Abstract
Context: Rheumatoid arthritis (RA) is a systemic autoimmune disease characterized by chronic inflammation in the synovial membrane of affected joints. It has been shown that several kinds of cytokine were increased in synovial fluid, while the underlying mechanism remains poorly understood.
Objectives: NF-κB activator 1 (Act1) is a recently identified protein binding to the IκB kinase complex. Our study aimed to investigate the expression of Act1 induced by cytokine IL-17 stimulation in SW982 cells.
Materials and methods: The human synovial sarcoma cell line SW982 and primary cultured RA fibroblast-like synovial cells were used. RT-PCR and Western blot assays were selected to investigate the genetic and protein expression of Act1. Additionally, four independent Act1 small interfering RNA (siRNA) oligonucleotides were designed and obtained according to the GenBank cDNA, the sequence of Act1 (Traf3ip2). Finally, enzyme-linked immunosorbent assay (ELISA) double antibody sandwich was used to assay supernatant IL-6 and IL-8 concentrations.
Results: The Act1 mRNA expression level increased significantly after stimulation with IL-17 (5–100 ng/ml) in SW982 cells. Additionally, the level of Act1 mRNA expression correlated positively with the concentration of IL-17 (p < 0.01). IL-17 induced IL-6 and IL-8 in SW982 cells was in a concentration- and time-dependent way. Furthermore, ELISA assay revealed that IL-17 (20 ng/ml) significantly increased IL-6 (1927.4 ± 288.77 versus 786.5 ± 172.42 ng/ml, p < 0.01) and IL-8 levels (984.8 ± 95.09 ng/ml versus 307.1 ± 90.83 ng/ml, p < 0.01) compared with control group after stimulation for 24 h. However, transfection of Traf3ip2 siRNA markedly decreased IL-6 (995.9 ± 115.30 ng/ml versus 1816.1 ± 273.27 ng/ml, p < 0.01) and IL-8 levels (575.6 ± 65.96 ng/ml versus 929.4 ± 124.39 ng/ml, p < 0.01) compared to transfection negative control. These findings suggested that IL-6 and IL-8 level induced by IL-17 in SW982 cells could be reversed by down-regulation of Act1 expression level with Traf3ip2 siRNA.
Conclusion: Our results suggested that Act1 might play a key role in the pathophysiology and the treatment of RA.
Introduction
Rheumatoid arthritis (RA) is a systemic chronic autoimmune disease that is characterized by chronic inflammation in the synovial membrane of the affected joints and causes irreversible destruction of cartilage and bones. The transcription factor nuclear factor-kappa B (NF-κB) has been well recognized as a pivotal regulator of inflammation in RA (Makarov, Citation2001). Activated NF-κB has been detected in synovium in humans and animal models of RA (Asahara et al., Citation1995; et al., Citation1998; Handel et al., Citation1995; Marok et al., Citation1996). NF-κB is a collective name for dimeric transcription factors comprised of the Rel family of proteins that include RelA (p65), c-Rel, RelB, NF-κB1 (p50) and NF-κB2 (p52) (Baldwin, Citation1996). NF-κB translocates into the nucleus to activate transcription of target genes, including cell adhesion molecules, cytokines, chemokines and anti-apoptotic factors (Pahl, Citation1999).
Act1, a 60-kD protein that activates IκB kinases (IKK), was first identified and characterized in a HEK293 cell line by Li et al. (Citation2000). It has been reported that Act1 mRNA was in a wide variety of normal human tissues and organs (Li et al., Citation2000; Rothwarf et al., Citation1998), according to Northern blot analysis results, with high levels in thymus, kidney and placenta; moderate levels in heart, skeletal muscle, colon, liver, lung and small intestine; and a very low levels in brain, spleen and peripheral blood leukocytes. Li et al. (Citation2000) suggested that Act1 activates IKK, subsequently releases NF-κB from its complex with IκB and activates NF-κB.
