Analysis of mitochondrial permeability transition on neosergeolide-induced cell death
To corroborate the central role of mitochondria in the apoptotic process induced by neosergeolide, cells were pretreated or not for 30 min with CsA at 5 μM [a blocker of mitochondrial permeability transition (MPT)] prior to the neosergeolide exposure and cotreated for 3 h. After exposure time, mitochondrial depolarization, internucleosomal DNA fragmentation, active caspases (9, 3, and 7) detection, and morphological analysis of cell death (AO/EB staining) were performed as described above.
Measurement of intracellular reactive oxygen species
Intracellular reactive oxygen species (ROS) were estimated after treatment with neosergeolide using 2′,7′-dichlorofluorescein diacetate (H2DCFDA) as fluorescence probe. The H2DCFDA diffuses through the cell membrane readily and is hydrolyzed by intracellular esterases to non-fluorescent dichlorofluorescein (DCFH), which is then rapidly oxidized to highly fluorescent DCFH (2′,7′-dichlorofluorescein) by a broad range of intracellular oxidative stresses other than H2O2 (CitationCrow, 1997; CitationHempel et al., 1999). Therefore, increased mean fluorescence intensity of DCF represents a probe of oxidation by a broad range of oxidative events and not only reaction with H2O2. At different exposure times (3 and 24 h), the culture medium was replaced by fresh serum-free medium containing 20 μM H2DCFD. DCF fluorescence intensity was detected by flow cytometry using a Guava EasyCyte Mini (Guava Technologies, Inc., Hayward, CA, USA) and Guava Express Plus software. The DCF fluorescence intensity is proportional to the amount of intracellularly formed ROS (CitationLeBel et al., 1992).
DNA damage analysis
Micronuclei assay
After pulse treatment (3 h), HL-60 cells were incubated in complete medium for 48 h and Cyt-B (3 μg/mL) was treated at 24 h. Cells were harvested and re-suspended in a hypotonic solution (0.075 M KCl) for 10 min. Afterward, HL-60 cells were harvested again and Carnoy’s fixative was added gently. Then, cells were dropped onto clean slides and air-dried. These slides were stained with 8% Giemsa solution (pH 6.8) and then observed under a light microscope. Micronuclei (MN) were reported per 1,000 binucleated cells (BNC) with well-preserved cytoplasm (CitationEckhardt et al., 1994). To verify the genotoxic potential of neosergeolide against PBMC, the same protocol used for leukemia cells was applied.
Alkaline comet assay
The comet assay was conducted under alkaline conditions as described by CitationSingh et al. (1988) with modifications (CitationKlaude et al., 1996) and following the recommendations of the International Workshop on Genotoxicity Test Procedures (CitationTice et al., 2000). After pulse treatment (3 h), HL-60 cells were collected and processed for the assay as follows. Briefly, 15 μL of the cell suspension were mixed with 90 μL of 0.75% low melting point agarose in PBS at 37°C; 100 µL of the cell suspension were spread on a glass slide previously coated with a layer of 1.5% normal melting point agarose in PBS, covered with a glass coverslip and placed at 4°C for 15 min. The coverslip was gently removed and the slide was submerged into ice-cold lysing solution (2.5 M NaCl, 10 mMTris, 0.1 mM EDTA, 1% sodium sarcosinate, 1% Triton X-100, and 10% DMSO, pH 10) at 4°C for at least 1 h. After lysis, the slides were placed in a horizontal gel electrophoresis chamber with freshly prepared alkaline buffer (300 mM NaOH and 1 mM EDTA, pH >13.0). The slides were kept in this solution for 20 min at 4°C to allow unwinding of the DNA and expression of alkali-labile sites. Then, the samples were subjected to electrophoresis in the same solution at 300 mA, 0.81 V/cm for 20 min at 4°C. After electrophoresis, the slides were rinsed gently three times (5 min each time) with 0.4 M Tris-HCl (pH 7.5). Each slide was stained with 50 μL of ethidium bromide (20 μg/mL) and covered with a coverslip. The analysis of the cells was performed by a visual scoring system (CitationMiyamae et al., 1998). Briefly, fluorescently stained nucleoids were scored visually using an epifluorescence microscope (Olympus, Tokyo, Japan) with an excitation filter of 510–560 nm and a barrier filter of 590 nm at 400× magnification.
A total of 300 randomly selected cells (100 cells from each of the three replicate slides) were analyzed for each concentration of test substance. Cells were scored visually according to tail length into five classes: (1) class 0: undamaged cells having no tail; (2) class 1: cells having a tail shorter than the diameter of the head (nucleus); (3) class 2: cells having a tail length 1–2 times the diameter of the head; (4) class 3: cells having a tail longer than 2 times the diameter of the head; (5) class 4: comets having no heads. A value (damage index, DI) was assigned to each comet according to its class, using the formula:
DI = (0 × n0) + (1 × n1) + (2 × n2) + (3 × n3) + (4 × n4),
where n = number of cells in each class analyzed. Damage index thus ranged from 0 (completely undamaged: 100 cells × 0) to 400 (with maximum damage: 100 cells × 4) (CitationCollins et al., 1995; CitationSilva et al., 2000). Doxorubicin (0.6 μM) was used as positive control. To assess DNA damage in PBMC caused by neosergeolide, the same protocol used for leukemia cells, as described above, was applied.
Statistical analysis
Data are presented as means ± S.E.M. IC50 values and 95% confidence intervals (CI 95%) were obtained by nonlinear regression using the GRAPHPAD program (Intuitive Software for Science, San Diego, CA, USA). For all experiments, data were analyzed by one-way analysis of variance (ANOVA) followed by the Newman–Keuls test.
Results
Neosergeolide reduces both proliferation and viability of HL-60 cells
The MTT-based assay showed that neosergeolide exhibited elevated cytotoxicity to HL-60 cells after 24 h. Data from two independent experiments carried out in triplicate provided an IC50 value of 0.1 μM (IC95 0.06–0.15 μM). Neosergeolide was not cytotoxic to PBMC (IC50 > 4.0 μM) at tested concentrations. Doxorubicin was used as positive control and displayed potent cytotoxicity against HL-60 cells (IC50 0.04 μM, CI 95% 0.02–0.04 μM) and PBMC (IC50 0.33 μM, CI95% 0.20-0.44 μM). Subsequent experiments were conducted at concentrations corresponding to 1/2 × IC50, IC50, and 2 × IC50 (0.05, 0.1, and 0.2 μM, respectively).
To further study the inhibition of proliferation, the incorporation of the nucleotide BrdU into DNA was evaluated by direct peroxidase immunocytochemistry in treated and untreated HL-60 cells. After 24 h, neosergeolide at the concentrations of 0.05, 0.1, and 0.2 μM inhibited BrdU incorporation by 48.00 ± 2.30, 89.67 ± 1.45, and 94.67 ± 1.45%, respectively, in HL-60 cells (; p < 0.001).
