
Abstract
Context: Toll-like receptor 4 (TLR4)-independent inflammatory and apoptosis responses contribute to β-cell failure in diabetes mellitus (DM). Berberine (BBR), a bioactive isoquinoline derivative alkaloid, ameliorates the inflammatory response in DM.
Objective: This study explored the protective mechanisms of BBR on TLR4-independent inflammation response in β cells.
Materials and methods: Lipopolysaccharide (LPS; 100 ng/ml) was used to induce the inflammatory response in NIT-1 and rat insulinoma (INS-1) cells for 24 h. 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay and colony formation assays were used for the determination of cell viability. The levels of monocyte chemoattractant protein (MCP-1), interleukin 6 (IL-6), tumor necrosis factor α (TNF-α) and insulin in cultured supernatant were detected by enzyme-linked immunosorbent assay kits. Western blot analysis was performed for the expression of p-c-Jun N-terminal kinase (JNK) and p65 NF-κB in NIT-1 cells, and p65 NF-κB in INS-1 cells.
Results: BBR (1.25, 2.5 and 5 μM) or TLR4 inhibitor (TAK-242, 1 μM) increased remarkably NIT-1 cell viability by 72.6 ± 5.0, 85.9 ± 9.3, 94.7 ± 7.1 and 92.6 ± 8.4%. The EC50 of BBR was 1.14 μM. Colony formation assay showed that BBR increased the number of colonies of NIT-1 and INS-1 cells. BBR, TAK-242 or SP-600125 (1 μM) could significantly reduce the levels of MCP-1, IL-6 and TNF-α, insulin and JNK and NF-κB phosphorylation in NIT-1 cells, as well as the p65 NF-κB in INS-1 cells.
Discussion and conclusion: BBR could ameliorate LPS-induced β-cell injury through the TLR4-independent JNK/NF-κB pathway. Thus, this pathway may be a potential target for the prevention and treatment of DM.
Introduction
Diabetes mellitus (DM) has been characterized by progressive β-cell dysfunction and insulin resistance, which are associated with the inflammatory response (Lontchi-Yimagou et al., Citation2013). Metabolic disorders contribute to β-cell dysfunction and insulin resistance in DM patients and induce the increase of inflammatory cytokines such as C-reactive protein and interleukin 6 (IL-6) (Edalat et al., Citation2013). A growing body of evidence supports the fact that the inflammatory response may alter the normal structure of β-cells and induce insulin resistance as well as decrease insulin secretion (Akash et al., Citation2013). Thus, it is beneficial for the improvement of β-cell function to attenuate the inflammation response.
Many cytokines have been found to contribute to the occurrence and development of β-cell dysfunction. Toll-like receptor 4 (TLR4), a type I transmembrane protein and one of the main receptors of the TLR family, has been directly implicated in proinflammatory signaling in β-cell dysfunction (Chavez et al., Citation2011; Ma et al., Citation2012). The activation of TLR4 is essential for the activation of NF-κB signaling downstream of receptor such as c-Jun N-terminal kinase (JNK) (Hamidi et al., Citation2012). Studies confirmed that the JNK/NF-κB signaling pathway was associated closely with the activation of TLR4 and contributed to the release of inflammatory mediators, such as tumor necrosis factor α (TNF-α), IL-6, interferon-β and monocyte chemoattractant protein (MCP-1) (Song et al., Citation2013).
Berberine (BBR), one of the main alkaloids of Rhizoma coptidis, has been proven to have anti-diabetic potential (Wu et al., Citation2012) (). Investigation of the molecule mechanism showed that adenosine monophosphate-activated protein kinase might contribute to the action of BBR in type 2 DM (Shen et al., Citation2012). In diabetic rats, BBR, prior to the administration of lipopolysaccharide (LPS), could increase the levels of inflammatory cytokines, and could also activate TLR4 and NF-κB (Zhang et al., Citation2011). Furthermore, BBR might also inhibit JNK activation in Th1 T-cell differentiation in type 1 diabetic mice (Cui et al., Citation2009). However, it is unclear whether BBR attenuates the inflammatory response in β-cell dysfunction via the TLR4-independent pathway.