Interleukin (IL)-17, a proinflammatory cytokine mainly produced by the Th17 cell line, is required by the host to defend against bacterial and fungal infection. IL-17 plays a critical role in the pathogenesis of inflammatory and autoimmune diseases such as RA, systemic lupus erythematosus, multiple sclerosis and psoriasis (Annunziato et al., Citation2009; Di Cesare et al., Citation2009; Miossec et al., Citation2009). The expression of IL-17 (also named IL-17A) can recruit neutrophils to sites of inflammation (Korn et al., Citation2009; Xu & Cao, Citation2010). Previous reports (Chang et al., Citation2006) revealed that IL-17 receptors (IL-17Rs) use Act1 as a membrane-proximal factor to initiate the signaling. Act1 that was stimulated by IL-17 has been shown to be recruited to IL-17R via SEFIR-SEFIR domain interaction. Deficiency of Act1 in fibroblasts leads to the impairment of NF-κB and abolishes IL-17-induced cytokine and chemokine expression, indicating that Act1 is an essential adaptor of IL-17 receptor in induction of inflammatory genes. Therefore, Act1 might play a critical role in IL-17-mediated inflammatory responses and autoimmune diseases.
Animal studies indicate that IL-17 acts as an important mediator of experimentally induced autoimmune and inflammatory disorder, such as collagen-induced arthritis (CIA) in mice (Lubberts et al., Citation2004; Nakae et al., Citation2003). In humans, IL-17 plays a key role in the regulation of inflammatory processes in RA (Honorati et al., Citation2001). It was found that Act1-deficient mice on a C57BL/6(B6) background were less susceptible to the development of myelin oligodendrocyte glycoprotein-induced experimental autoimmune encephalomyelitis (EAE) (Qian et al., Citation2007). Moreover, tissue-specific deletion of Act1 in the gut ameliorates disease progression in dextran sodium sulfate-induced colitis, both of which are IL-17-dependent inflammatory diseases. In both cases, the absence of Act1 correlates with lower inflammatory cytokine production by astrocytes (EAE model) or by intestinal epithelial cells (colitis model).
In the current study, we aim to investigate whether Act1 is required for IL-17-dependent inflammation and whether IL-17 can induce production of IL-6 and IL-8 through the Act1 pathway and whether the aberrant expression of Act1 gene is involved in the pathogenesis of RA. The results will provide the evidence for the further research on the pathogenesis of RA and will represent a new direction for the improved treatment of RA.
Materials and methods
Cell line
The human synovial sarcoma cell line SW982 (ATCC No. HTB-93) was purchased from Changsha Wanbai Biotechnology Co., Ltd. (Changsha, Hunan Province, China). The primary cultured RA fibroblast-like synovial cells were provided by Prof. Hu Yunxia, Tianjin Medical University. The cells were cultured in the medium consisting of 90% RPMI-1640 Medium, 10% fetal calf serum, 200 kU/l benzylpenicillin and 100 mg/l streptomycin in a humidified incubator with 5% CO2, and 37 °C for 3–4 days.
Transfection of small interfering RNA
Transfection of small interfering RNA (siRNA) using DharmaFECT™1 was conducted to silence homeobox gene Act1 expression in SW982 cell. The sequences of four different siRNA plasmids are TRAF3IP2-si 1: -5′-GAGCAUGGCUUACAUACUA-3′; TRAF3IP2-si 2: -5′-GUAAAUGGCUUCCAAACUG-3′; TRAF3IP2-si 3: -5′-GAUAAGACCGUGAUGAUAA-3′; TRAF3IP2-si 4: -5′-GACCAAAGUUUACCUAAUG-3′. The sequence for control-siRNA is -5′-UAGCGACUAAACACAUCAAUU-3′, which cannot encode the target gene. The transfection procedure was adapted from the instruction of DharmaFECT™.