Neosergeolide alters cell morphology, inducing both apoptosis and necrosis in HL-60 cells
To determine whether growth inhibition was related to the induction of apoptosis and necrosis, morphological analysis of neosergeolide-treated HL-60 cells was carried out using May-Grünwald-Giemsa staining.
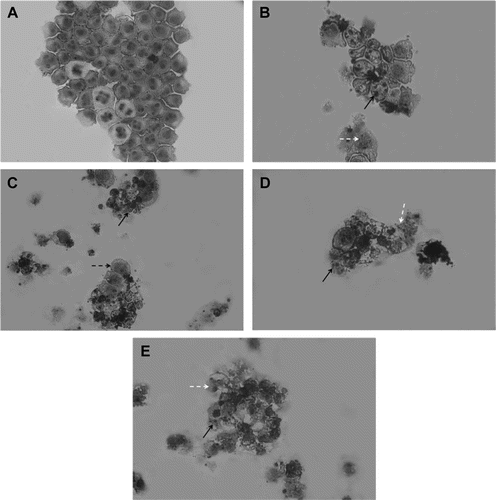
Analysis of May-Grünwald-Giemsa stained neosergeolide-treated and untreated HL-60 cells revealed several drug-induced morphological changes. Control cells exhibited a typical non-adherent and vacuolization round morphology after 24 h in culture (). Neosergeolide at all concentrations induced DNA fragmentation, reduction in cell volume, and destabilization of the plasma membrane. Progression toward cell death in a dose-dependent manner was observed (). Also, pyknotic nuclei were observed at all concentrations. The positive control substance doxorubicin (0.6 µM) also induced reduction in cell volume, besides nuclear fragmentation and destabilization of the plasma membrane ().
Figure 2. Morphological features induced by neosergeolide. Microscopic analysis of untreated (A) and neosergeolide-treated HL-60 cells (C-0.05 µM, D-0.1 µM and E-0.2 µM). Doxorubicin (0.6 µM) was used as positive control (B). Cells were incubated for 24 h and stained by May-Grünwald-Giemsa. Black arrows: nuclei pyknotic and nuclear fragmentation, white dashed arrows: debris, and black dashed arrow: membrane damage. Magnification: 400×.
Effects of neosergeolide on cell membrane integrity, cell proliferation, internucleosomal DNA fragmentation, and Δψm by flow cytometry
Proliferation and cell membrane integrity were determined by the exclusion of PI ( and ). In HL-60 cell cultures treated with 0.05 and 0.1 μM neosergeolide, loss of membrane integrity was observed only after 24 h (p < 0.05). At 0.2 μM, neosergeolide induced early loss of membrane integrity, which began after 6 h of exposure. Neosergeolide reduced the number of viable cells in a time- and concentration-dependent manner. Cultures exposed to neosergeolide at low concentration (0.05 μM) showed a decrease in cell viability only after 12 and 24 h of treatment (p < 0.05). However, at the highest concentration (0.2 μM), neosergeolide reduced cell viability at all exposure times evaluated. Doxorubicin reduced the number of viable cells () without membrane damage ().
Figure 3. Effect of neosergeolide on HL-60 cell membrane integrity (A), proliferation (B), internucleosomal DNA fragmentation (C), and mitochondrial transmembrane potential (D), determined by flow cytometric analysis after 3, 6, 12, and 24 h of incubation. The vehicle (0.1% DMSO) used for diluting the test substance was used as negative control. Doxorubicin at 0.6 µM was used as positive control. A total of 5,000 events were analyzed in each experiment. *p < 0.001; **p < 0.05 compared to control by ANOVA followed by Newman–Keuls test. Data are presented as mean values ± S.E.M. from three independent experiments in triplicate.
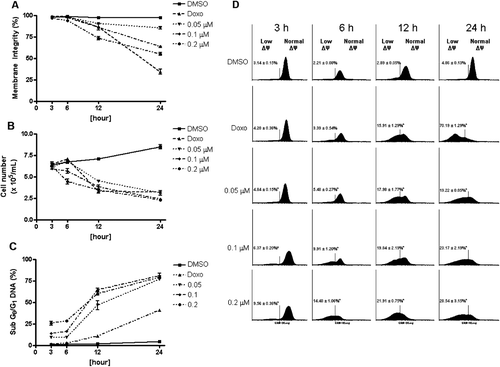
At all concentrations tested, neosergeolide caused cell shrinkage and nuclear condensation as evidenced by decrease in forward light scattering and transient increase in side scattering, respectively. Both of these morphological modifications are compatible with the presence of apoptotic cells. All subdiploid-sized DNA (sub-G0/G1) was considered to be due to internucleosomal DNA fragmentation. Increased neosergeolide-induced apoptotic sub-G0/G1 peaks represent apoptotic cells having fractional DNA content and were observed at all concentrations 3, 6, 12, and 24 h after treatment (; p < 0.001). Doxorubicin also induced apoptotic effects.
Neosergeolide-induced mitochondrial depolarization in HL-60 cells was evidenced by Rho-123 incorporation after 3, 6, 12, and 24 h of incubation (). These data suggest that neosergeolide induces apoptosis in HL-60 cells by triggering an intrinsic mitochondrial pathway.
Protection by CsA against neosergeolide-induced cell killing in HL-60
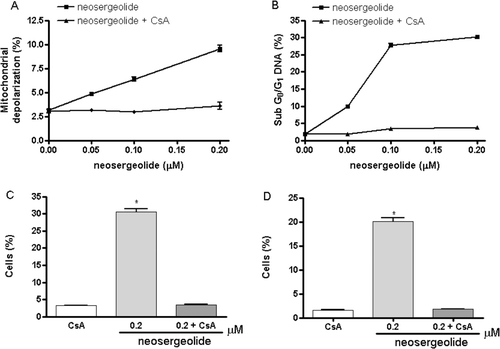
As MPT and caspases play crucial roles in the process of apoptosis, the effects of CsA, a specific inhibitor of MPT, on neosergeolide-induced apoptosis was investigated. After 3 h exposure, neosergeolide reduces the mitochondrial depolarization () and causes an increase in the DNA fragmentation (sub-G0/G1 peaks) () as well in the number of cells with activate caspase-9 () and caspases-3 and -7 (). To implicate a role of the MPT in neosergeolide-induced cell killing, HL-60 cells were pretreated with 5 μM CsA before exposure to neosergeolide. The CsA completely blocked neosergeolide-induced apoptosis ().