Figure 1. The structure of BBR (chemical formula: C20H18NO4, molecular weight: 336.36).
The purpose of this study was conducted to investigate the potential effect of BBR on the TLR4-dependent pathway, which was responsible for LPS-induced inflammation and apoptosis responses in pancreatic β-cell dysfunction.
Materials and methods
General
Ultra sensitive mouse insulin enzyme-linked immunosorbent assay (ELISA) kit was purchased from Crystal Chem Inc. (Downers Grove, IL). MCP-1, IL-6 and TNF-α ELISA kits were ordered from Boster Biological Engineering Co. Ltd. (Wuhan, China). TLR4 inhibitor, TAK-242 (greater than 98% purity), was from Chembest Co. Ltd. (Shanghai, China). Antibodies to TLR4, p-JNK and NF-κB p65 were from Santa Cruz Biotechnology (Santa Cruz, CA). JNK selective inhibitor (SP-600125) was provided by Shanghai Haoran Bio Technologies Co. Ltd. (Shanghai, China). BBR (purity > 99%) was purchased from Nanjing Zelang Pharmaceutical Technology Co. Ltd. (Nanjing, China). 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) and LPS L2880 were obtained from Sigma (St. Louis, MO). All the other biochemicals and chemicals used in the experiment were of analytical grade.
Cell culture
The pancreatic β-cell NIT-1 cell line and rat insulinoma (INS-1) β-cell line were purchased from American Type Culture Collection. NIT-1 cells were cultured in Dulbecco Minimum Essential Medium (DMEM) supplemented with 25 mM glucose, 2 mM l-glutamine, 100 U/l penicillin, 100 mg/l streptomycin and 10% heat-inactivated fetal calf serum. INS-1 β cells were incubated in RPMI 1640 medium supplemented with 10% FBS, 1% penicillin/streptomycin, 11.1 mM glucose and 2 mM l-glutamine. Cells were cultured in 25 cm2 culture flasks and maintained in a humidified atmosphere of 5% (v/v) CO2 incubator at 37 °C. Cells were passaged by 0.25% trypsin and 0.01% ethylenediaminetetraacetic acid (EDTA) for 2–3 min. Culture medium was exchanged every two days.
MTT assay for cell viability
NIT-1 cells (1 × 105 cells/ml) were seeded into the wells of a 96-well plate and cultured in DMEM for 24 h. Cells were pre-treated with BBR (0.1813, 0.3625, 0.625, 1.25, 2.5, 5.0 and 10 μM) for 1 h prior to exposure to LPS (1 μg/ml for cell viability) stimulation for another 24 h (Hussey et al., Citation2012). Medium was used for vehicle control for 1 h. At the end of the incubation, the cultured supernatants were taken for ELISA assay and then new serum-free medium was added. Simultaneously, fresh MTT solution (5 mg/ml in phosphate-buffer saline; PBS) of 20 μl/well was added and incubated for another 4 h. Sequentially, the supernatant of per well was discarded and DMSO (100 μl/well) was added to dissolve the formed crystals. The solutions were mixed in a shaker for 10 min. The optical density (OD) of samples was measured in a microplate spectrophotometer at 570 nm. The relative cell viability was calculated by following formula: (OD of samples/OD of blank control) × 100%.
Colony formation assays
The reduction in colony number of cells was closely related to the cell cycle arrest or cell death. Colony formation assays were typically used for evaluating the cloning capability of cells. In this study, NIT-1 cells or INS-1 cells (200 cells/well) were trypsinized, counted and seeded into per well in a 12-well plate for a colony formation assay. After being treated with drugs for 10 d, 200 µl of crystal violet staining solution was added to stain the colonies purple and co-incubated with cells at room temperature for 20 min. The number of colonies purple was counted for cell viability.