Preparation of total RNA and RT-PCR
SW982 cells were seeded in six-well plastic plates in the growth medium, the cells were harvested and the total RNA was extracted using Trizol reagent, following the instructions provided by the manufacturer. RNA concentration was estimated by absorbance at 260 nm using a UV spectrophotometer (Shimadzu, Kyoto, Japan). RNA purity, estimated by the ratio of absorbance at 260 nm to absorbance at 280 nm, was found to be between 1.8 and 2.0, thus fulfilling the criteria for use in the next step. RT-PCR was done according to the instructions provided by manufacturer. Total RNA 1 μg was added into 50 μl RT-PCR reaction system containing Mg2+ 0.5 mM and a pair of primers 0.3 μg × 2. Sequences of the Act1 primers specific to humans were: -5′-ACAGAGAGACTGCTCCTTTCC-3′ and -5′-GTATTTGGGGCTGATTGCT-3′. The expected size of Act1 was 415 bp. Sequences of β-actin primers were: -5′-AAACTGGAACGGTGAAGGTG-3′ and -5′-GTGGCTTTTAGGATGGCAAG-3′. The reaction mixtures were incubated for 5 min at 95°C to denature the template, and then 30 cycles (45 s at 95°C, 45 s at 55°C and 45 s at 72°C) and a final cycle (5 min at 72°C) were performed. The RT-PCR products were stored at 4°C. RT-PCR product 10 μl and EDNA (ECor I/Hind III restriction digest) marker were electrophoresed in 2% agarose gel, stained with 0.5 μg/ml ethidium bromide, visualized under an ultraviolet light and photographed with a UVP gel imaging system (Street Upland, CA). The amount of RT-PCR product was semi-quantitatively determined by measuring the density of the specific bands.
Western blot
SW982 cells were harvested and were homogenized with RIPA lysis buffer (Beyotime Biotechnology, Haimen, Jiangsu Province, China). The protein concentrations of all of the samples were determined using a BCA assay kit (Beyotime Biotechnology). The samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (10% acrylamide/0.27% N.N′-methylenebisacryalamide resolving gel) for approximately 30 min at 80 V in stacking gel and approximately 1 h at 120 V in resolving gel. Proteins were transferred electrophoretically to Immobilon-P transfer membranes (Millipore, Bedford, MA) at 0.28 A for 1 h. The membranes were washed with TBST (Tris-buffered saline plus 0.05% Tween-20, pH 7.4) before dipping in blocking buffer (5% skimmed dry milk in TBST) overnight at 4 °C. The membranes were then incubated for 1 h at room temperature with primary antibody against human Act1 (1:500, Cell Signaling, Danvers, MA) and β-actin (1:2000, A5316; Sigma, St. Louis, MO) in TBST plus 5% bovine serum albumin. After the membranes were shaken in 4 × 6 min washes in TBST buffer, the blots were incubated for 45 min at room temperature with horseradish peroxidase-conjugated secondary antibody (rabbit anti-human IgG; Santa Cruz Biotechnology, Santa Cruz, CA, and Vector Labs, Burlingame, CA, respectively) diluted 1:5000 in blocking buffer. The blots were then shaken in 4 × 6 min washes in TBST. Afterward, the blots were incubated with a layer of Super Signal-enhanced chemiluminescence substrate mixture (Pierce Biotechnology, Rockford, IL) for 1 min at room temperature. Finally, the blots were exposed against X-ray film (Eastman Kodak Company, Rochester, NY). Band intensities were quantified using Quantity One software (version 4.0.3) from Bio-Rad (Hercules, CA).
Enzyme-linked immunosorbent assay
The SW982 cells were seeded at a density of 5 × 105/ml and were classified into the following four groups (n = 9 per group): control, IL-17 (20 ng/ml), control-siRNA + IL-17 (20 ng/ml), Traf3ip2-siRNA IL-17 (20 ng/ml). After incubation for another 24 h, the medium was collected and the IL-6 and IL-8 levels were determined with the enzyme-linked immunosorbent assay (ELISA) assay kit (Jingmei Biotech Co., Ltd, Shenzhen, Guangdong Province, China). The enzyme substrate 3′,5,5′-tetramethylbenzidine (Sigma-Aldrich, St. Louis, MO) produces a colored end product that can be read spectrophotometrically at 450 nm.
Data analysis
The data are expressed as mean ± SEM. The statistical analyses were performed using a one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test (see “Results” section for details). Values of p < 0.05 were considered statistically significant.
