
Abstract
Context: The widely used antineoplastic drug cyclophosphamide causes pulmonary toxicity by inducing oxidative stress. Selenium, a dietary micronutrient, has been found to protect various organs from oxidative injuries.
Objective: This study was designed to investigate the protective efficacy of an organoselenium compound 2-(5-selenocyanato-pentyl)-benzo[de]isoquinoline 1,3-dione against cyclophosphamide-induced pulmonary toxicity in Swiss albino mice.
Materials and methods: Cyclophosphamide (25 mg/kg b.w.) was administered intraperitoneally for 10 d and the organoselenium compound (3 mg/kg b.w.) was given by oral gavage in concomitant and pretreatment schedules. Various biochemical parameters related to oxidative stress and antioxidant enzymes along with histology of lungs were evaluated to assess the effect of the test compound.
Results: The oral LD50 of the test compound was more than 1000 mg/kg b.w. in Swiss albino mice. The test compound substantially ameliorated cyclophosphamide-induced pulmonary injury by reducing the levels of reactive oxygen species, reactive nitrogen species, and lipid peroxidation, respectively, by 14.88, 18.54, and 21.10% in concomitant treatment schedule and by 23.89, 35.73, and 30.76% in the pretreatment schedule as well as by restoring the level of reduced glutathione and activities of glutathione-S-transferase, superoxide dismutase, catalase, and glutathione peroxidase, respectively, by 36.88, 42.43, 38.0, 35.0, and 34.06% in the concomitant treatment schedule and by 66.02, 59.29, 57.23, 71.59, and 57.22% in the pretreatment schedule. The test compound also attenuated cyclophosphamide-induced histological alterations of lung tissue.
Discussion and conclusion: The test compound emerged as an efficient antioxidant protecting lungs tissue from cyclophosphamide-induced injury.
Introduction
Cyclophosphamide (CP), an alkylating agent, is one of the most commonly used anticancer and immunosuppressant drugs. It is used for the treatment of chronic and acute leukemias, multiple myeloma, lymphomas, rheumatic arthritis, and also in the preparation for bone marrow transplantation (Goldberg et al., Citation1986). CP is administered as a prodrug that requires activation by hepatic cytochrome P450 (Zhang et al., Citation2006). Phosphoramide mustard and acrolein are the two active metabolites of CP formed by hepatic microsomal cytochrome P450 mixed functional oxidase system (Ludeman, Citation1999). The antineoplastic effects of CP are associated with the phosphoramide mustard, while the acrolein is linked with its toxic side effects (Kern & Kehrer, Citation2002). Acrolein interferes with the tissue antioxidant defense system (Arumugam et al., Citation1997), produces highly reactive oxygen free radicals (Mythili et al., Citation2004), and is mutagenic to mammalian cells (Kawanishi et al., Citation1998). Due to the highly reactive nature, free radicals can readily combine with other molecules, such as enzymes, receptors, and ion pumps, causing their oxidation, and resulting in the alteration or inactivation of their normal functions (Davies, Citation1993). Free radicals produced by CP-metabolites. In addition, they are potential DNA damaging agents, which can lead to a number of pathological conditions, including cancer (Richter et al., Citation1988). Several studies indicate that the development of oxidative stress after CP administration leads to a decrease in the activities of antioxidant enzymes and an increase in lipid peroxidation (LPO) in liver and lungs of mice and rats (Kaya et al., Citation1999; Premkumar et al., Citation2001). In addition, due to the absence of two important detoxifying enzymes, namely aldehyde oxidase and aldehyde dehydrogenase, which catalyze the conversion of toxic aldehydes to less toxic carboxylic acids, in the lung tissues, CP has been reported to cause selective pulmonary injury (Sulkowska et al., Citation2002). Moreover, increased reactive oxygen species (ROS) production by metabolites of CP may cause the over production of neutrophils, monocytes, and macrophages in the lung resulting in the development of pulmonary fibrosis (Chaudhary et al., Citation2006). CP was also found to enhance the process of pulmonary metastasis by the synergistic effect of matrix-degrading proteases and adhesion proteins (Man et al., Citation2008). Epidemiological study has also documented CP-induced pulmonary injury in humans. A comprehensive retrospective study at a tertiary referral center identified six patients over 20 years. In those patients, CP was found to be the only factor that contributed to pulmonary injury (Limper, Citation2004). Clinical features of CP-induced pulmonary toxicity include dyspnea, fever, cough, gas exchange abnormalities, parenchymal infiltrates, and pleural thickening. Two patterns of CP-induced lung toxicity were observed in that series, early-onset pneumonitis and late-onset pneumonitis. The early-onset pneumonitis occurred within the first 1–6 months of the therapy. This early-onset pneumonitis responded to withdrawal of CP and the patients returned to their previous condition. The late-onset pneumonitis developed months or years after chemotherapy cessation, and was associated with progressive lung fibrosis and bilateral pleural thickening. This late-onset variety showed no response to withdrawal of CP (Limper, Citation2004).
Effects due to toxic metabolites of CP could be avoided by detoxifying with agents that are able to conjugate or quench these toxic metabolites (Fouladi et al., Citation2001). Therefore, there is a need for a novel agent which would protect the normal tissue from chemotherapy-induced toxicity without causing any negative effect on the antitumor properties of the therapeutic drugs.
The increasing amount of information in recent years on the role of reactive oxygen species in pathology has brought new ideas for the therapy of a variety of diseases. Several reports have appeared describing the antioxidant activity of organoselenium compounds (Meotti et al., Citation2004; Nogueira et al; Citation2004; Sk et al., Citation2010) in different experimental models. In fact, selenium is an essential trace element required for animals and humans for its possible role as a potent antioxidant against oxidative stress induced by xenobiotic compounds of diverse nature (Ozardali et al., Citation2004) protecting cell membranes from free radical-induced damage (Olivieri et al., 1996). This trace element is present in the active site of antioxidant enzymes like glutathione peroxidase and thioredoxin reductase, which is essential for the catalytic activity of these antioxidant enzymes. Studies suggest that low selenium status may increase the risk of oxidative damage and cancer (Rayman, Citation2000). As the organoselenium compounds have greater bioavailability and less toxicity than their inorganic counterparts, a great deal of attention has been directed by the researchers towards the synthesis of novel organoselenium compounds with chemoprotective and chemoehancing properties. Among organoselenium compounds, organoselenocyanates are of particular interest, because some of the organoselenium compounds like benzyl selenocyanate (Fiala et al., Citation1997), diphenylmethyl selenocyanate (Das et al., Citation2004, Citation2005), and phenylenebis(methylene)selenocynate (Tanaka et al., Citation1997) have been reported to possess cancer chemopreventive properties against lung, skin, liver, colon, and mammary gland cancer.
