Abstract
Background. There is controversy on whether estrogen receptors are present and functioning in the myocardium. Aims. To explore if after myocardial infarction (MI) estrogen receptors α (ERα) and β (ERβ) are upregulated in myocardial tissue and to explore if the presence/ absence of ERα or ERβ influences angiogenesis after MI. Methods. MI was induced by ligation of the left anterior descending artery in knockout (KO) mice, ERαKO and ERβKO, respectively, and non-KO littermate-controls, C57Bl/6 mice. The hearts were harvested after 12 days. A part of the periinfarct tissue was collected for ERα and ERβ mRNA expression determination by real-time polymerase chain reaction. Using immunohistochemistry, ERα and ERβ protein expression and capillary and arteriolar densities were blindly determined in the periinfarct area. Results. In myocardium disrupted mRNA was upregulated in both ERαKO and ERβKO, (p < 0.005) and did not change after MI. There was no change in mRNA expression of ERα or ERβ in wild type mice after MI. Expression of ERβ in ERαKO and of ERα in ERβKO did not change. Following MI ERα or ERβ could not be demonstrated by immunohistochemistry in either wild type or ERαKO or ERβKO. The capillary and arteriolar densities after MI did not differ between the groups in the periinfarct area. Conclusions. Although disrupted ER mRNA is upregulated in myocardium of ER knockout mice, no change in these or native receptors occurs following MI. At least in this model ER therefore seems not to have a role in myocardial arteriogenesis and angiogenesis after MI.
The age-matched incidence of cardiovascular disease is greater in men than in premenopausal women. After menopause the incidence increases also for women, indicating that estrogen plays a protecting role in the cardiovascular system (Citation1). Several clinical trials have tried this hypothesis in giving hormone replacement therapy to postmenopausal women in order to prolong the protection against ischemic heart disease, but an increased cancer risk and adverse cardiovascular outcomes in subgroups prompt that a better understanding of the cardiovascular effects of estrogens is needed (Citation2–4).
Estrogen is a steroidal hormone produced in the ovaries and the mammary glands, during pregnancy also by the placenta. In males the production of estrogen takes place in the testes. In both genders there is also production in the liver and adrenal glands. The effects of the estrogen hormone is mediated by two subtype receptors, estrogen receptor α (ERα) and estrogen receptor β (ERβ). These receptors are predominantly nuclear hormone receptors acting as ligand-dependent transcription factors. In addition 2–3% of ERs are situated on the cell membrane and act through a non-genomic pathway, suggested to involve eNOS for rapid vasodilator effects (Citation5,Citation6). In human adult tissue both ERα and ERβ have been identified in the myocardium, aorta, coronary artery, carotid artery, inferior vena cava, and also in endothelial cells of the thyroid and uterine cervix (Citation7). Functional ERα- and ERβ-proteins have been reported to be expressed in cardiomyocytes, cardiac fibroblasts, vascular smooth muscle cells and endothelial and epithelial cells of the kidney (Citation8,Citation9). Cardiovascular ERs have been associated with the prevention of apoptosis (Citation10), with an antihypertrophic effect in the heart (Citation11,Citation12), and with the control of cell to cell interactions by targeting the adherence molecules both in endothelial and myocardial cells (Citation13,Citation14). Furthermore estrogen is reported to stimulate VEGF production in cholangiocarcinoma cells (Citation15), and in myometrial microvascular endothelial cells (Citation16).
In a recent study in male and female mice ERα expression was higher in the ventricles compared to the atria, whereas expression of ERβ and androgen receptor was similar in both heart chambers (Citation17). The role of estrogens in cardiac remodeling post MI has been assessed in several animal studies. Experimental studies suggest that ERβ may have a protective role with improved myocardial function in female hearts following acute ischemia (Citation18). Other studies report contradictory effects of estrogen after MI, with both increased infarct size and mortality, but long-term protection against left ventricular remodeling (Citation19), or increased mortality in spite of decreased infarct size (Citation20).
Contrary to these studies Förster et al. (Citation21) reported the absence of ERs in cardiac structures challenging that cardiovascular protective effects are direct cardiac effects and rather suggesting indirect effects by modulation of the peripheral cardiovascular system.
The aims of this investigation were to explore if myocardial ERα and ERβ are upregulated after MI and if the presence/ absence of ERα or ERβ influences angiogenesis after MI.
Material and methods
Animals
ERαKO (Citation22) and ERβKOmice (Citation23) (Jan-Åke Gustafsson, Novum, Sweden) and C57BL/6 mice weighing around 25 g (Taconic, Denmark) were used. Both ERαKO and ERβKO mice were made by “insertion” of a neocassette into exon2 (ERαKO) and exon 3 (ERβKO). Thus the genes produced non-functional proteins; and the promoters of the genes were intact. The study was approved by the Stockholm Southern Ethics Review Board for Animal Experiments. The animals were treated in accordance with the Institutional Guidelines for care of laboratory animals.