ELISA assay
NIT-1 cells were treated with BBR (1.25, 2.5 and 5.0 μM), TLR4 inhibitor TAK-242 (1 μM) or JNK selective inhibitor SP-600125 (1 μM) prior to 100 ng/ml LPS stimulation (Hussey et al., Citation2012). The levels of inflammation factors and insulin secretion were measured using ELISA kits. For measuring insulin released from NIT-1 cells, the amount of LPS-stimulated insulin in the culture medium was measured according to the manufacturer’s protocols. The ultra sensitive ELISA kit has specific reaction with mature mouse insulin, while no reaction with other forms, such as pro-insulin. For inflammatory factors, the levels of MCP-1, IL-6 and TNF-α in cell culture medium were measured. After being mixed for 5 min in a shaker, the OD values of all samples were determined in a microplate reader at 450 nm wavelength. The protein concentrations of insulin, MCP-1, IL-6 and TNF-α were calculated according to the standard curve.
Flow cytometric analysis on cell cycle
In order to explore the effects of BBR on LPS-induced cell cycle distribution, NIT-1 cells (1 × 105 cells/ml) were pre-treated with BBR (2.5 and 5.0 μM), TAK-242 (1 μM) or SP-600125 (1 μM) following 100 ng/ml LPS stimulation and then cultured for 24 h. At the end of incubation, these cells were harvested and centrifuged (3000 g for 10 min) and then washed with 5 ml ice-cold PBS for three times. After being fixed by 70% ethanol overnight, these cells were further washed twice with cold PBS and treated with RNase (10 μg/ml) at 37 °C. Finally, these cells were stained with 10 μg/ml propidium iodide for 30 min at room temperature in the dark. The cell cycle was analyzed by fluorescent-activated cell sorting flow cytometry (Becton-Dickinson, Franklin Lakes, NJ).
Western blot analysis for JNK and NF-κB protein expression
After being treated with drugs, NIT-1 cells and INS-1 β-cells were washed with phosphate buffered saline (PBS; pH = 7.2) and lysed in an ice-cold lysis buffer containing 10% glycerol, 1% Triton X-100, 1 mM Na3VO4, 1 mM EGTA, 10 mM NaF, 1 mM Na4P2O7, 20 mM Tris buffer (pH = 7.4), 100 mM β-glycerophosphate, 137 mM NaCl and 5 mM EDTA as described previously (Zohn et al., Citation1995). Cells were harvested, boiled and centrifuged at 12 000 g for 30 min at 4 °C. Orderly, equal amounts of proteins (50 μg) in supernatant were loaded onto 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis and then transferred to PVDF membranes. Sequentially, the membranes were blocked with 5% skim milk for 2 h at room temperature. After incubation, the membranes were probed by primary antibodies p-JNK (1:1000) and p65 NF-κB (1:1000) in 5% skim milk for another 2 h at 37 °C. After being washed three-times, membranes were incubated with HRP-conjugated IgG secondary antibodies for 1 h at 37 °C. The visualizations of brands were developed by enhanced chemiluminescence reagent. β-Actin was used for the internal reference. The relative intensities of brands were quantified by Image pro plus software (Media Cybernetics, Silver Spring, MD).
Statistical analysis
Data were presented as mean ± standard deviation (SD). The differences among the groups were analyzed using one-way analysis of variance with SPSS 16.0 statistic software (Chicago, IL). p < 0.05 was considered statistically significant.
Results
Effects of BBR on cell viability in NIT-1
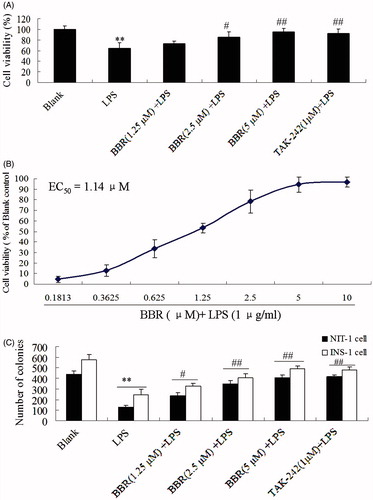
LPS of 1 μg/ml could inhibit significantly the cell viability of NIT-1 cells, compared with blank control (p < 0.01). However, the treatment with BBR at a concentration of 1.25, 2.5 and 5.0 μM markedly increased the LPS-reduced cell viability in a concentration-dependent manner (p < 0.05 and p < 0.01) (). The EC50 of BBR on reducing LPS-induced damage was 1.14 μM (). In addition, TLR4 inhibitor, TAK-242 (1 μM), might also attenuate LPS-induced cell injury. The colony formation assay was performed to detect the growth ability of NIT-1 and INS cells (). As shown in , both BBR (1.25, 2.5 and 5.0 μM) TAK-242 (1 μM) showed a significant increase in NIT-1 cells as compared to LPS group. These above data demonstrated that BBR has a potential protective activity on LPS-induced damage in NIT-1 cells.