The present study was designed to evaluate the protective efficacy of a novel naphthalimide-based organoselenium compound 2-(5-selenocyanato-pentyl)-benzo[de]isoquinoline 1,3-dione (compound 4) towards the antioxidant defensive mechanism in CP-induced pulmonary toxicity in experimental mice.
Materials and methods
Animals
Adult (5–6 weeks) Swiss albino female mice (25 ± 2 g), bred in the animal colony of Chittaranjan National Cancer Institute, Kolkata, were used for this study. The mice were maintained at controlled temperature under alternating light and dark conditions. Standard food pellets (EPIC rat and mice pellet) from Kalyani Feed Milling Plant, Kalyani, West Bengal, India, and drinking water were provided ad libitum. The experiments were carried out strictly following the guidelines of Institutional Animal Ethics Committee [Committee for the Purpose of Control and Supervision of Experiment on Animals (CPCSEA registration. no. 175/99/CPCSEA), India].
Chemicals
Cyclophosphamide was purchased from Cadila Health Care Limited, Kundaim Industrial Estate, Ponda, Goa, India. 1,8-Naphthalic anhydride, 5-amino-1-pentanol [H2N(CH2)5OH], phosphorous tribromide (PBr3), ethyl acetate, potassium selenocyanate, HEPES, 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA), vanadium chloride (VCl3), 1-chloro-2,4-dinitrobenzene (CDNB), N-(1-naphthyl) ethylenediamine dihydrochloride (NEDD), sulfanilamide, sodium nitrite, ethylene diamine tetraacetic acid (EDTA), reduced glutathione (GSH), pyrogallol, 5,5′-dithio-bis (2-nitro benzoic acid) (DTNB), sodium dodecyl sulphate (SDS), bovine serum albumin (BSA), β-nicotinamide adenine dinucleotide phosphate (reduced) (NADPH), glutathione reductase, and sodium azide (NaN3) were obtained from Sigma-Aldrich Chemicals Private Limited, Bangalore, India. Hydrogen peroxide 30% (H2O2), thiobarbituric acid (TBA), propylene glycol, sodium carbonate, copper sulfate, sodium hydroxide, potassium-sodium tartrate, sucrose, TRIS, dithiothreitol (DTT), disodium hydrogen phosphate, sodium dihydrogen phosphate, acetic acid, n-butanol, pyridine, formaldehyde (37%), paraffin (58–60°C), calcium chloride, ortho-phosphoric acid, xylene, and DPX mounting medium were obtained from Merck (India) Limited, Mumbai, India. Chloroform, zinc sulfate (ZnSO4), sodium bicarbonate, glucose, and Folin-phenol reagent were purchased from Sisco Research Laboratories Private Limited Mumbai, India. Dipotassium hydrogenphosphate and potassium dihydrogenphosphate were obtained from Spectrochem Private Limited, Mumbai, India. Magnesium chloride (MgCl2) was purchased from Glaxo Laboratories (India) Ltd, Mumbai, India. Hematoxylin and eosin were obtained from Qualigens Limited, Mumbai, India. Egg albumin was prepared in our laboratory. Conventional microscope slides and cover glass (no. 1, 24 × 60 mm) were purchased from Blue Star, Andhra Pradesh, India.
Synthesis of compound 4
Compound 4 was prepared following a literature procedure (Hossain et al., Citation2005). In short, the desired compound was prepared by the reaction of respective bromoalkyl naphthalimide intermediate (3) with KSeCN in acetone at room temperature. The intermediate compound 3 was successively synthesized from respective 1,8-naphthalic anhydride by refluxing with 5-amino-1-pentanol followed by bromination with PBr3 in ethyl acetate at 70 °C ().
Figure 1. Synthesis of compound 4, reagents, and conditions: (i) H2N(CH2)5OH, water, reflux, (ii) PBr3, ethyl acetate, and (iii) KSeCN, acetone, room temperature.
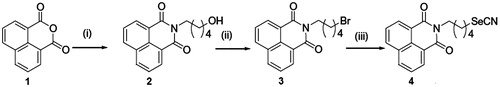
Yield: 68%. M.p.: 126–127 °C. It was crystallized from diethyl ether–light petroleum 60–80 °C to afford compound 4 of analytical grade m.p.: 154–155 °C IR (KBr) νmax cm−1: 2932.6, 2156.91 (CN), 1694.78, 1648.71, 1590.35. 1H NMR (300 MHz, CDCl3, TMS = 0.00): 1.61 (q, 2H, J = 7.8 Hz), 1.8 (q, 2H, J = 7.3 Hz), 2.0 (q, 2H, J = 7.1 Hz), 3.1 (t, 2H, J = 7.3 Hz), 4.2 (t, 2H, J = 7.2 Hz), 7.8 (t, 2H, Ar-H, J = 7.6 Hz), 8.22 (d, 2H, Ar-H, J = 7.9 Hz), 8.60 (d, 2H, Ar-H, J = 7.0 Hz). 13CNMR (75 MHz, CDCl3): 26.45, 27.14, 29.27, 30.33, 39.78, 101.45, 122.46, 126.87, 128.02, 131.17, 131.48, 133.93, 164.1. FABHRMS m/z: (M+ + 1, 373). 80Se correct isotope pattern. Anal. Found: C, 58.27; H, 4.3; N, 7.21. Calcd. For C18H16O2N2Se: C: 58.23; H: 4.34; N: 7.54.