Results
Act1 expresses in human synovioblast cells and the SW982 cells
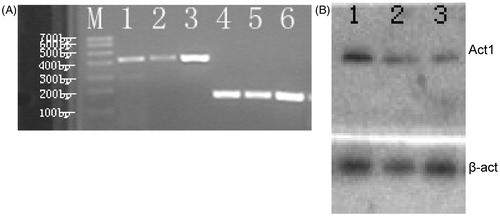
The levels of Act1 mRNA and protein expression were measured in primary cultures of human synoviocyte in RA patients after the stimulation by IL-17 (20 ng/ml). The results of RT-PCR showed that Act1 mRNA was stimulated in human synovioblast and the SW982 cell line by administration of IL-17 (). Similarly, the Act1 protein level was also induced in human synovioblast and SW982 cells by IL-17 as determined by Western blot assay ().
Figure 1. Act1 mRNA expressed in synovioblast cells and SW982 cells. (A) Gel image of RT-PCR product for Act1 mRNA. Lines 1, 2: Act1 mRNA (415 bp) in primary cultured RA fibroblast-like synovial cells; line 3: Act1 mRNA in SW982 cells; lines 4–6: β-actin mRNA (220 bp) in SW982 cells; M: Trans DNA Marker I. (B) Representative Western blot bands of Act1 and β-actin are shown. Line 1: SW982; lines 2–3: primary cultured RA fibroblast-like synovial cells.
Changes of Act1 mRNA expression level were induced by different concentrations of IL-17 in SW982 cells
The Act1 mRNA expression level increased significantly after stimulation with IL-17 (5–100 ng/ml) in SW982 cells (). The level of Act1 mRNA expression correlated positively with the concentration of IL-17 (p < 0.01, ). These data indicated that increased IL-17 concentration was attributable to elevated levels of Act1 mRNA in SW982 cells. These findings suggested that IL-17 increased Act1 mRNA levels in a concentration-dependent way.
Figure 2. Act1 mRNA levels are induced by different concentrations and different incubation times of IL-17 in SW982 cells. (A) Gel image of RT-PCR product for Act1 mRNA 4 h after stimulation by IL-17. Lines 1–6: Act1 mRNA in 100, 50, 20, 10, 5, 0 ng/ml IL-17; lines 1′–6′: the corresponding β-actin mRNA. (B) The quantitive analysis for Act1 mRNA in SW982 cells. The Act1 mRNA level was increased with the elevated concentration of IL-17. The data are expressed as mean ± SEM (n = 9 per group). **p < 0.01, ***p < 0.001, compared with 0 ng/ml IL-17. (C) Gel image of RT-PCR product for Act1 mRNA after stimulation by 20 ng/ml IL-17. Lines 1–6: Act1 mRNA in 0 min, 15 min, 30 min, 1 h, 2 h, 4 h after induction by IL-17; lines 1′–6′-: the corresponding β-actin mRNA. (D) The quantitative analysis for Act1 mRNA in SW982 cells. The Act1 mRNA level was increased with the stimulation time of IL-17. The data are expressed as mean ± SEM (n = 9 per group). *p < 0.05, **p < 0.01, ***p < 0.001, compared with IL-17 at 0 min.
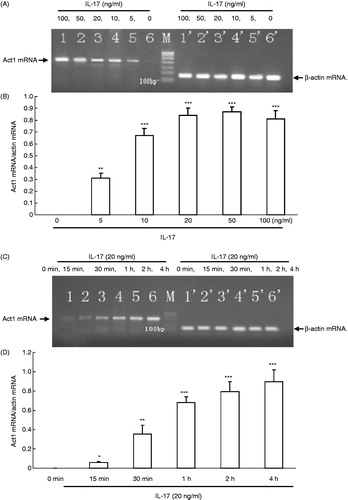
Changes of Act1 mRNA expression level were induced by IL-17 at different timepoints in SW982 cells
RT-PCR data revealed that Act1 mRNA expression level was significantly increased by IL-17 after incubation for 15 min, 30 min, 1 h, 2 h and 4 h at the concentration of 20 ng/ml (). The level of Act1 mRNA is dependent on the different incubation time of IL-17. Additionally, the results showed that the Act1 mRNA levels in SW982 cells were time-dependent on the stimulation of IL-17 (p < 0.01, ).