Figure 2. Effect of BBR on LPS-induced celll viability in NIT-1 or INS-1 cells. Cells were treated with BBR at a concentration of 0.1813, 0.3625, 0.625, 1.25, 2.5, 5.0 and 10.0 μM in the presence of 1 μg/ml LPS. MTT assay was preformed in NIT-1 cells (A and B) while colony formation assays were preformed in NIT-1 cells and INS-1 cells (C). The OD value of samples was detected by MTT method at 570 nm. Data are expressed as mean ± SD (n = 18). **p < 0.01 versus blank; #p < 0.05, ##p < 0.01 versus LPS.
Effects of BBR on inflammatory cytokines
In order to evaluate the regulation of BBR on inflammation response, ELISA assay was performed to determine the levels of inflammatory cytokines, such as MCP-1, IL-6 and TNF-α in cell supernatant. After being stimulated for 24 h, LPS of 100 ng/ml increased significantly MCP-1, IL-6 and TNF-α levels. However, BBR (2.5 and 5.0 μM) decreased the levels of these inflammatory cytokines (p < 0.05 and p < 0.01). Interestingly, there was no significant difference between BBR (1.25 μM) and LPS; however, BBR at low concentration, 1.25 μM, could also reduce the mean levels of MCP-1, IL-6 and TNF-α (). More importantly, TLR4 inhibitor, TAK-242, or JNK selective inhibitor, SP-600125, could also reduce significantly the levels of these inflammatory cytokines, compared with LPS alone (p < 0.01). These results suggested that BBR could attenuate LPS-induced inflammation response contributing to NIT-1 cells dysfunction via TLR4/JNK pathway.
Table 1. Effect of BBR on LPS-induced inflammatory cytokines and insulin level (n = 12).
BBR decreases LPS-induced insulin level
When NIT-1 cells were exposed to LPS (100 ng/ml), the insulin level in cell supernatant was elevated (). However, the presence of BBR (1.25, 2.5 and 5.0 μM) exhibited significant inhibition effects on this elevation, under compared with LPS alone (p < 0.05 and p < 0.01). BBR inhibited insulin secretion in a concentration-dependent manner. More importantly, TAK-242 or SP-600125 also reduced remarkably insulin level after treatment for 24 h (p < 0.01). These results showed that the inhibition of BBR on LPS-induced insulin level might be associated with the blockade on the TLR4/JNK pathway.
BBR reduces LPS-induced apoptosis of β cells
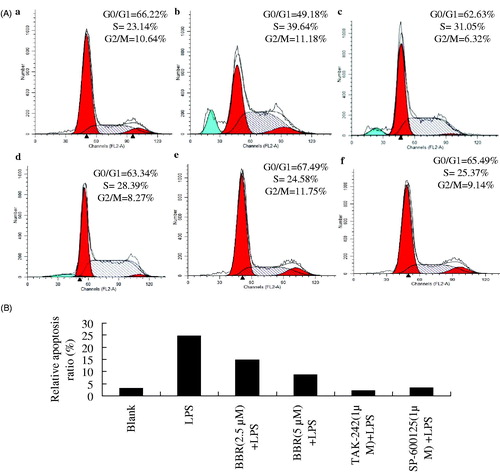
Apoptosis has been regarded as one mechanism contributing to the inflammatory response of β cells in DM. As depicted in , after being stimulated with LPS (100 ng/ml) for 24 h, the percentage of S-phase was significantly increased to 39.64% from blank control of 23.14%. However, a significant decrease on S-phase while an increase on G0/G1 phase was observed after being treated with BBR (2.5 and 5.0 μM), TAK-242 or SP-600125. In addition, the obvious LPS-induced apoptosis ratio (24.61%) was decreased evidently by the pretreatment with BBR (2.5 and 5.0 μM), TAK-242 as well as SP-60012. These data suggested a fact that BBR could attenuate LPS-induced apoptosis of β cells. This protective effect of BBR was similar to the action of TLR4 inhibitor, TAK-242, as well as JNK selective inhibitor, SP-60012.