Figure 4. Effect of neosergeolide after 3 h exposure in the presence or absence of CsA (5 µM) on HL-60 mitochondrial transmembrane potential (A), internucleosomal DNA fragmentation (B), percentage of cells with active caspase-9 (C), and caspases-3 and -7 (D) determined by flow cytometric analysis.
Neosergeolide induces phosphatidylserine externalization and activation of caspases-3 and -7 after 3 h (pulse treatment)
To verify if neosergeolide-induced cytotoxicity was a reversible process, the cells were treated for 3 h; after short-exposure time, the drug was removed, and then the cultures were reincubated for 21 h. After 3 h pulse treatment, neosergeolide caused a significant increase in the number of early (AnnV+/PI−) and late (AnnV+/PI+) apoptotic HL-60 cells when compared to the control group (). For AnnV/PI experiments, we did not observe a significant concentration–response relationship in the number of necrotic cells (AnnV−/PI+): 0.12 ± 0.01, 0.04 ± 0.01, 0.46 ± 0.02, 3.40 ± 0.98, and 1.90 ± 0.22%, for negative control (vehicle) cultures, doxorubicin-treated cultures, and 0.05, 0.1, and 0.2 μM neosergeolide-treated cultures, respectively. Corroborating with our AnnV/PI data, neosergeolide increases the percentage of apoptotic (early and late stages) HL-60 cells with caspases-3 and -7 activated, and a slight increase of necrotic cells population was observed: 0.70 ± 0.31 (vehicle cultures), 2.67 ± 0.25 (doxorubicin cultures), and 0.67 ± 0.21, 2.30 ± 0.56, and 3.46 ± 1.15% for 0.05, 0.1, and 0.2 μM neosergeolide-treated cultures, respectively ().
Figure 5. Effect on PS externalization after HL-60 cells treated with 0.05 (C), 0.1 (D) and 0.2 µM (E) neosergeolide. The PS externalization was determined by flow cytometry using AnnV-FITC (YLW-HLog) and PI (RED-HLog) after pulse treatment (3 h of neosergeolide exposure following 21 h reincubation period without drug). Viable cells are plotted at lower left quadrant, cells in early and late apoptosis with PS externalized are plotted at lower right and upper right quadrants, respectively, and necrotic cells are plotted at upper left quadrant. Negative control (A) was treated with the vehicle (0.1% DMSO) used for diluting the test substance. Doxorubicin (B) at 0.6 µM was used as positive control. A total of 5,000 events were analyzed in each experiment. *p < 0.05 compared to control by ANOVA followed by Newman–Keuls test. Data are presented as mean values ± S.E.M. from two independent experiments in triplicate.
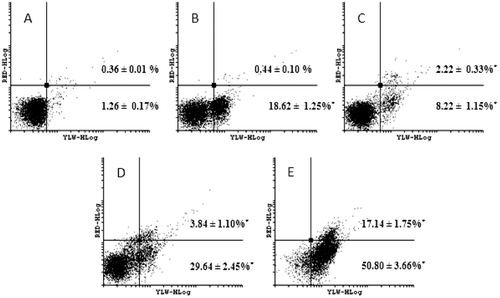
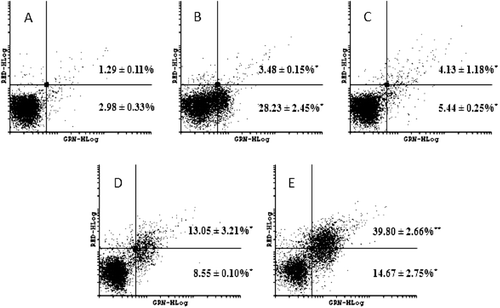
Figure 6. Effect on caspases-3 and -7 activation after HL-60 cells treated with 0.05 (C), 0.1 (D) and 0.2 µM (E) neosergeolide. Caspases activity was determined by flow cytometry using PI (RED-HLog) and fluorescent-labeled inhibitor of caspases, FLICATM (GRN-HLog) after pulse treatment (3 h of neosergeolide exposure following 21 h reincubation period without drug). Viable cells are plotted at lower left quadrant, cells in early and late apoptosis with active caspases-3 and -7 are plotted at lower right and upper right quadrants, respectively, and necrotic cells are plotted at upper left quadrant. Negative control (A) was treated with the vehicle (0.1% DMSO) used for diluting the test substance. Doxorubicin (B) at 0.6 µM was used as positive control. A total of 5,000 events were analyzed in each experiment. *p < 0.001; **p < 0.05 compared to control by ANOVA followed by Newman–Keuls test. Data are presented as mean values ± S.E.M. from two independent experiments in triplicate.
Neosergeolide induces changes in the HL-60 cell cycle and DNA damage after 3 h (pulse treatment)
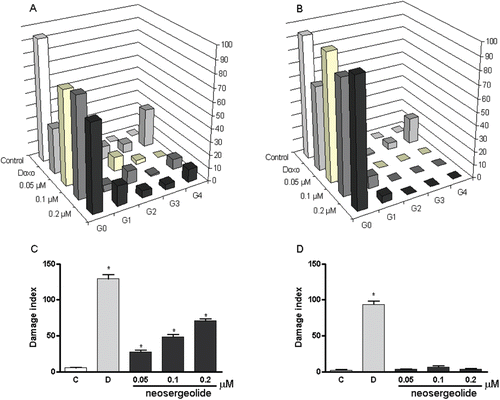
shows the effect of neosergeolide on the HL-60 cell cycle. In general, neosergeolide at all concentrations lowered the number of cells at G1, S, and G2/M phases, which suggests that neosergeolide interfered in a non-specific manner in the HL-60 cell cycle. As expected, neosergeolide treatment also induces a reduction in cell proliferation, mitochondrial depolarization, and internucleosomal DNA fragmentation (sub-G0/G1 peaks). Also, no intracellular ROS production was detected 3–24 h after treatment with neosergeolide (data not shown).
Table 1. Effect of neosergeolide on cell cycle distribution in HL60 cells by flow cytometric using PI after 3 h pulse treatment.
In neosergeolide-treated HL-60 cells, the DNA damage indexes were higher than in the control cells (). On the other hand, no increase on DNA migration was observed in neosergeolide-treated PBMC (). The MN frequencies were significantly increased by neosergeolide treatment at all concentrations tested (p < 0.05) and neosergeolide decreased cytokinesis-block proliferation of leukemia cells (; p < 0.05). However, the DNA damage concentrations for HL-60 cells were not genotoxic for PBMC and did not decrease the proliferation ratio of PBMC ().
Table 2. Effect of neosergeolide on HL-60 and PBMC micronucleated cell (MN) frequency in the micronucleus test after 3 h pulse treatment.