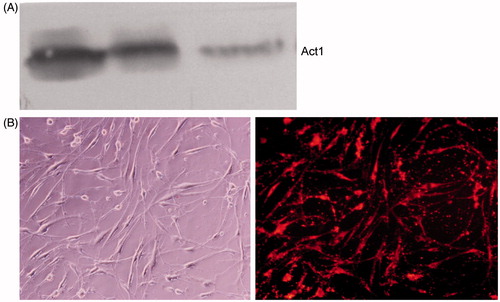
Traf3ip2 siRNA decreased Act1 protein level in SW982 cells
As shown in , Traf3ip2 siRNA significantly inhibited the expression level of Act1 protein compared with negative control in SW982 cells. Western blot assay revealed that Traf3ip2 siRNA transfection can effectively silence Act1 gene as shown by the reduced Act1 protein level. The DY-547-labeled SW982 cells after transfection with Traf3ip2 gene for 48 h were shown in .
Figure 3. Traf3ip2 siRNA significantly decreased Act1 protein expression level in SW982 cells. (A) Representative Western blot bands of Act1 are shown. Lines 1–2: control-siRNA; line 3: Traf3ip2-siRNA. (B) The DY-547-labeled SW982 cells after transfection with Traf3ip2 gene for 48 h. Left: common microscopy; right: fluorescence microscopy (×400).
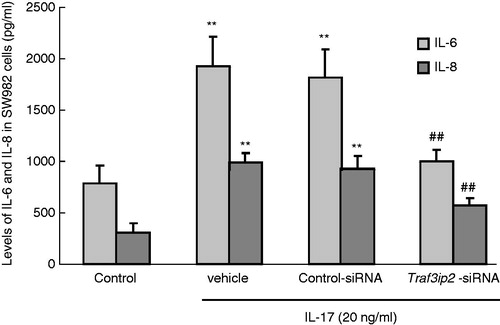
Effects of Traf3ip2 siRNA on the level of IL-6 and IL-8 stimulated by IL-17 in SW982 cells
Data from the ELISA assay revealed that IL-17 (20 ng/ml) significantly increased IL-6 levels compared with the control group (1927.4 ± 288.77 versus 786.5 ± 172.42 ng/ml, one-way ANOVA, p < 0.01) after stimulation for 24 h. However, transfection of Traf3ip2 siRNA markedly decreased IL-6 levels compared to the transfection negative control (995.9 ± 115.30 ng/ml versus 1816.1 ± 273.27 ng/ml, p < 0.01). These results suggest that inhibition of Act1 expression level through Traf3ip2 siRNA reduced the production of IL-6 level stimulated by IL-17 in SW982 cells (). Furthermore, we also measured IL-8 levels in SW982 cells using ELISA analysis. The results showed that IL-17 (20 ng/ml) significantly increased IL-8 levels compared with the control group (984.8 ± 95.09 ng/ml versus 307.1 ± 90.83 ng/ml, p < 0.01) after stimulation for 24 h. Consistent with the changes of IL-6, transfection of Traf3ip2 siRNA markedly decreased IL-8 levels compared to transfection negative control (575.6 ± 65.96 ng/ml versus 929.4 ± 124.39 ng/ml, p < 0.01, ). These findings suggested that IL-8 levels induced by IL-17 in SW982 cells could be reversed by down-regulation of Act1 expression level with Traf3ip2 siRNA.
Figure 4. IL-6 and IL-8 were induced in SW982 cells with various treatments. IL-17 (20 ng/ml) significantly increased IL-6 and IL-8 in SW982 cells, while transfection of Traf3ip2 siRNA markedly decreased IL-6 and IL-8 level compared to transfection control-siRNA group. The data are expressed as mean ± SEM (n = 9 per group). **p < 0.01 compared with the control group; ##p < 0.01 compared with the control-siRNA group.