Figure 3. Effects of BBR on cell cycle distribution and apoptosis in cultured NIT-1 cells. Flow cytometric analysis of the DNA content in control (Aa), LPS-treated model group (Ab), BBR of 2.5 μM (Ac) and 5.0 μM (Ad), TAK-242 of 1 μM (Ae) or SP-600125 of 1 μM (Af). (A) PI staining and flow cytometric analysis of cell cycle distribution in treated NIT-1 cells. (B) Relative apoptosis ratio.
BBR blocks LPS-induced cells injury via the TLR4-independent JNK-NF-κB pathway
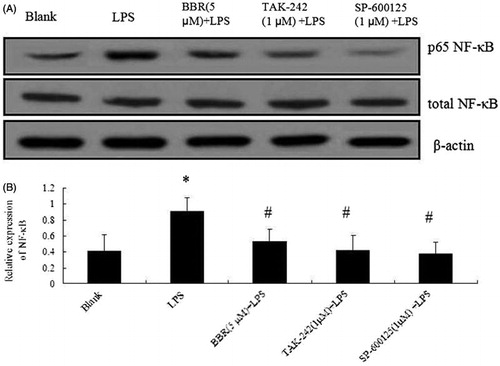
In order to further explore the mechanism of BBR on LPS-induced NIT-1 cell injury, p-JNK and p65 NF-κB phosphorylations were determined by Western blot analysis. As depicted in and , p-JNK and p65 NF-κB phosphorylations were increased by LPS alone (100 ng/ml). However, they were decreased significantly by the treatment with BBR (2.5 and 5.0 μM) (p < 0.05 and p < 0.01). When NIT-1 cells were pre-incubated with TLR4 inhibitor, TAK-242, or JNK inhibitor, SP-600125, the up-regulated phosphorylations were decreased dramatically. In order to evaluate the effect of BBR on LPS-induced β cells injury, INS-1 β cell line was also performed by Western blot analysis in this study. As shown in , a significant increase of p65 NF-κB was observed in LPS-treated cells while all BBR (5 μM), TAK-242 (1 μM) or SP-600125 (1 μM) reduced this phosphorylation in INS-1 cells. These results indicated that BBR inhibited LPS-induced β cells injury including NIT-1 cells and INS-1 cells through intervening with the TLR4/JNK/NF-κB pathway.
Figure 4. BBR attenuated JNK/NF-κB pathway in NIT-1 cells involving the inflammation response. NIT-1 cells were treated with BBR (1.25 μM, 2.5 μM and 5.0 μM), TAK-242 (1 μM) or SP-600125 (1 μM) before LPS stimulation (100 ng/ml). p-JNK (1:1000) and p65 NF-κB (1:1000) expression was detected by Western blot. β-Actin (1:1000) was used for internal reference. (A) The brands of p-JNK and p65 NF-κB; (B) the relative level of p-JNK and p65 NF-κB. Data are taken from individual experiments and expressed as mean ± SD (n = 3). **p < 0.01 versus blank control; #p < 0.05, ##p < 0.01 versus LPS alone; $$p < 0.01 versus LPS alone.