Figure 7. Effects of neosergeolide after 3 h pulse treatment on the distribution of damaged cells in alkaline comet assay into damage grades (G; grades 0–4) on HL-60 (A) and PBMC (B), its effects on HL-60 (C), and PBMC (D) DNA damage index. Bars represent the mean ± S.E.M. of three independent experiments. Negative control (C): cells were treated with the vehicle used for diluting the tested substance. Doxorubicin (0.6 µM) was used as positive control (D). *p < 0.001 compared to control by ANOVA followed by Newman–Keuls test.
Discussion
Previous reports on the cytotoxicity of neosergeolide (CitationSilva et al., 2009) demonstrated that, as observed to other quassinoids (CitationKupchan et al., 1976; CitationLee et al., 1982; CitationLumonadio et al., 1991; CitationImamura et al., 1993; CitationMata-Greenwood et al., 2001; CitationJiang et al., 2008; CitationLau et al., 2009), it strongly inhibited the proliferation of tumor cells in spite of their histological origin. The present study was designed to evaluate the selectivity of neosergeolide to tumor cells in comparison to normal lymphocytes, and moreover, to elucidate the underlying mechanism of action. MTT analysis confirmed the strong cytotoxicity of neosergeolide to leukemia cells (IC50 = 0.1 μM after 24 h of exposure), while it suggest a good selectivity for this compound, since no cytotoxicity was observed to proliferating lymphocytes at tested concentrations. A balance between therapeutic and toxicological effects of a candidate compound is important for establishing applicability as a pharmaceutical. Also, when considering the side effects of chemotherapy, it is very important to determine whether a drug has a harmful effect on normal dividing cells, such as proliferating lymphocytes (CitationZucot et al., 2002; CitationAnazetti et al., 2003). Brucein D, a quassinoid isolated from Brucea javanica (L.) Merr. (Simaroubaceae), also demonstrated selectivity against pancreatic tumor cells in comparison to nontumorigenic cells (CitationLau et al., 2009).
In the present study, cytotoxic activity was also evaluated through the loss of membrane integrity as shown by results of flow cytometry analyses, especially after 24 h of exposure. During early stages of apoptosis, cell membrane becomes impermeable to vital dyes, such as trypan blue (CitationPiacentini et al., 1991) or PI (CitationVan Cruchten & Van Den Broeck, 2002), and opposite situation occurs during late apoptosis or necrosis. So, other assays are needed to evaluate cells undergoing early apoptosis (i.e., annexin detection and caspases activation).
Proliferation capacity is equivalent to cell growth and was measured by incorporation of BrdU, a thymidine analog that is incorporated into DNA during the S phase and can be detected by immunocytochemistry (CitationHolm et al., 1998). Over a period of 24 h, neosergeolide at low concentration decreased the number of BrdU-positive cells. The lower BrdU uptake by cell DNA after neosergeolide exposure also corroborates the result obtained from the MTT assay. In addition, our results are in general agreement with the antiproliferative properties of other quassinoids, such as brusatol and bruceantin, which were shown to inhibit the proliferation of several established leukemia cell lines, including HL-60 cells, in an assay based on the incorporation of 3H-thymidine (CitationMata-Greenwood et al., 2002).
Induction of apoptosis by quassinoids has been reported before (CitationMata-Greenwood et al., 2002; CitationCuendet et al., 2004; CitationRosati et al., 2004; Citationvon Bueren et al., 2007; CitationLau et al., 2009). In this study, several sensitive methods for detecting apoptosis were used, based on the different morphological or biochemical features of apoptosis and necrosis. The results demonstrate that neosergeolide induces apoptosis in HL-60 cells at micromolar concentrations as evidenced by flow cytometric analyses and morphological alterations (May-Grünwald-Giemsa staining). DNA fragmentation during apoptosis could lead to extensive loss of DNA content and a distinct sub-G0/G1 peak when analyzed by flow cytometry. In the present study, apoptosis was analyzed by the determination of sub-G0/G1 cells. Our analysis revealed that neosergeolide stimulation increased the percentage of sub-G0/G1 peaks (hypodiploid DNA) in HL-60 cells in a time- and concentration-dependent manner.
Apoptosis and necrosis represent only the extreme ends of a wide range of possible morphological and biochemical deaths and can occur simultaneously in tissues and cell cultures exposed to the same stimulus (CitationNicotera et al., 1999). However, there is no clear biochemical definition of necrotic cell death and consequently no positive biochemical marker that unambiguously discriminate necrosis from apoptosis (late apoptosis features). Another problem is that even the interpretation of dying cell morphology may be complex, because in the absence of phagocytosis apoptotic cells proceed to a stage called secondary necrosis, which shares many features of primary necrosis (CitationKerr et al., 1994; CitationKroemer et al., 1998; CitationKrysko et al., 2008).
One of the earliest manifestations of apoptosis, regardless of the initiating stimulus, is the redistribution of phospholipids in the plasma membrane that leads to the exposure of phosphatidylserine (PS) at the cell surface (CitationFadok et al., 1992; CitationKoopman et al., 1994; CitationMartin et al., 1995). AnnV is a Ca2+ dependent phospholipid-binding protein that has a high affinity for PS, and binds to cells with exposed PS (CitationKrysko et al., 2008). Combining AnnV with PI can help to distinguish between apoptosis (early and late stages) and necrosis. The data showed that after pulse treatment (3 h of neosergeolide exposure following 21 h reincubation period without drug), many cells were in early and late apoptosis with active caspases-3 and -7, and after a short period exposure (3 h), activation of caspases-3 and -7 was also observed. These results showed that the activation of apoptotic mechanisms occurs earlier and do not depend on extended periods of exposure.
When mitochondrial membrane potential is dissipated, a cell begins an irreversible apoptotic process (CitationGao et al., 2006; CitationHsu & Yen, 2007). Detection of mitochondrial membrane potential changes can, therefore, be useful as a probe for the onset of apoptosis. The correlation between the loss of the mitochondrial membrane potential and DNA fragmentation indicates that the reduction of Δψm constitutes an obligate and irreversible step of ongoing HL-60 death. In this study, neosergeolide increased potential loss in the mitochondrial membrane thus providing evidence for the activation of an intrinsic apoptosis pathway in HL-60 cells. These results are in general agreement with the findings of CitationRosati et al. (2004) who demonstrated that quassinoids induce mitochondrial depolarization and caspase-3 activation. Also, according to CitationMata-Greenwood et al. (2002), treatment of leukemic cells with bruceantin induces a decrease in c-myc mRNA and protein which in turn may be responsible for some of the pro-apoptotic effects of quassinoids (CitationCuendet & Pezzuto, 2004).