Determination of LD50 value of compound 4
For the determination of the oral LD50 of the synthesized compound 4, different doses of the compound were given by oral gavage once as a suspension in aqueous solution of 5.5% propylene glycol and the animals were observed for 30 d. The number of animals died each day was noted. LD50 value of compound 4 was found to be more than 1000 mg/kg b.w. (Roy et al., Citation2010)
A non-toxic dose (3 mg/kg b.w.) of compound 4 (Roy et al., Citation2010) was used as a suspension in 5.5% propylene glycol in water. It was prepared each day just before treatment.
Experimental groups
Animals were distributed into five groups containing six animals in each group. The day of beginning of CP administration was considered as day 1.
Vehicle-treated group (Gr. I): Each animal received 5.5% propylene glycol in water orally for consecutive 10 d (from day 1 to day 10).
Organoselenium compound-treated group (Gr. II): Animals were treated with compound 4 only at a dose of 3 mg/kg b.w. throughout the experimental period.
CP-treated group (Gr. III): CP was administered intraperitoneally at a dose of 25 mg/kg b.w. in water for consecutive 10 d (from day 1 to day 10).
Concomitant treatment group (Gr. IV): Compound 4 was administered orally at a dose of 3 mg/kg b.w. from day 1 to day 10 and CP was also given at a dose of 25 mg/kg b.w. from day 1 to day 10.
Pretreatment group (Gr. V): Compound 4 was administered orally at a dose of 3 mg/kg b.w. 15 d prior to CP treatment and then continued up to day 10 and CP was given at a dose of 25 mg/kg b.w. from day 1 to 10.
The mice were sacrificed on day 11, 24 h after the last treatment and the parameters described below were studied.
Biochemical estimation
Determination of reactive oxygen species (ROS) generation
ROS generation was measured in lung homogenate using DCFH-DA following a simplified protocol with slight modifications (Lu et al., Citation2010; Shinomol & Muralidhara, Citation2007). DCFH-DA is a non-fluorescent probe that is hydrolyzed by mitochondrial esterase to form 2′,7′-dichlorodihydrofluorescein (DCFH), which is then oxidized by ROS to form the fluorescent compound 2′,7′-dichlorofluorescein (DCF). Lung tissues were homogenized in Locke’s Buffer (pH 7.4 containing 140 mM NaCl, 5 mM KCl, 10 mM HEPES, 1 mM CaCl2, 1 mM MgCl2, 10 mM glucose) to yield a 10% w/v homogenate. 250 μL of tissue homogenate in whole was taken and then loaded with 30 µL of 10 µM DCFH-DA to make a final volume of 3 mL. The samples were incubated in dark for 45 min to allow the formation of DCF and then analyzed for fluorescence (excitation 485 nm/emission 530 nm) using spectrofluorimeter (Varian Cary Eclipse, Walnut Creek, CA). Values were expressed as fluorescence intensity/mg of protein.
Measurement of NO production
NO production in lung homogenate was determined by estimating the level of stable NO metabolites, namely nitrate () and nitrite (
) ions (Coşkun et al., Citation2007; Green et al., Citation1982). The nitrate content in tissue homogenate was reduced to nitrite by VCl3. The reaction is followed by colorimetric detection of nitrite as an azo dye product of the Griess reaction. Lung tissues were homogenized with phosphate buffer (pH 7.5, containing 137 mM NaCl, 27 mM KCl, 10 mM Na2HPO4, and 1.8 mM KH2PO4) to yield a 20% w/v homogenate. The total nitrite level was determined by reaction with the Griess reagent (1% sulfanilamide, 5% phosphoric acid, and 0.1% NEDD) using sodium nitrite as a standard. The absorbance was taken at 545 nm and expressed as micromol nitrite/mg of protein.
Quantitative estimation of LPO level
LPO level was estimated in lung microsomal fraction. The level of lipid peroxides formed was measured using TBA and expressed as nmol of thiobarbituric acid reactive substances (TBARS) formed/mg of protein using the extinction co-efficient of 1.56 × 105 M−1 cm−1 (Okhawa et al., Citation1979).
Estimation of reduced GSH level
GSH level was estimated in lung cytosol spectrophotometrically by the determination of DTNB reduced by ‐SH groups by measuring the absorbance at 412 nm. The level of GSH was expressed as nmol/mg of protein (Sedlack & Lindsay, Citation1968).
Estimation of GST activity
GST activity was measured in the lung cytosol. The enzyme activity was determined from the increase in absorbance at 340 nm with CDNB as the substrate and specific activity of the enzyme was expressed as the formation of CDNB-GSH conjugate min−1 mg −1 of protein (Habig et al., Citation1974).
Estimation of SOD activity
SOD activity in lung was determined by quantification of pyrogallol auto-oxidation inhibition and expressed as unit/mg of protein. One unit of enzyme activity is defined as the amount of enzyme necessary for inhibiting the reaction by 50%. Auto-oxidation of pyrogallol in Tris–HCL buffer (50 mM, pH 7.5) is measured by an increase in absorbance at 420 nm (Marklund & Marklund, Citation1974; McCord & Fridovich, Citation1969).
Estimation of CAT activity
CAT activity in lung cytosol was determined spectrophotometrically at 240 nm and expressed as unit/mg of protein, where the unit is the amount of enzyme that liberates half the peroxide oxygen from H2O2 in second at 25 °C (Luck, Citation1963).
Estimation of GPx activity
GPx activity in lung was measured by NADPH oxidation using a coupled reaction system consisting of GSH, glutathione reductase, and H2O2 (Paglia & Valentine, Citation1967). Briefly, 100 µL of enzyme sample was incubated for 10 min with 800 μL reaction mixture (0.25 M potassium phosphate buffer containing 2.5 mM EDTA and 2.5 mM NaN3, 10 mM GSH, 2.5 mM NADPH, and 2.4 units of glutathione reductase). The reactions started on adding 100 μL H2O2 and follow the decrease in NADPH absorbance at 340 nm for 3 min. The enzyme activity was expressed as micromol NADPH utilized. min−1 mg−1 of protein, using extinction co-efficient of NADPH at 340 nm as 6200 M−1 cm−1.