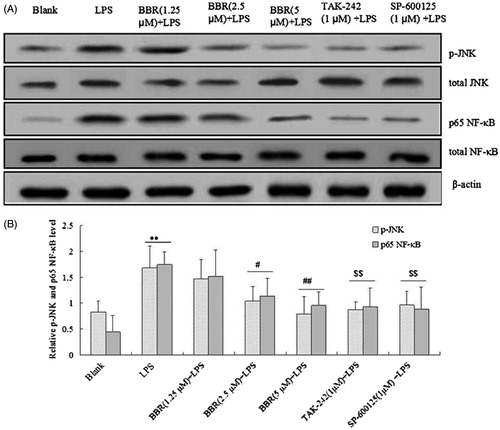
Figure 5. Effect of BBR on NF-κB expression in INS-1 cells. Cells were treated with BBR (5μM), TAK-242 (1 μM) and SP-600125 (1 μM) prior to LPS stimulation (100 ng/ml). (A) Expression of NF-κB in INS-1 cells by Western blotting. (B) Relative expression ratio of NF-κB. Data are expressed as mean ± SD (n = 3). *p < 0.05 versus blank; #p < 0.05 versus LPS.
Discussion
The inflammation response has been regarded as one mechanism associated with insulin signal transduction (Du et al., Citation2013). The secretion of inflammatory cytokines can contribute to the alterations to both insulin signaling and insulin sensitivity. Recent accumulating studies showed that the TLR4-dependent inflammation response was central to the regulation of proinflammatory markers and might lead to several types of inflammatory disorders (Ahmad et al., Citation2012). In this study, the protective activity and its underlying mechanism of BBR on LPS-induced inflammation response in NIT-1 cells were explored. Our findings indicated that BBR suppressed the LPS-induced inflammation response through the TLR4/JNK/NF-κB pathway.
TLR4, one of the members of the Toll-like receptor family, has been found to be involve in a variety of diseases associated with inflammation, such as type 2 diabetes (Li et al., Citation2013). Studies have shown that TLR4 regulates the overall function of islet β-cells and is closely related with insulin resistance in type 2 diabetes. The toxicity of glucose and lipid stimulates pancreatic β-cells to generate and mediate its downstream signaling cascade events (Hellemans et al., Citation2007). In this study, it was observed that the stimulation of LPS could increase inflammatory cytokines including MCP-1, IL-6 and TNF-α. The treatment with TLR4 inhibitor TAK-242 or BBR suppressed the expression of inflammatory cytokines in NIT-1 cells challenged with LPS. These results indicated that BBR might attenuate LPS-induced inflammation injury by blocking the early intracellular events in the TLR4 signaling.
Pro-inflammatory cytokines are important mediators of β-cell demise in type 1 and 2 diabetes (Russell et al., Citation2013). The accumulation of inflammatory cytokines can result in the loss of β-cell viability in diabetes (Mangan & Wahl, Citation1991). In this study, BBR might ameliorate LPS-induced loss in a concentration-dependent manner. It was likely that the actions of BBR in NIT-1 cells might be associated with its anti-inflammation. After being treated with TAK-242, the loss of cell viability was significantly inhibited and enhanced to a normal level. MTT assay revealed that BBR attenuated LPS-induced cell viability and inflammation response with a similar action.
Inflammatory cytokines, such as MCP-1, induced β-cell dysfunction through JNK and NF-κB related signaling pathways, contributing to the insulin resistance implicated in the development of diabetes (Cai et al., Citation2011). In this study, we examined the effect of TLR4 inhibitor TAK-242 or JNK inhibitor SP-600125 on JNK and NF-κB phosphorylations by Western blot analysis. Our findings suggested that the blockade on TLR4 or JNK could improve LPS-stimulated JNK and NF-κB phosphorylation. Interestingly, the administration of BBR at concentrations of 1.25, 2.5 and 5.0 μM could down-regulate JNK and NF-κB phosphorylation in NIT-1 cells. These results implied that selective targeting of TLR4-mediated signaling components could provide a means to ameliorate β-cell dysfunction.