Mitochondria play a decisive role in the apoptotic pathway mediated by certain agonists (CitationGreen, 1998; CitationReed et al., 1998; CitationSmaili et al., 2003). Disruption of the inner and outer mitochondrial membrane and opening of the MPT pore, which is regulated by members of the Bcl-2 family as well as the redox and energy state of the cell, result in a collapse of Δψm, and in the exit of soluble proteins, such as cytochrome c and apoptosis-inducing factor (Citationvan Loo et al., 2002). This MPT from the intermembrane space can trigger an activation of downstream caspases (CitationZoratti & Szabo, 1995; CitationThornberry & Lazebnik, 1998). The classic MPT is dependent upon the [Ca2+]i, is energy dependent, and is accompanied by mitochondrial swelling and depolarization (CitationZoratti & Szabo, 1995), which CsA inhibits MPT pore opening (CitationArmstrong, 2006). The CsA binds to Cyp-M, a cyclophilin-family protein associated with the MPT pore, causing it to dissociate from the pore complex, and this increases the probability of MPT pore closure and thus prevents the Δψm disruption and block cytochrome c release (CitationLemasters, 1999; CitationJiang et al., 2001; CitationBrustovetsky et al., 2002). On the contrary, it has been reported that the nonclassic MPT, which is insensitive to CsA, occurs without swelling and depolarization of the mitochondria (CitationSultan & Sokolove, 2001).
Consistent with these observations, neosergeolide-induced apoptosis is dependent of the classic MPT mechanism. Cotreatment with CsA prevents neosergeolide-induced caspases-9, -3 and -7 activation suggesting that blocking of MPT prevents the leakage of cytochrome c and consequently prevents the activation of caspase-9 (caspase-dependent cytochrome c release) and apoptotic protease activating factor 1 (Apaf-1). Caspase-9 is thought to be the initiator caspase involved in the mitochondrial-initiated apoptotic pathway, and it activates downstream caspases, such as caspases-3, -6, and -7 (CitationStrasser et al., 2000). Caspases-3 and -7 are two well-known “executioner caspases.” Their activation is believed to be responsible for the morphological changes seen in apoptosis, including DNA fragmentation, chromatin condensation, and the formation of apoptotic bodies (CitationMarcelli et al., 1999; CitationNicholson, 1999).
Several mechanisms exist by which neosergeolide could potentially exert to achieve the observed apoptogenic effects in cancer cells. It was evaluated whether the ROS generation or direct DNA damage could be related to neosergeolide activity. Apoptosis induced by many chemical genotoxins is a consequence of blockage of DNA replication, which leads to collapse of replication forks and DNA double-strand breaks formation, which, the latter, is thought to be crucial downstream for apoptosis-triggering lesions (CitationRoos & Kaina, 2006). Genotoxic DNA damaging agents may activate both membrane death receptors and the endogenous mitochondrial damage pathway leading to cell death via apoptosis (CitationKaina, 2003). DNA damage, such as DNA strand breakage and induction of MN, may be important features of neosergolide’s cytotoxic mechanisms. Thus, HL-60 cultures treated with neosergeolide exhibit strong reduction in cytokinesis-block proliferation which is a biological parameter for the detection of cellular toxicity or cell cycle delay (CitationSurrallés et al., 1995). Also, neosergeolide induces DNA damage after a short incubation period (3 h) as evidenced by a significant increase in grades 3 and 4 comets in comparison to negative control (). The occurrence of comets with no heads and with nearly all DNA in the tail (grade 4) is an indication of the cytotoxic effect (CitationHartmann & Speit, 1997). Interestingly, neosergeolide induces DNA damages in HL-60 cells which are p53 null (CitationShimizu & Pommier, 1997) but not in PBMC (wild-type p53), suggesting that this effect is p53 independent. The tumor suppressor protein p53 is considered to be a major player in the apoptotic response to genotoxins. Some experiments, trying to elucidate in more detail the role of p53 in DNA damage-triggered apoptosis, have shown that some primary and established cell (mouse fibroblasts) lines deficient for p53 were clearly more sensitive than the corresponding wild-type after exposure to UV-C and alkylating agent (methyl methanesulfonate), supporting the view that p53 is not required for inducing apoptosis in these cells (CitationLackinger & Kaina, 2000; CitationLackinger et al., 2001). However, the pro- or anti-apoptotic effect of p53 appears to be a cell type-specific phenomenon since lymphoblastoid cells wild-type for p53 proved to be more sensitive to alkylating agents and UV-C than the p53 mutated counterparts (CitationKarran & Stephenson, 1990). The factor(s) involved in making the decision between protection against or stimulation of the apoptotic process by p53 remains unknown (CitationKaina, 2003).
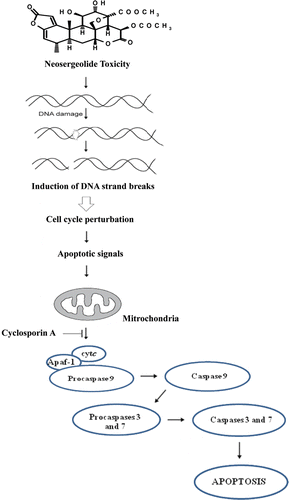
Intracellular ROS production is associated with a number of cellular events, including activation of NADH oxidase and xanthine oxidase, and the functioning of the mitochondrial respiratory chain (CitationPerez-Ortiz et al., 2007). The NADH oxidase is inhibited by several known potential antitumor agents, such as sulfonylurea, adriamycin, and capsaicin (Citationdel Castillo-Olivares et al., 1998). Interestingly, CitationMorré et al. (1998) showed that the cytotoxicity of the quassinoid glaucarubolone to HeLa cells was associated with NADH oxidase inhibition. CitationZhao et al. (2008) demonstrated that inhibition of NADPH oxidase activity by diphenyleneiodonium suppressed free radical production and inhibited cell growth of B16 melanoma cells. The present study provides evidence that the mechanisms of cell growth inhibition, cell death, and DNA-damage of neosergeolide do not depend on the production of ROS. These data are consistent with the previous report in which it was shown that the cytotoxicity of neosergeolide and another isolated quassinoid (isobrucein B) toward cancer cell lines is not related to oxidative stress (CitationSilva et al., 2009). A working mechanistic model is developed based on these findings and is summarized in a schematic diagram ().
Figure 8. Summarized model of neosergeolide antiproliferative effects.
Conclusions
The present study provided experimental evidence to support the underlying mechanism of action involved in the neosergeolide-mediated apoptosis. Taken together, the results indicated that neosergeolide leads to DNA damage triggering intrinsic pathways for apoptosis induction. In addition, no antiproliferative effect or DNA damage effect of neosergeolide was evident in PBMC, which is evidence of its therapeutic potential.