Estimation of protein
Total protein content in tissue homogenate during biochemical analysis was measured through the Lowry method using the Folin-phenol reagent (Lowry et al., Citation1951). The absorbance of the color was measured against the colorless blank sample at 660 nm using the Varian Cary 100 UV–visible spectrophotometer (Varian Cary Eclipse, Walnut Creek, CA).
Tissue section preparation and histopathological evaluation
Lungs were collected from all groups of mice and washed in phosphate buffered saline (PBS) and soaked on blotting paper to remove blood. Tissues were then fixed in 10% neutral buffered formalin for 24 h. The tissue samples were dehydrated in ascending concentrations of ethanol, cleared in xylene, and embedded in paraffin to prepare the block. Lung tissues were sectioned, mounted on slides, and stained. The 5 μm serial sections were used for staining with hematoxylin–eosin. Stained sections were evaluated by observing the arrangement of lung architecture with a light microscope (Leica DM 1000, Leica Microsystems, Bellows Falls, VT). Photomicrographs were taken with the software Las EZ (Leica Microsystems, Bellows Falls, VT).
Statistical analysis
All data were presented as mean ± SD, n = 6 animals per group. One way ANOVA followed by Tukey's multiple comparison test using Graph Pad Prism software (GraphPad Software Inc., San Diego, CA) was performed for comparisons among groups. Significant difference was indicated when the p value was <0.05.
Results
Compound 4 did not produce any significant change in the parameters investigated when administered alone as compared with the vehicle control group.
Inhibition of ROS level
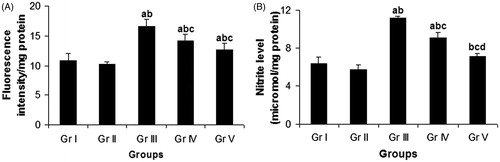
The ROS level in lung was found to increase significantly (p < 0.05) by 52.48% after the administration of CP (Gr. III), compared with the vehicle control group (Gr. I) (). Concomitant treatment (Gr. IV) with compound 4 reduced the pulmonary ROS level by 14.88% in comparison with the CP-treated group (Gr. III). When the test compound was given in pretreatment schedule (Gr. V), the inhibition in pulmonary ROS level was found to be 23.89% in comparison with the CP-treated group (Gr. III).
Figure 2. Effect of compound 4 on pulmonary ROS level (A) and pulmonary NO level (B) after administration of CP. Data are represented as mean ± SD. (a) Significant (p < 0.05) as compared with Gr. I; (b) significant (p < 0.05) as compared with Gr. II; (c) significant (p < 0.05) as compared with Gr. III; (d) significant (p < 0.05) as compared with Gr. IV.
Modulation of NO level
Intraperitoneal administration of CP (Gr. III) resulted in a significant (p < 0.05) enhancement in pulmonary NO level by 74.53% in comparison with the vehicle control group (Gr. I) (). Oral administration of compound 4 caused a significant depletion of CP-induced elevated pulmonary NO level by 18.54% in case of concomitant treatment schedule (Gr. IV) and by 35.73% in case of pretreatment schedule (Gr. V) in comparison with the CP-treated group (Gr. III).
Attenuation of LPO level
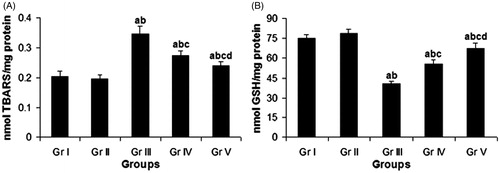
Intraperitoneal administration of CP (Gr. III) significantly (p < 0.05) elevated the LPO level in lung by 70.61%, compared with the vehicle control group (Gr. I) (). Concomitant treatment (Gr. IV) with compound 4 significantly reduced the pulmonary LPO level by 21.10%, whereas pretreatment (Gr. V) with the same compound inhibited the pulmonary LPO level much more efficiently by 30.76% in comparison with the CP-treated group (Gr. III).
Figure 3. Effect of compound 4 on pulmonary LPO level (A) and pulmonary GSH level (B) after administration of CP. Data are represented as mean ± SD. (a) Significant (p < 0.05) as compared with Gr. I; (b) significant (p < 0.05) as compared with Gr. II; (c) significant (p < 0.05) as compared with Gr. III; (d) significant (p < 0.05) as compared with Gr. IV.
Elevation of GSH level
The GSH content in lung was found to decline significantly (p < 0.05) by 46.03% after CP administration (Gr. III) as compared with the vehicle control group (Gr. I) (). Concomitant treatment (Gr. IV) and pretreatment (Gr. V) with the test compound resulted in a significant elevation of GSH content by 36.88 and 66.02%, respectively, in lung in comparison with the CP-treated group (Gr. III).
Restoration of GST activity
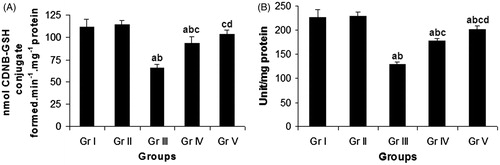
A significant (p < 0.05) reduction of GST activity by 41.41% in lung was observed in the CP-treated group (Gr. III) in comparison with the vehicle control group (Gr. I) (). Concomitant administration of compound 4 (Gr. IV) significantly increased the GST activity in lung by 42.43% compared with the CP-treated group (Gr. III). In the pretreatment schedule (Gr. V), the same compound raised the enzyme activity in lung by 59.29% in comparison with the CP-treated group (Gr. III).
Figure 4. Effect of compound 4 on pulmonary GST activity (A) and pulmonary SOD activity (B) after administration of CP. Data are represented as mean ± SD. (a) Significant (p < 0.05) as compared with Gr. I; (b) significant (p < 0.05) as compared with Gr. II; (c) significant (p < 0.05) as compared with Gr. III; (d) significant (p < 0.05) as compared with Gr. IV.