Discussion
In the current study, we found that Act1 mRNA and protein levels were increased by stimulation with IL-17 in human synovioblast. The increased IL-17 concentration and incubation time contribute to the elevated levels of Act1 mRNA in SW982 cells, suggesting that IL-17 increases Act1 mRNA levels in a concentration- and time-dependent way. In addition, Traf3ip2 siRNA transfection can effectively silence Act1 gene expression level. Finally, inhibition of Act1 expression by Traf3ip2 siRNA decreased IL-6 and IL-8 levels induced by IL-17 in SW982 cells. These results suggest that Act1, a NF-κB-activating protein, might be mediated in the pathophysiology of RA and could be the treatment target of this disorder.
The transcription factor NF-κB has been well recognized as a pivotal regulator of inflammation in RA. It has been evidenced that NF-κB is involved in abnormal apoptosis and proliferation of RA fibroblast-like synovial cells, as well as differentiation and activation of bone resorbing activity of osteoclasts (Makarov, Citation2001). Therefore, NF-κB is identified as one of the central downstream targets regarding the pathobiology of RA. In addition, NF-κB inhibition may be a potentially important therapeutic approach for RA. Studies in animal models of RA have demonstrated the high therapeutic efficacy of specific inhibitors of NF-κB pathway, suggesting the feasibility of anti-NF-κB therapy for human disease. Many conventional anti-inflammatory and anti-rheumatic drugs can effectively inhibit NF-κB activation.
NF-κB also plays a pivotal role in numerous cellular processes, including stress response, inflammation and protection against apoptosis. Basal NF-κB activity is required for cell normal development, particularly for the protection of liver against apoptosis (Alonzi et al., Citation1998). Furthermore, inactivation of the NF-κB pathway may be associated with detrimental side effects. Therefore, the activity of NF-κB needs to be tightly regulated. May et al. (Citation2000) designed a peptide derived from NEMO/IKKγ to block the assembly of IKK signalsome. Act1 is able to bind the regulatory subunit NEMO/IKKγ and to activate NF-κB. Additionally, inhibition of Act1 overexpression maybe also suppresses cytokine-inducible NF-κB activation without interfering with basal NF-κB activity. Act1 is able to bind to tumor necrosis factor receptor-associated factor3 (TRAF3) and functions as a receptor proximal-adaptor that is involved in CD40-mediated NF-κB and JNK activation, leading to the inhibition of CD40L-induced apoptosis in epithelial cells (Qian et al., Citation2002). Additionally, the expression of Act1 in Act1-null C33A cell confers CD40-mediated NF-κB activation and protects them from CD40L-induced apoptosis. One hallmark of RA synovial tissue is pronounced hyperplasia. Rheumatoid synovial cells overgrowth is a result of more cell proliferation than apoptosis.
Leonardi et al. used NEMO/IKKγ as bait and identified a NEMO/IKKγ-interacting protein in a yeast two-hybrid screen, they called it CIKS (connection to IKK and stress activated protein kinase(SAPK)/JNK) (Li et al., Citation2000). CIKS stimulated IKK, p38, and Jun kinase (JNK), and transactivated an NF-κB-dependent reporter, thus connecting upstream signaling events to IKK, SAPK and JNK. It has been demonstrated that CIKS and Act1 have the similar sequence domain (Novatchkova et al., Citation2003). Act1 does not have any known enzymatic domain; instead it contains a helix-loop-helix motif at the amino terminus and a coiled coil at the carboxyl terminus (Li et al., Citation2000; Rothwarf et al., Citation1998). Forced expression of Act1 activates NF-κB and JNK/SAPK pathways, suggesting that Act1 may act as an adaptor protein, linking upstream signaling pathways to the NF-κB and JNK/SAPK pathways. IKK is composed with two catalytic subunits, IKKα and IKKβ (Zandi et al., Citation1997), and a regulatory subunit called NF-κB essential modulator (NEMO) or IKKγ (Rothwarf et al., Citation1998). Several proteins have been demonstrated to interact with NEMO/IKKγ, and to regulate the activation of the NF-κB pathway in response to different stimuli, one of which is the cytoplasmic adaptor protein Act1 (Leonardi et al., Citation2000; Li et al., Citation2000).