Acknowledgments
The authors thank the National Cancer Institute (Bethesda, MD, USA) for the donation of the leukemia cell line used in this study. The authors also thank Silvana França dos Santos for technical assistance and are also grateful for scientific support and assistance from BioTechCell LTDA (Brazil).
Declaration of interest
The authors declare no conflict of interest, and they wish to thank CNPq (PNOPG 550.260/01-3 e PPG-7 557.106/05-2), MCT/PPBIO 480.002/04-5, CAPES, Instituto Claude Bernard, PRONEX, FUNCAP, Banco do Nordeste, and FINEP for financial support in the form of grants and fellowship awards.
References
- Anazetti MC, Melo PS, Durán N, Haun M. (2003). Comparative cytotoxicity of dimethylamide-crotonin in the promyelocytic leukemia cell line (HL60) and human peripheral blood mononuclear cells. Toxicology, 188, 261–274.
- de Andrade-Neto VF, Pohlit AM, Pinto AC, Silva EC, Nogueira KL, Melo MR, Henrique MC, Amorim RC, Silva LF, Costa MR, Nunomura RC, Nunomura SM, Alecrim WD, Alecrim MG, Chaves FC, Vieira PP. (2007). In vitro inhibition of Plasmodium falciparum by substances isolated from Amazonian antimalarial plants. Mem Inst Oswaldo Cruz, 102, 359–365.
- Armstrong JS. (2006). The role of the mitochondrial permeability transition in cell death. Mitochondrion, 6, 225–234.
- Arseneau JC, Wolter JM, Kuperminc M, Ruckdeschel JC. (1983). A Phase II study of bruceantin (NSC-165, 563) in advanced malignant melanoma. Invest New Drugs, 1, 239–242.
- Bertani S, Houël E, Stien D, Chevolot L, Jullian V, Garavito G, Bourdy G, Deharo E. (2006). Simalikalactone D is responsible for the antimalarial properties of an Amazonian traditional remedy made with Quassia amara L. (Simaroubaceae). J Ethnopharmacol, 108, 155–157.
- Brustovetsky N, Brustovetsky T, Jemmerson R, Dubinsky JM. (2002). Calcium-induced cytochrome c release from CNS mitochondria is associated with the permeability transition and rupture of the outer membrane. J Neurochem, 80, 207–218.
- Collins AR, Ma AG, Duthie SJ. (1995). The kinetics of repair of oxidative DNA damage (strand breaks and oxidised pyrimidines) in human cells. Mutat Res, 336, 69–77.
- Collins SJ. (1987). The HL-60 promyelocytic leukemia cell line: proliferation, differentiation, and cellular oncogene expression. Blood, 70, 1233–1244.
- Crow JP. (1997). Dichlorodihydrofluorescein and dihydrorhodamine 123 are sensitive indicators of peroxynitrite in vitro: implications for intracellular measurement of reactive nitrogen and oxygen species. Nitric Oxide, 1, 145–157.
- Cuendet M, Christov K, Lantvit DD, Deng Y, Hedayat S, Helson L, McChesney JD, Pezzuto JM. (2004). Multiple myeloma regression mediated by bruceantin. Clin Cancer Res, 10, 1170–1179.
- Cuendet M, Pezzuto JM. (2004). Antitumor activity of bruceantin: An old drug with new promise. J Nat Prod, 67, 269–272.
- Cury-Boaventura MF, Pompéia C, Curi R. (2004). Comparative toxicity of oleic acid and linoleic acid on Jurkat cells. Clin Nutr, 23, 721–732.
- del Castillo-Olivares A, Yantiri F, Chueh PJ, Wang S, Sweeting M, Sedlak D, Morré DM, Burgess J, Morré DJ. (1998). A drug-responsive and protease-resistant peripheral NADH oxidase complex from the surface of HeLa S cells. Arch Biochem Biophys, 358, 125–140.
- Eckhardt SG, Dai A, Davidson KK, Forseth BJ, Wahl GM, Von Hoff DD. (1994). Induction of differentiation in HL60 cells by the reduction of extrachromosomally amplified c-myc. Proc Natl Acad Sci USA, 91, 6674–6678.
- Fadok VA, Voelker DR, Campbell PA, Cohen JJ, Bratton DL, Henson PM. (1992). Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol, 148, 2207–2216.
- Gao SY, Wang QJ, Ji YB. (2006). Effect of solanine on the membrane potential of mitochondria in HepG2 cells and [Ca2+]i in the cells. World J Gastroenterol, 12, 3359–3367.
- Green DR. (1998). Apoptotic pathways: The roads to ruin. Cell, 94, 695–698.
- Guo Z, Vangapandu S, Sindelar RW, Walker LA, Sindelar RD. (2005a). Biologically active quassinoids and their chemistry: Potential leads for drug design. Curr Med Chem, 12, 173–190.
- Guo Z, Vangapandu S, Nimrod A, Walker LA, Sindelar RD. (2005b). Synthesis of A/B-ring partial analogs of bruceantin as potential antimalarial agents. Med Chem, 1, 3–11.
- Hartmann A, Speit G. (1997). The contribution of cytotoxicity to DNA-effects in the single cell gel test (comet assay). Toxicol Lett, 90, 183–188.
- Hempel SL, Buettner GR, O’Malley YQ, Wessels DA, Flaherty DM. (1999). Dihydrofluorescein diacetate is superior for detecting intracellular oxidants: Comparison with 2′,7′-dichlorodihydrofluorescein diacetate, 5(and 6)-carboxy-2′,7′-dichlorodihydrofluorescein diacetate, and dihydrorhodamine 123. Free Radic Biol Med, 27, 146–159.
- Holm M, Thomsen M, Høyer M, Hokland P. (1998). Optimization of a flow cytometric method for the simultaneous measurement of cell surface antigen, DNA content, and in vitro BrdUrd incorporation into normal and malignant hematopoietic cells. Cytometry, 32, 28–36.
- Hsu CL, Yen GC. (2007). Effects of capsaicin on induction of apoptosis and inhibition of adipogenesis in 3T3-L1 cells. J Agric Food Chem, 55, 1730–1736.
- Imamura K, Fukamiya N, Okano M, Tagahara K, Lee KH. (1993). Bruceanols D, E, and F three new cytotoxic quassinoids from Brucea antidysenterica. J Nat Prod, 56, 2091–2097.
- Itokawa H, Ibraheim ZZ, Qiao YF, Takeya K. (1993). Anthraquinones, naphthohydroquinones and naphthohydroquinone dimers from Rubia cordifolia and their cytotoxic activity. Chem Pharm Bull, 41, 1869–1872.
- Jiang D, Sullivan PG, Sensi SL, Steward O, Weiss JH. (2001). Zn(2+) induces permeability transition pore opening and release of pro-apoptotic peptides from neuronal mitochondria. J Biol Chem, 276, 47524–47529.