Augmentation of SOD activity
Intraperitoneal administration of CP (Gr. III) significantly (p < 0.05) decreased pulmonary SOD activity by 43.16%, as compared with the vehicle control group (Gr. I) (). Concomitant treatment (Gr. IV) with compound 4 significantly enhanced the SOD activity in lung by 38.0%, while pretreatment (Gr. V) with the same compound increased the SOD activity in lung much more efficiently by 57.23% in comparison with the CP-treated group (Gr. III).
Enhancement of CAT activity
The pulmonary CAT activity was found to decrease significantly (p < 0.05) by 51.89% in the CP-treated group (Gr. III) in comparison with the vehicle control group (Gr. I) (). Concomitant administration of compound 4 (Gr. IV) resulted in a significant enhancement in pulmonary CAT activity by 35.0% in comparison with the CP-treated group (Gr. III). Pretreatment (Gr. V) with the same compound increased the pulmonary CAT activity significantly by 71.59% as compared with the CP-treated group (Gr. III).
Figure 5. Effect of compound 4 on pulmonary CAT activity (A) and pulmonary GPx activity (B) after administration of CP. Data are represented as mean ± SD. (a) Significant (p < 0.05) as compared with Gr. I; (b) significant (p < 0.05) as compared with Gr. II; (c) significant (p < 0.05) as compared with Gr. III; (d) significant (p < 0.05) as compared with Gr. IV.
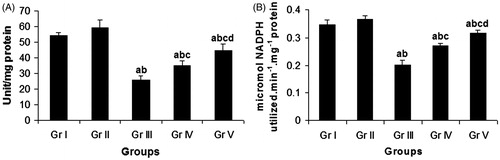
Modulation of GPx activity
Intraperitonial administration of CP (Gr. III) caused a significant (p < 0.05) reduction in the GPx activity in lung by 41.77% in comparison with the vehicle control group (Gr. I) (). Compound 4 in the concomitant treatment schedule (Gr. IV) resulted in a significant enhancement of the GPx activity by 34.06% in pneumocytes in comparison with the CP-treated group (Gr. III), while in the pretreatment schedule (Gr. V), the same compound significantly elevated the pulmonary GPx activity by 57.22%, as compared with the CP-treated group (Gr. III).
Histopathology of lung tissue
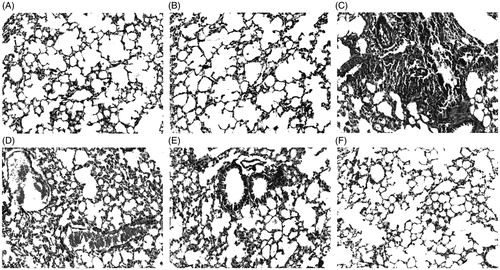
Histology of lung from different groups of mice is shown in . Histopathological evaluation was used as confirmatory diagnostic test to prove the pneumotoxic effect of CP as well as pneumoprotective potential of the test compound. Histological examination showed a normal architecture of lung tissue in vehicle control animals. On the administration of CP, the histopathological study showed distinct cellular and architectural changes of lung tissue including lymphocytic interstitial pneumonia, extensive fibrous tissue proliferation, and thickened alveolar septa. Such lesions were successfully attenuated by the administration of compound 4, where the most effective protection was exerted by the administration of test compound in the pretreatment schedule.
Figure 6. Photomicrographs of lung sections of mice stained with hematoxylin and eosin, 200 × ; (A) normal histology of lung with normal alveolar architecture without any epithelial damage in Gr. I; (B) normal histological features of lung in Gr. II; (C) alveolar damage, thickening of alveolar septa, and fibrous tissue proliferation in Gr. III; (D) pulmonary congestion and pulmonary inflammation in Gr. III; (E) mild to moderate thickening of alveoli, moderate reduction in alveolar distortion, and fibrous tissue proliferation in Gr. IV; (F) almost normal appearance of lung histology with no or very slight thickening of alveolar septa and no indication of fibrous tissue proliferation in Gr. V.
Discussion
The usage of anticancer drugs for killing cancer cells is a compromise between necessity and undesirable toxicity to normal host cells. These drugs fail to discriminate normal cells from cancerous cells, resulting in the killing of normal proliferating cells of the body. CP is a widely used antitumor agent. High doses of CP can cause an acute form of lethality within 10 d of its administration (Gharib & Burnett, Citation2002). The mechanism of CP mediated lung injury is not completely understood. It is suggested that the early deleterious effect of CP on the rat lungs is connected with acrolein generation during drug metabolism and severe inflammatory reactions connected with accumulation of neutrophils, generation of ROS, and enhancement of LPO (Tannehill & Mehta, Citation1996). It is known that enhanced generation of ROS cause changes in cellular redox balance, which lead to oxidative stress, causing healthy cell damage and ultimately gives rise to many diseases including cancer (Matés et al., Citation1999). It has been proven that oxidative stress leads to injury of bone marrow, gastrointestinal tract, kidneys, urinary bladder, lungs, nervous system, and vascular system of healthy cells (Siems et al., Citation2002). Oxidative stress and free radical-induced damages have been implicated in the etiology of several toxic effects caused by CP (Fadillioğlu & Erdoğan, Citation2003). NO is an important signaling molecule and is also involved in acute and chronic inflammation. It is released during the metabolism of l-arginine by a family of isoenzymes called NO synthases (NOSs) (Förstermann et al., Citation1991). NO is produced in high concentrations during infection, acute lung injury, and adult respiratory distress syndrome suggesting its direct role in inflammatory lung diseases (Ricciardolo et al., Citation2004). It has been shown that increased production of NO during chemotherapy using anticancer drugs is associated with inflammation in the lungs (Sayed-Ahmed et al., Citation2004). Furthermore, NO may react with superoxide to form peroxynitrite which may cause acute lung injury (Gurujeyalakshmi et al., Citation2000). Therefore, suppression of NO production may be beneficial against conditions caused due to its increased production. In the present study, the pulmonary ROS and NO levels were found to increase after administration of CP. The test compound successfully ameliorated this enhancement in pulmonary ROS and NO levels. The test compound in the pretreatment schedule was found to be more effective in attenuating the pulmonary ROS and NO levels. This result suggests the ability of the test compound in preventing CP-induced oxidative and nitrosative stress.