The Act1-mediated activation of NF-κB might protect the synovial cells from apoptosis in RA. Inhibition of the expression of Act1reduced synovial cell apoptosis. Hence, selective targeting of Act1 in RA patients could be considered as a novel therapeutic strategy. RNA interference (RNAi), a remarkable new research tool to control gene expression, is a recently characterized gene silencing pathway by which specific mRNAs are either degraded or translationally suppressed (Zamore & Haley, Citation2005). The therapeutic application of RNAi has been performed against a variety of diseases. The potential application of RNAi-based therapy in rheumatoid disorders is evaluated. The effects that TNF-α was silenced by local and systemic delivery of siRNA have been evaluated in experimental arthritic models (Inoue et al., Citation2005; Schiffelers et al., Citation2005). The siRNA designed to silence TNF-α, was delivered into joints by electroporation against experimental arthritis in animal models and strongly inhibited joint inflammation by inhibition of the expression of TNF-α. The current results indicated that the siRNA designed to inhibit Act1 is a feasible therapeutic tool for treatment of joint disorders in clinic. These findings have important implications for future research on pathophysiological pathways of RA and treatment.
Pisitkun et al. (Citation2010) found that Act1-mediated signaling is necessary for the pathogenesis of CIA. Their findings suggest that Act1 plays a critical role in the development of arthritis in the CIA model, including the formation of CII antibodies. Act1-mediated signaling is responsible for inflammatory cell infiltration into joints, for bone erosion, and for cartilage damage, and significantly contributes to CII-specific antibody production. The total dependence of pathologic changes in the joint on Act1 mediated signaling may reflect multiple contributions of IL-17 family cytokines to the development of arthritis. Studies from OA and skin fibroblasts showed that IL-17 enhanced the production of IL-6 and IL-8 (Katz et al., Citation2001). Overproduction of IL-17 can elevate the production of proinflammatory mediators, such as TNF-α, IL-1β, IL-6 and IL-8 in a variety of cells (Katz et al., Citation2001). Of these proinflammatory cytokines, IL-6 and IL-8 are most likely major instigators of RA joint inflammation with the evidence that IL-6 or IL-8 gene knockout leads to protection against arthritis (Alonzi et al., Citation1998).
Conclusions
The present results showed that the expression of Act1 mRNA and protein was elevated after stimulation with IL-17 in SW982 cells. Transfection of siRNA led to silence homeobox gene Act1 expression and reduced the levels of IL-6 and IL-8 in SW982 cell. IL-17 stimulation on Act1 deficient cells resulted in the reduction of IL-6 and IL-8. These results suggested that elevated expression of IL-6 and IL-8 induced by IL-17 may be involved in the mechanisms of RA via the Act1 pathway. Therefore, inhibition of the expression of Act1 may be a therapeutic target of RA, and Act1 regulates the inflammation process in RA as a potentially adaptor. Taken together, this evidence revealed that Act1 plays a key role in the pathogenesis of RA. Act1 and related signaling pathways may provide highly sensitive targets for therapeutic intervention in the development of RA. Most probably, blocking the expression of Act1 may be a new therapeutic target for RA.
Declaration of interest
The authors declare that they do not have any conflicts of interest.