- Jiang G, Albihn A, Tang T, Tian Z, Henriksson M. (2008). Role of Myc in differentiation and apoptosis in HL60 cells after exposure to arsenic trioxide or all-trans retinoic acid. Leuk Res, 32, 297–307.
- Jiwajinda S, Santisopasri V, Murakami A, Sugiyama H, Gasquet M, Riad E, Balansard G, Ohigashi H. (2002). In vitro anti-tumor promoting and anti-parasitic activities of the quassinoids from Eurycoma longifolia, a medicinal plant in Southeast Asia. J Ethnopharmacol, 82, 55–58.
- Kaina B. (2003). DNA damage-triggered apoptosis: critical role of DNA repair, double-strand breaks, cell proliferation and signaling. Biochem Pharmacol, 66, 1547–1554.
- Karran P, Stephenson C. (1990). Mismatch binding proteins and tolerance to alkylating agents in human cells. Mutat Res, 236, 269–275.
- Kerr JF, Winterford CM, Harmon BV. (1994). Apoptosis. Its significance in cancer and cancer therapy. Cancer, 73, 2013–2026.
- Klaude M, Eriksson S, Nygren J, Ahnström G. (1996). The comet assay: Mechanisms and technical considerations. Mutat Res, 363, 89–96.
- Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. (1994). Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood, 84, 1415–1420.
- Kroemer G, Dallaporta B, Resche-Rigon M. (1998). The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol, 60, 619–642.
- Krysko DV, Vanden Berghe T, D’Herde K, Vandenabeele P. (2008). Apoptosis and necrosis: detection, discrimination and phagocytosis. Methods, 44, 205–221.
- Kupchan SM, Britton RW, Ziegler MF, Sigel CW. (1973). Bruceantin, a new potent antileukemic simaroubolide from Brucea antidysenterica. J Org Chem, 38, 178–179.
- Kupchan SM, Lacadie JA, Howie GA, Sickles BR. (1976). Structural requirements for biological activity among antileukemic glaucarubolone ester quassinoids. J Med Chem, 19, 1130–1133.
- Lackinger D, Kaina B. (2000). Primary mouse fibroblasts deficient for c-Fos, p53 or for both proteins are hypersensitive to UV light and alkylating agent-induced chromosomal breakage and apoptosis. Mutat Res, 457, 113–123.
- Lackinger D, Eichhorn U, Kaina B. (2001). Effect of ultraviolet light, methyl methanesulfonate and ionizing radiation on the genotoxic response and apoptosis of mouse fibroblasts lacking c-Fos, p53 or both. Mutagenesis, 16, 233–241.
- Lau ST, Lin ZX, Liao Y, Zhao M, Cheng CH, Leung PS. (2009). Bruceine D induces apoptosis in pancreatic adenocarcinoma cell line PANC-1 through the activation of p38-mitogen activated protein kinase. Cancer Lett, 281, 42–52.
- LeBel CP, Ischiropoulos H, Bondy SC. (1992). Evaluation of the probe 2′,7′-dichlorofluorescin as an indicator of reactive oxygen species formation and oxidative stress. Chem Res Toxicol, 5, 227–231.
- Lee KH, Okano M, Hall IH, Brent DA, Soltmann B. (1982). Antitumor agents XLV: Bisbrusatolyl and brusatolyl esters and related compounds as novel potent antileukemic agents. J Pharm Sci, 71, 338–345.
- Lemasters JJ. (1999). V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol, 276, G1–G6.
- Lumonadio L, Atassi G, Vanhaelen M, Vanhaelen-Fastre R. (1991). Antitumor activity of quassinoids from Hannoa klaineana. J Ethnopharmacol, 31, 59–65.
- Marcelli M, Cunningham GR, Walkup M, He Z, Sturgis L, Kagan C, Mannucci R, Nicoletti I, Teng B, Denner L. (1999). Signaling pathway activated during apoptosis of the prostate cancer cell line LNCaP: overexpression of caspase-7 as a new gene therapy strategy for prostate cancer. Cancer Res, 59, 382–390.
- Martin SJ, Reutelingsperger CP, McGahon AJ, Rader JA, van Schie RC, LaFace DM, Green DR. (1995). Early redistribution of plasma membrane phosphatidylserine is a general feature of apoptosis regardless of the initiating stimulus: inhibition by overexpression of Bcl-2 and Abl. J Exp Med, 182, 1545–1556.
- Mata-Greenwood E, Daeuble JF, Grieco PA, Dou J, McChesney JD, Mehta RG, Kinghorn AD, Pezzuto JM. (2001). Novel esters of glaucarubolone as inducers of terminal differentiation of promyelocytic HL-60 cells and inhibitors of 7,12-dimethylbenz[a]anthracene-induced preneoplastic lesion formation in mouse mammary organ culture. J Nat Prod, 64, 1509–1513.
- Mata-Greenwood E, Cuendet M, Sher D, Gustin D, Stock W, Pezzuto JM. (2002). Brusatol-mediated induction of leukemic cell differentiation and G(1) arrest is associated with down-regulation of c-myc. Leukemia, 16, 2275–2284.
- Militão GC, Dantas IN, Pessoa C, Falcão MJ, Silveira ER, Lima MA, Curi R, Lima T, Moraes MO, Costa-Lotufo LV. (2006). Induction of apoptosis by pterocarpans from Platymiscium floribundum in HL-60 human leukemia cells. Life Sci, 78, 2409–2417.
- Miyamae Y, Yamamoto M, Sasaki YF, Kobayashi H, Igarashi-Soga M, Shimoi K, Hayashi M. (1998). Evaluation of a tissue homogenization technique that isolates nuclei for the in vivo single cell gel electrophoresis (comet) assay: A collaborative study by five laboratories. Mutat Res, 418, 131–140.
- Morré DJ, Grieco PA, Morré DM. (1998). Mode of action of the anticancer quassinoids–inhibition of the plasma membrane NADH oxidase. Life Sci, 63, 595–604.
- Mosmann T. (1983). Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J Immunol Methods, 65, 55–63.
- Murakami C, Fukamiya N, Tamura S, Okano M, Bastow KF, Tokuda H, Mukainaka T, Nishino H, Lee KH. (2004). Multidrug-resistant cancer cell susceptibility to cytotoxic quassinoids, and cancer chemopreventive effects of quassinoids and canthin alkaloids. Bioorg Med Chem, 12, 4963–4968.
- Nicholson DW. (1999). Caspase structure, proteolytic substrates, and function during apoptotic cell death. Cell Death Differ, 6, 1028–1042.