Lung injury and its pathogenesis involve peroxidative breakdown of polyunsaturated fatty acids, which affects the membrane function, inactivates membrane bound receptors, and increases tissue permeability. Aldehydes which are generated during the process of LPO have been associated with oxidative stress in tissue (Uchida et al., Citation1999). Lipid peroxides are important in signal transduction process and are responsible for inflammatory changes in the lung tissue (Uchida et al., Citation1999). Free radical-induced LPO has been suggested to alter the membrane structure and function causing cellular abnormalities such as mutations and cell death (Singal et al., Citation1988; Singh et al., Citation1995). In the present study, due to the induction of LPO by CP, the level of TBARS was found to increase in lung tissue of mice. This might be due to the increased production of free radicals or decreased antioxidant status. Following the administration of test compound, the levels of TBARS were maintained to near normal status which indicates the reduced level of LPO. From this, it may be concluded that the oxidative stress induced by CP was prevented by the test compound due to its antioxidant and membrane stabilizing properties.
GSH plays an important role in the regulation of a variety of cell functions and in cell protection from oxidative injury. It is a non-enzymatic antioxidant and acts as an important scavenger of electrophiles such as acrolein (Alarcon, Citation1976) and has been shown to be a critical factor in the toxicity of CP (DeLeve, Citation1996). It is well known that depletion of GSH results in enhanced LPO and excessive LPO can cause increased GSH consumption, as observed in the present study. The marked decline in GSH level in CP-treated animals may have resulted from the direct conjugation of CP-metabolites with free or protein bound –SH groups (Yousefipour et al., Citation2005) or due to the enhanced utilization of GSH to detoxify the ROS, produced by the metabolites of CP (Roy et al., Citation2012). In GSH-depleted condition, oxidative stress develops in the cells due to the overwhelming production of ROS, which leads to the declined activities of the antioxidant enzymes like CAT, SOD, and GPx, making the cells more prone to oxidative damages (Rajasekaran et al., Citation2002). Administration of compound 4 elevated the GSH level in lung tissue of CP-treated mice thereby increased the ability of the experimental animals to cope with the free radicals produced after CP administration. This observation also suggested the protective effect of the test compound against CP-induced toxicity. GST isozymes catalyze the conjugation of GSH to several electrophilic compounds, including polyaromatic hydrocarbons (Perera et al., Citation1995). GST also catalyzes the conjugation of various CP-metabolites including acrolein with GSH to form less toxic ones (Zhang et al., Citation2006). CP administration in the present investigation resulted in a decreased activity of this GSH metabolizing enzyme in lung tissue. The oxidative modification in the native protein structure of GST and its increased utilization in the detoxification processes that involve GSH may be the most probable explanation of the reduced GST activity in the CP-treated mice. Treatment with compound 4 enhanced the activity of GST, attributing to the ability of the compound in amelioration of CP-induced toxicity.
The antioxidant enzymes SOD, CAT, and GPx act in coordination to combat the ROS. Out of these, GPx is of particular interest as it is a selenoenzyme and is involved in protecting various organisms from oxidative stresses by catalyzing the reduction of hydroperoxides at the expense of GSH (Stadtman, Citation1991). The GPx super family contains four types of enzymes, all of which require selenium in their active sites for the catalytic activity (Stadtman, Citation1991). The reactivity of these enzymes differs considerably depending on the hydroperoxides and thiol cofactor. The GPx catalytic site includes a selenocysteine residue in which the selenium undergoes a redox cycle involving the selenol as the active form that reduces H2O2 and organic peroxides (Brigelius-Flohé, Citation1999). Cellular defense against the intermediates of dioxygen reduction (, OH and H2O2) are done by the combined activity of SOD, CAT, and GPx (Senthilkumar et al., Citation2006). The reduction in the activities of these antioxidant enzymes in the lung tissue of the CP administered mice is probably connected with damage to their protein structures by ROS or aldehydes generated by the metabolites of CP. Cellular proteins are susceptible to free radical-induced oxidative modification which ultimately results in impaired cellular function. Treatment with compound 4 resulted in an improved activities of SOD, CAT, and GPx than the CP only treated group; pretreatment showed better result. These findings evidenced that the ROS-mediated inactivation of these antioxidant enzymes was prevented by the test compound and thus protecting the pneumocytes from oxidative damage.
The better results showed by the pretreatment group than the concomitant group may be explained by the fact that during pretreatment with the test compound, the antioxidant enzymes might already be in an enhanced level which may have been persisted even after CP treatment, as the treatment with the test compound was continued throughout the entire experimental period, whereas during the concomitant treatment, the levels of enzymes were also enhanced but most of it was utilized to counter the oxidative stress induced by CP.
In the histological study, it was found that CP administration caused various types of alterations in the histological architecture of the lung tissue. The most important pathological alterations were the interstitial pneumonia, pulmonary fibrosis, thickened alveolar septa, pulmonary congestion, and pulmonary inflammation. These histopathological alterations of the lung were attenuated after administration of the test compound, and the best result was obtained in the pretreatment group. This observation indicates the test compound has the potential to protect lung tissues even at the histological level. As the reactive oxygen species (ROS) and reactive nitrogen species (RNS) are the major sources of lung inflammatory response (Kinnula et al., Citation1995), the test compound by directly scavenging ROS and RNS and by restoring the activities of antioxidant enzymes, prevented oxidative damage of pneumocytes and finally exerted protection towards the histological architecture of the lung tissue.
Conclusions
The results point out that the test compound successfully protects healthy lung cells against CP-induced injury. It can be expected that administration of CP together with the test compound will not change the antineoplastic activity of CP, simultaneously alleviating its toxic effects. The findings of this study seem to be particularly important for their clinical purposes and for the possibility to use them during chemotherapy.