Overall, these results suggest that BBR inhibits the LPS-induced inflammation response in NIT-1 cells through intervening with the TLR4/JNK/NF-κB pathway, at least in part. BBR may serve as a promising agent for the development of TLR-dependent inflammatory diseases, such as DM.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Ahmad R, Al-Mass A, Atizado V, et al. (2012). Elevated expression of the toll like receptors 2 and 4 in obese individuals: Its significance for obesity-induced inflammation. J Inflamm (Lond) 9:48
- Akash MS, Rehman K, Chen S. (2013). Role of inflammatory mechanisms in pathogenesis of type 2 diabetes mellitus. J Cell Biochem 114:525–31
- Cai K, Qi D, Hou X, et al. (2011). MCP-1 upregulates amylin expression in murine pancreatic β cells through ERK/JNK-AP1 and NF-κB related signaling pathways independent of CCR2. PLoS One 6:e19559
- Chavez SA, Martinko AJ, Lau C, et al. (2011). Development of β-amino alcohol derivatives that inhibit Toll-like receptor 4 mediated inflammatory response as potential antiseptics. J Med Chem 54:4659–69
- Cui G, Qin X, Zhang Y, et al. (2009). Berberine differentially modulates the activities of ERK, p38 MAPK, and JNK to suppress Th17 and Th1 T cell differentiation in type 1 diabetic mice. J Biol Chem 284:28420–9
- Du P, Fan B, Han H, et al. (2013). NOD2 promotes renal injury by exacerbating inflammation and podocyte insulin resistance in diabetic nephropathy. Kidney Int 84:265–76
- Edalat B, Sharifi F, Badamchizadeh Z, et al. (2013). Association of metabolic syndrome with inflammatory mediators in women with previous gestational diabetes mellitus. J Diabetes Metab Disord 12:8
- Hamidi A, von Bulow V, Hamidi R, et al. (2012). Polyubiquitination of transforming growth factor β (TGFβ)-associated kinase 1 mediates nuclear factor-κB activation in response to different inflammatory stimuli. J Biol Chem 287:123–33
- Hellemans K, Kerckhofs K, Hannaert JC, et al. (2007). Peroxisome proliferator-activated receptor alpha-retinoid X receptor agonists induce beta-cell protection against palmitate toxicity. FEBS J 274:6094–105
- Hussey SE, Liang H, Costford SR, et al. (2012). TAK-242, a small-molecule inhibitor of Toll-like receptor 4 signalling, unveils similarities and differences in lipopolysaccharide- and lipid-induced inflammation and insulin resistance in muscle cells. Biosci Rep 33:37–47
- Li J, Wang X, Zhang F, Yin H. (2013). Toll-like receptors as therapeutic targets for autoimmune connective tissue diseases. Pharmacol Ther 138:441–51
- Lontchi-Yimagou E, Sobngwi E, Matsha TE, Kengne AP. (2013). Diabetes mellitus and inflammation. Curr Diab Rep 13:435–44
- Ma L, Dong F, Zaid M, et al. (2012). ABCA1 protein enhances Toll-like receptor 4 (TLR4)-stimulated interleukin-10 (IL-10) secretion through protein kinase A (PKA) activation. J Biol Chem 287:40502–12
- Mangan DF, Wahl SM. (1991). Differential regulation of human monocyte programmed cell death (apoptosis) by chemotactic factors and pro-inflammatory cytokines. J Immunol 147:3408–12
- Russell MA, Cooper AC, Dhayal S, Morgan NG. (2013). Differential effects of interleukin-13 and interleukin-6 on Jak/STAT signaling and cell viability in pancreatic β-cells. Islets 5:95–105
- Shen N, Huan Y, Shen ZF. (2012). Berberine inhibits mouse insulin gene promoter through activation of AMP activated protein kinase and may exert beneficial effect on pancreatic β-cell. Eur J Pharmacol 694:120–6
- Song Y, Dou H, Gong W, et al. (2013). bis-N-Norgliovictin, a small-molecule compound from marine fungus, inhibits LPS-induced inflammation in macrophages and improves survival in sepsis. Eur J Pharmacol 705:49–60
- Wu D, Wen W, Qi CL, et al. (2012). Ameliorative effect of berberine on renal damage in rats with diabetes induced by high-fat diet and streptozotocin. Phytomedicine 19:712–18
- Zhang Q, Piao XL, Piao XS, et al. (2011). Preventive effect of Coptis chinensis and berberine on intestinal injury in rats challenged with lipopolysaccharides. Food Chem Toxicol 49:61–9
- Zohn IE, Yu H, Li X, et al. (1995). Angiotensin II stimulates calcium-dependent activation of c-Jun N-terminal kinase. Mol Cell Biol 15:6160–8