- Nicotera P, Leist M, Ferrando-May E. (1999). Apoptosis and necrosis: different execution of the same death. Biochem Soc Symp, 66, 69–73.
- Okano M, Fukamiya N, Aratani T, Juichi M, Lee KH. (1985). Antitumor agents, 74. Bruceanol-A and -B, two new antileukemic quassinoids from Brucea antidysenterica. J Nat Prod, 48, 972–975.
- Okano M, Fukamiya N, Lee KH. (1990). Biologically active compounds from simaroubaceous plants. In: Atta-ur-Rahman, ed. Studies in Natural Products Chemistry. Amsterdam: Elsevier, 369–404.
- Okano M, Fukamiya N, Tagahara K, Cosentino M, Lee T, Morris-Natschke S, Lee KH. (1996). Anti-HIV activity of quassionoids. Bioorg Med Lett, 6, 701–706.
- Pera F, Mattias P, Detzer K. (1977). Methods for determining the proliferation kinetics of cells by means of 5-bromodeoxyuridine. Cell Tissue Kinet, 10, 255–264.
- Pérez-Ortiz JM, Tranque P, Burgos M, Vaquero CF, Llopis J. (2007). Glitazones induce astroglioma cell death by releasing reactive oxygen species from mitochondria: Modulation of cytotoxicity by nitric oxide. Mol Pharmacol, 72, 407–417.
- Piacentini M, Fesus L, Farrace MG, Ghibelli L, Piredda L, Melino G. (1991). The expression of “tissue” transglutaminase in two human cancer cell lines is related with the programmed cell death (apoptosis). Eur J Cell Biol, 54, 246–254.
- Polonsky J, Varon Z, Moretti C, Pettit GR, Herald CL, Rideout JA, Saha SB, Khastgir HN. (1980). The antineoplastic quassinoids of Simaba cuspidata spruce and Ailanthus grandis Prain. J Nat Prod, 43, 503–509.
- Reed JC, Jurgensmeier JM, Matsuyama S. (1998). Bcl-2 family proteins and mitochondria. Biochim Biophys Acta, 1366, 127–137.
- Roos WP, Kaina B. (2006). DNA damage-induced cell death by apoptosis. Trends Mol Med, 12, 440–450.
- Rosati A, Quaranta E, Ammirante M, Turco MC, Leone A, De Feo V. (2004). Quassinoids can induce mitochondrial membrane depolarisation and caspase 3 activation in human cells. Cell Death Differ, 11 Suppl 2, S216–S218.
- Shimizu T, Pommier Y. (1997). Camptothecin-induced apoptosis in p53-null human leukemia HL60 cells and their isolated nuclei: Effects of the protease inhibitors Z-VAD-fmk and dichloroisocoumarin suggest an involvement of both caspases and serine proteases. Leukemia, 11, 1238–1244.
- Silva J, Freitas TRO, Heuser V, Marinho JR, Erdtmann B. (2000). Genotoxicity biomonitoring in coal regions using wild rodent Ctenomys torquatus by comet assay and micronucleus test. Environ Mol Mutagen, 35, 270–278.
- Silva EC, Cavalcanti BC, Amorim RC, Lucena JF, Quadros DS, Tadei WP, Montenegro RC, Costa-Lotufo LV, Pessoa C, Moraes MO, Nunomura RC, Nunomura SM, Melo MR, Andrade-Neto VF, Silva LF, Vieira PP, Pohlit AM. (2009). Biological activity of neosergeolide and isobrucein B (and two semi-synthetic derivatives) isolated from the Amazonian medicinal plant Picrolemma sprucei (Simaroubaceae). Mem Inst Oswaldo Cruz, 104, 48–56.
- Singh NP, McCoy MT, Tice RR, Schneider EL. (1988). A simple technique for quantitation of low levels of DNA damage in individual cells. Exp Cell Res, 175, 184–191.
- Smaili SS, Hsu YT, Carvalho AC, Rosenstock TR, Sharpe JC, Youle RJ. (2003). Mitochondria, calcium and pro-apoptotic proteins as mediators in cell death signaling. Braz J Med Biol Res, 36, 183–190.
- Strasser A, O’Connor L, Dixit VM. (2000). Apoptosis signaling. Annu Rev Biochem, 69, 217–245.
- Sultan A, Sokolove PM. (2001). Palmitic acid opens a novel cyclosporin A-insensitive pore in the inner mitochondrial membrane. Arch Biochem Biophys, 386, 37–51.
- Surrallés J, Xamena N, Creus A, Catalán J, Norppa H, Marcos R. (1995). Induction of micronuclei by five pyrethroid insecticides in whole-blood and isolated human lymphocyte cultures. Mutat Res, 341, 169–184.
- Thornberry NA, Lazebnik Y. (1998). Caspases: Enemies within. Science, 281, 1312–1316.
- Tice RR, Agurell E, Anderson D, Burlinson B, Hartmann A, Kobayashi H, Miyamae Y, Rojas E, Ryu JC, Sasaki YF. (2000). Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ Mol Mutagen, 35, 206–221.
- Tischler M, Cardellina JH 2nd, Boyd MR, Cragg GM. (1992). Cytotoxic quassinoids from Cedronia granatensis. J Nat Prod, 55, 667–671.
- Van Cruchten S, Van Den Broeck W. (2002). Morphological and biochemical aspects of apoptosis, oncosis and necrosis. Anat Histol Embryol, 31, 214–223.
- van Loo G, Saelens X, van Gurp M, MacFarlane M, Martin SJ, Vandenabeele P. (2002). The role of mitochondrial factors in apoptosis: A Russian roulette with more than one bullet. Cell Death Differ, 9, 1031–1042.
- von Bueren AO, Shalaby T, Rajtarova J, Stearns D, Eberhart CG, Helson L, Arcaro A, Grotzer MA. (2007). Anti-proliferative activity of the quassinoid NBT-272 in childhood medulloblastoma cells. BMC Cancer, 7, 19.
- Wiseman CL, Yap HY, Bedikian AY, Bodey GP, Blumenschein GR. (1982). Phase II trial of bruceantin in metastatic breast carcinoma. Am J Clin Oncol, 5, 389–391.
- Zhao Y, Liu J, McMartin KE. (2008). Inhibition of NADPH oxidase activity promotes differentiation of B16 melanoma cells. Oncol Rep, 19, 1225–1230.
- Zoratti M, Szabò I. (1995). The mitochondrial permeability transition. Biochim Biophys Acta, 1241, 139–176.
- Zuco V, Supino R, Righetti SC, Cleris L, Marchesi E, Gambacorti-Passerini C, Formelli F. (2002). Selective cytotoxicity of betulinic acid on tumor cell lines, but not on normal cells. Cancer Lett, 175, 17–25.