Acknowledgements
The authors would like to thank Prof. (Dr.) Jaydip Biswas, Director, Chittaranjan National Cancer Institute, for his support in this study. Arin Bhattacharjee and Abhishek Basu gratefully acknowledge Indian Council of Medical Research, New Delhi, India for Senior Research Fellowship (no. 45/36/2008/PHA-BMS and no. 3/2/2/58/2011/NCD-III, respectively). Somnath Singha Roy gratefully acknowledges Council of Scientific and Industrial Research, New Delhi, India for Fellowship (01(2160)/07/EMRII).
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Alarcon RA. (1976). Studies on the in vivo formation of acrolein: 3-Hydroxypropylmercapturic acid as an index of cyclophosphamide (NSC-26271) activation. Cancer Treat Rep 60:327–35
- Arumugam N, Sivakumar V, Thanislass J, Devaraj H. (1997). Effects of acrolein on rat liver antioxidant defense system. Indian J Exp Biol 35:1373–4
- Brigelius-Flohé R. (1999). Tissue-specific functions of individual glutathione peroxidases. Free Radic Biol Med 27:951–65
- Chaudhary NI, Schnapp A, Park JE. (2006). Pharmacologic differentiation of inflammation and fibrosis in the rat bleomycin model. Am J Respir Crit Care Med 173:769–76
- Coşkun S, Gönül B, Ozer C, et al. (2007). The effects of dexfenfluramine administration on brain serotonin immunoreactivity and lipid peroxidation in mice. Cell Biol Toxicol 23:75–82
- Das RK, Ghosh S, Sengupta A, et al. (2004). Inhibition of DMBA/croton oil-induced two-stage mouse skin carcinogenesis by diphenylmethyl selenocyanate. Eur J Cancer Prev 13:411–17
- Das RK, Hossain SK, Bhattacharya S. (2005). Diphenylmethyl selenocyanate inhibits DMBA-croton oil induced two-stage mouse skin carcinogenesis by inducing apoptosis and inhibiting cutaneous cell proliferation. Cancer Lett 230:90–101
- Davies KJ. (1993). Protein modification by oxidants and the role of proteolytic enzymes. Biochem Soc Trans 21:346–53
- DeLeve LD. (1996). Cellular target of cyclophosphamide toxicity in the murine liver: Role of glutathione and site of metabolic activation. Hepatology 24:830–7
- Fadillioğlu E, Erdoğan H. (2003). Effects of erdosteine treatment against doxorubicin-induced toxicity through erythrocyte and plasma oxidant/antioxidant status in rats. Pharmacol Res 47:317–22
- Fiala ES, Sohn OS, Li H, et al. (1997). Inhibition of 2-nitropropane-induced rat liver DNA and RNA damage by benzyl selenocyanate. Carcinogenesis 18:1809–15
- Förstermann U, Schmidt HH, Pollock JS, et al. (1991). Isoforms of nitric oxide synthase. Characterization and purification from different cell types. Biochem Pharmacol 42:1849–57
- Fouladi M, Stempak D, Gammon J, et al. (2001). Phase I trial of a twice-daily regimen of amifostine with ifosfamide, carboplatin, and etoposide chemotherapy in children with refractory carcinoma. Cancer 92:914–23
- Gharib MI, Burnett AK. (2002). Chemotherapy-induced cardiotoxicity: Current practice and prospects of prophylaxis. Eur J Heart Fail 4:235–42
- Goldberg MA, Antin JH, Guinan EC, Rappeport JM. (1986). Cyclophosphamide cardiotoxicity: An analysis of dosing as a risk factor. Blood 68:1114–18
- Green LC, Wagner DA, Glogowski J, et al. (1982). Analysis of nitrate, nitrite, and [15N] nitrate in biological fluids. Anal Biochem 126:131–8
- Gurujeyalakshmi G, Wang Y, Giri SN. (2000). Suppression of bleomycin-induced nitric oxide production in mice by taurine and niacin. Nitric Oxide 4:399–11
- Habig WH, Pabst MJ, Jacoby WB. (1974). Glutathione S-transferases, the first enzymatic step in marcapturic acid formation. J Biol Chem 249:7130–9
- Hossain SU, Sengupta S, Bhattacharya S. (2005). Synthesis and evaluation of antioxidative properties of a series of organoselenium compounds. Bioorg Med Chem 13:5750–8
- Kawanishi M, Matsuda T, Nakayama A, et al. (1998). Molecular analysis of mutations induced by acrolein in human fibroblast cells using supF shuttle vector plasmids. Mutat Res 417:65–73
- Kaya H, Oral B, Ozgüner F, et al. (1999). The effect of melatonin application on lipid peroxidation during cyclophosphamide therapy in female rats. Zentralbl Gynakol 121:499–502
- Kern JC, Kehrer JP. (2002). Acrolein-induced cell death: A caspase-influenced decision between apoptosis and oncosis/necrosis. Chem Biol Interact 139:79–95
- Kinnula VL, Crapo JD, Raivio KO. (1995). Generation and disposal of reactive oxygen metabolites in the lung. Lab Invest 73:3–19
- Limper AH. (2004). Chemotherapy-induced lung disease. Clin Chest Med 25:53–64
- Lowry OH, Rosenbrough NJ, Farr AL, Randall RJ. (1951). Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–76
- Lu J, Wu DM, Hu B, et al. (2010). Chronic administration of troxerutin protects mouse brain against d-galactose-induced impairment of cholinergic system. Neurobiol Learn Mem 93:157–64
- Luck HA. (1963). Spectrophotometric method for estimation of catalase. In: Bergmeyer HV, ed. Methods of Enzymatic Analysis. New York: Academic Press, 886–8
- Ludeman SM. (1999). The chemistry of the metabolites of cyclophosphamide. Curr Pharm Des 5:627–43
- Man S, Zhang Y, Gao W, et al. (2008). Cyclophosphamide promotes pulmonary metastasis on mouse lung adenocarcinoma. Clin Exp Metastasis 25:855–64