References
- Alonzi T, Fattori E, Lazzaro D, et al. (1998). Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med 187:461–8
- Annunziato F, Cosmi L, Liotta F, et al. (2009). Type 17 T helper cells-origins, features and possible roles in rheumatic disease. Nat Rev Rheumatol 5:325–31
- Asahara H, Asanuma M, Ogawa N, et al. (1995). High DNA-binding activity of transcription factor NF-kappa B in synovial membranes of patients with rheumatoid arthritis. Biochem Mol Biol Int 37:827–32
- Baldwin AS Jr. (1996). The NF-kappa B and I kappa B proteins: New discoveries and insights. Annu Rev Immunol 14:649–83
- Chang SH, Park H, Dong C. (2006). Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem 281:35603–7
- Di Cesare A, Di Meglio P, Nestle FO. (2009). The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J Invest Dermatol 129:1339–50
- Han Z, Boyle DL, Manning AM, Firestein GS. (1998). AP-1 and NF-kappa B regulation in rheumatoid arthritis and murine collagen-induced arthritis. Autoimmunity 28:197–208
- Handel ML, McMorrow LB, Gravallese EM. (1995). Nuclear factor-kappa B in rheumatoid synovium. Localization of p50 and p65. Arthritis Rheum 38:1762–70
- Honorati MC, Meliconi R, Pulsatelli L, et al. (2001). High in vivo expression of interleukin-17 receptor in synovial endothelial cells and chondrocytes from arthritis patients. Rheumatology (Oxford) 40:522–7
- Inoue A, Takahashi KA, Mazda O, et al. (2005). Electro-transfer of small interfering RNA ameliorated arthritis in rats. Biochem Biophys Res Commun 336:903–8
- Katz Y, Nadiv O, Beer Y. (2001). Interleukin-17 enhances tumor necrosis factor alpha-induced synthesis of interleukins 1, 6, and 8 in skin and synovial fibroblasts: A possible role as a “fine-tuning cytokine” in inflammation processes. Arthritis Rheum 44:2176–84
- Korn T, Bettelli E, Oukka M, Kuchroo VK. (2009). IL-17 and Th17 Cells. Annu Rev Immunol 27:485–517
- Leonardi A, Chariot A, Claudio E, et al. (2000). CIKS, a connection to Ikappa B kinase and stress-activated protein kinase. Proc Natl Acad Sci USA 97:10494–9
- Li X, Commane M, Nie H, et al. (2000). Act1, an NF-kappa B-activating protein. Proc Natl Acad Sci USA 97:10489–93
- Lubberts E, Koenders MI, Oppers-Walgreen B, et al. (2004). Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50:650–9
- Makarov SS. (2001). NF-kappa B in rheumatoid arthritis: A pivotal regulator of inflammation, hyperplasia, and tissue destruction. Arthritis Res 3:200–6
- Marok R, Winyard PG, Coumbe A, et al. (1996). Activation of the transcription factor nuclear factor-kappaB in human inflamed synovial tissue. Arthritis Rheum 39:583–91
- May MJ, D'Acquisto F, Madge LA, et al. (2000). Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science 289:1550–4
- Miossec P, Korn T, Kuchroo VK. (2009). Interleukin-17 and type 17 helper T cells. N Engl J Med 361:888–98
- Nakae S, Nambu A, Sudo K, Iwakura Y. (2003). Suppression of immune induction of collagen-induced arthritis in IL-17-deficient mice. J Immunol 171:6173–7
- Novatchkova M, Leibbrandt A, Werzowa J, et al. (2003). The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci 28:226–9
- Pahl HL. (1999). Activators and target genes of Rel/NF-kappa B transcription factors. Oncogene 18:6853–66
- Pisitkun P, Claudio E, Ren N, et al. (2010). The adaptor protein CIKS/ACT1 is necessary for collagen-induced arthritis, and it contributes to the production of collagen-specific antibody. Arthritis Rheum 62:3334–44
- Qian Y, Liu C, Hartupee J, et al. (2007). The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 8:247–56
- Qian Y, Zhao Z, Jiang Z, Li X. (2002). Role of NF kappa B activator Act1 in CD40-mediated signaling in epithelial cells. Proc Natl Acad Sci USA 99:9386–91
- Rothwarf DM, Zandi E, Natoli G, Karin M. (1998). IKK-gamma is an essential regulatory subunit of the IkappaB kinase complex. Nature 395:297–300
- Schiffelers RM, Xu J, Storm G, et al. (2005). Effects of treatment with small interfering RNA on joint inflammation in mice with collagen-induced arthritis. Arthritis Rheum 52:1314–18
- Xu S, Cao X. (2010). Interleukin-17 and its expanding biological functions. Cell Mol Immunol 7:164–74
- Zamore PD, Haley B. (2005). Ribo-gnome: The big world of small RNAs. Science 309:1519–24
- Zandi E, Rothwarf DM, Delhase M, et al. (1997). The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell 91:243–52