- Marklund S, Marklund G. (1974). Involvement of the superoxide anion radical in autooxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur J Biochem 47:469–74
- Matés JM, Pérez-Gómez C, Núñez de Castro I. (1999). Antioxidant enzymes and human diseases. Clin Biochem 32:595–603
- McCord JM, Fridovich I. (1969). Superoxide dismutase: An enzymatic function for erythrocuprein (hemoprotein). J Biol Chem 244:6049–55
- Meotti FC, Stangherlin EC, Zeni G, et al. (2004). Protective role of aryl and alkyl diselenides on lipid peroxidation. Environ Res 94:276–82
- Mythili Y, Sudharsan PT, Selvakumar E, Varalakshmi P. (2004). Protective effect of dl-alpha-lipoic acid on cyclophosphamide induced oxidative cardiac injury. Chem Biol Interact 151:13–19
- Nogueira CW, Zeni G, Rocha JB. (2004). Organoselenium and organotellurium compounds: Toxicology and pharmacology. Chem Rev 104:6255–85
- Okhawa H, Ohishi N, Yagi K. (1979). Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Annal Biochem 95:351–8
- Olivieri O, Girelli D, Stanzial AM, et al. (1996). Selenium, zinc, and thyroid hormones in healthy subjects: Low T3/T4 ratio in the elderly is related to impaired selenium status. Biol Trace Elem Res 51:31–41
- Ozardali I, Bitiren M, Karakilçik AZ, et al. (2004). Effects of selenium on histopathological and enzymatic changes in experimental liver injury of rats. Exp Toxicol Pathol 56:59–64
- Paglia DE, Valentine WN. (1967). Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med 70:158–69
- Perera FP, Estabrook A, Hewer A, et al. (1995). Carcinogen-DNA adducts in human breast tissue. Cancer Epidemiol Biomarkers Prev 4:233–8
- Premkumar K, Pachiappan A, Abraham SK, et al. (2001). Effect of Spirulina fusiformis on cyclophosphamide and mitomycin-C induced genotoxicity and oxidative stress in mice. Fitoterapia 72:906–11
- Rajasekaran NS, Devaraj H, Devaraj SN. (2002). The effect of glutathione monoester (GME) on glutathione (GSH) depleted rat liver. J Nutr Biochem 13:302–6
- Rayman MP. (2000). The importance of selenium to human health. Lancet 356:233–41
- Ricciardolo FL, Sterk PJ, Gaston B, Folkerts G. (2004). Nitric oxide in health and disease of the respiratory system. Physiol Rev 84:731–65
- Richter C, Park JW, Ames BN. (1988). Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc Natl Acad Sci USA 85:6465–7
- Roy SS, Ghosh P, Sk UH, et al. (2010). Naphthalimide based novel organoselenocyanates: Finding less toxic forms of selenium that would retain protective efficacy. Bioorg Med Chem Lett 20:6951–5
- Roy SS, Chakraborty P, Ghosh P, et al. (2012). Influence of novel naphthalimide-based organoselenium on genotoxicity induced by an alkylating agent: The role of reactive oxygen species and selenoenzymes. Redox Rep 17:157–66
- Sayed-Ahmed MM, Eissa MA, Kenawy SA, et al. (2004). Progression of cisplatin-induced nephrotoxicity in a carnitine-depleted rat model. Chemotherapy 50:162–70
- Sedlack J, Lindsay RN. (1968). Estimation of total protein bound and non-protein sulfhydryl groups in tissue with Ellman reagent. Annal Biochem 25:192–205
- Senthilkumar S, Ebenezar KK, Sathish V, et al. (2006). Modulation of the tissue defense system by squalene in cyclophosphamide induced toxicity in rats. Arch Med Sci 2:94–100
- Shinomol GK, Muralidhara. (2007). Differential induction of oxidative impairments in brain regions of male mice following sub chronic consumption of Khesari dhal (Lathyrus sativus) and detoxified Khesari dhal. Neurotoxicology 28:798–806
- Siems W, Quast S, Carluccio F, et al. (2002). Oxidative stress in chronic renal failure as a cardiovascular risk factor. Clin Nephrol 58:S12–9
- Singal PK, Petkau A, Gerrard JM, et al. (1988). Free radicals in health and disease. Mol Cell Biochem 84:121–2
- Singh N, Dhalla AK, Seneviratne C, Singal PK. (1995). Oxidative stress and heart failure. Mol Cell Biochem 147:77–81
- Sk UH, Sharma AK, Ghosh S, Bhattacharya S. (2010). Synthesis and biological evaluation of novel spiro 6-methoxytetralin-1,3′-pyrrolidine based organoselenocyanates against cadmium-induced oxidative and hepatic damage in mice. Eur J Med Chem 45:3265–73
- Stadtman TC. (1991). Biosynthesis and function of selenocysteine-containing enzymes. J Biol Chem 266:16257–60
- Sulkowska M, Skrzydlewska E, Sobaniec-Lotowska M, et al. (2002). Effect of cyclophosphamide-induced generation of oxygen forms on ultrastructure of liver and lung. Bull Vet Inst Pulawy 46:239–46
- Tanaka T, Makita H, Kawabata K, et al. (1997). 1,4-Phenylenebis(methylene)selenocyanate exerts exceptional chemopreventive activity in rat tongue carcinogenesis. Cancer Res 57:3644–8
- Tannehill SP, Mehta MP. (1996). Amifostine and radiation therapy: Past, present, and future. Semin Oncol 23:69–77
- Uchida K, Shiraishi M, Naito Y, et al. (1999). Activation of stress signaling pathways by the end product of lipid peroxidation. 4-Hydroxy-2-nonenal is a potential inducer of intracellular peroxide production. J Biol Chem 274:2234–42
- Yousefipour Z, Ranganna K, Newaz MA, Milton SG. (2005). Mechanism of acrolein-induced vascular toxicity. J Physiol Pharmacol 56:337–53
- Zhang J, Tian Q, Zhou SF. (2006). Clinical pharmacology of cyclophosphamide and ifosfamide. Curr Drug Ther 1:55–84