Myocardial infarction model
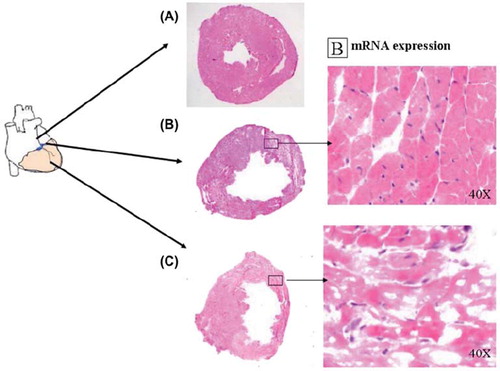
The mice were anesthetized with an intraperitoneal injection of Midazolam (Dormicum, 5 mg/kg) (F. Hoffmann-La Roche Ltd, Switzerland), Medetomidin hydrochloride (Domitor vet, 0.1 mg/kg) (Orion Corp., Espoo, Finland), Fentanyl (0.3 mg/kg) (B. Braun Medical AG, Seesatz, Switzerland) and subsequently endotracheally intubated, and ventilated using a Zoovent ventilator (Model CWC600AP, BK Universal, UK). The heart was exposed via a thoracotomy, and the left anterior descending artery (LAD) was ligated with a 7.0 polypropylene suture. Myocardial infarction (MI) was confirmed by a color change and dyskinesia of the anterior wall. The anesthesia was reversed by an intraperitoneal injection of Flumazenil (Lanexat, 0.1 mg/kg) (F. Hoffmann-La Roche Ltd, Switzerland), Atipamezol hydrochloride (Antisedan vet 5 mg/kg) (Orion Corp., Espoo, Finland) and Buprenorphin hydrochloride (Temgesic, 0.1 mg/kg) (Schering-Plough Corp., Kenilworth, UK). Sham operated animals (n = 5) subjected to thoracotomy only were used as non-infarcted controls as regards myocardial vascular densities. The animals were sacrificed and hearts were harvested for further analysis. The harvested hearts were divided into three parts and the basal part with part of the MI was frozen in OCT (Histolab, Sweden) and stored at −70°C for later immunohistochemical analysis, and a cross sectional circular slice from the infarcted part was snap frozen and kept at −70°C for mRNA extraction and polymerase chain reaction (PCR) analysis. Eosin/hematoxylin staining verified that the samples chosen came from the periinfarct area ().
Figure 1. Transmural myocardial infarction (C) in murine left ventricle 12 days after ligation of the LAD. Immunostainings and mRNA analyses were done on cross sections at the periinfarct level (B). Eosin hematoxylin staining. (A) indicates non-infarcted myocardium.
The initial design of the study was to have equal groups as regards gender (one female and one male group with five to six animals in each) for control, ERαKO and ERβKO. A total of 63 mice were operated with a mortality of 41%. Analyses were blinded including histological diagnosis of MI scar that was observed in 25 (68%) of the survivors. When the blinding was opened after analysis the groups consisted of: Controls (n = 10, five females), ERαKO (n = 9, six females) and ERβKO (n = 5, one female). As there was no statistical difference between gender controls and female ERβKO and male ERαKO we considered that the overall results were not influenced by the gender skewness in the ERαKO and ERβKO groups and that the gender groups could be put together for further analysis and characterization.
mRNA extraction and PCR analysis
RNA extraction and reverse transcription. Total RNA was prepared with a homogenizer in Trizol (Invitrogen, Sweden) and quantified spectrophotometrically by absorbance at 260 nm. Integrity of total RNA was determined by 1% agarose-gel electrophoresis. Two micrograms of RNA were reverse transcribed by Superscript reverse transcriptase (Life Technologies, Stockholm, Sweden) using random hexamer primers (Roche Diagnostics GmbH, Mannheim, Germany) in a total volume of 20 μl.
mRNA measurements. Real-time PCR was used to measure mRNA (ABI-PRISM 7700 Sequence Detector, Perkin-Elmer Applied Biosystems Inc, Foster City, CA, USA). Hprt was used as an endogenous control to correct for potential variation in RNA loading or efficiency of the amplification. Hprt values were at an even level with ct values 20–25 with no values of 26 or more. Primers and probes were supplied as a TaqMan® Reagents kit, Hprt (Tamra) Esr1 (Mm00433149_m1) (ERα) and Esr2 (Mm00599819_m1) (ERβ) from Applied Biosystems.
All reactions were performed in 96-well Micro-Amp Optical plates. Amplification mixes (25 μl) contained the sample 5 μl cDNA diluted 1:100 and TaqMan Universal PCR Mastermix. Thermal cycling conditions included 2 min at 50°C and 10 min at 95°C before the PCR cycles began. Thermal cycling consisted of 40 cycles at 95°C for 15 s and 65°C for 1 min. For each individual, all samples were simultaneously analyzed in one assay run. Measurements of the relative distribution of Esr1 and Esr2 were performed for each individual, a ΔCT value was obtained by subtracting hprt CT values from respective target CT values. The expression of each target was then evaluated by 2−Δ−ΔCT (Citation24).
Immunohistochemistry
ER immunostaining. The OCT embedded tissue was cut in 5 μm thick sections by cryostat. The sections were fixed and then blocked with endogenous peroxide using 0.3% hydrogen peroxide (30 min). After washing, slides were incubated during 30 min in PBS (phosphate buffered solution, 5 min each) with 0.01% Tween 20 and then incubated (one hour) with 10% normal goat serum. After washing, sections were incubated overnight with polyclonal rabbit antibodies against mouse ERα and ERβ diluted in PBS-Tween (1:100 dilution; ERα sc-542 and ERβ sc-8974, Santa Cruz Biotechnology). The specificity of these antibodies has been described previously (Citation25). In addition to these antibodies, presence of ERα was tested with antibodies iD5, DAKO and sc787, Santa Cruz Biotechnology and ERβ with antibody 503 (gift from M. Warner, Medical Nutrition, Karolinska University Hospital, Sweden). Samples were rinsed four times, incubated with goat anti-rabbit secondary antibody (Vector Laboratories, Burlingam, CA, USA, 1:200, one hour), washed and incubated with avidin–biotin complexed with peroxidase (Vector Laboratories, Burlingam, CA, USA, one hour), followed by the application of 3,3-diaminobenzidine in H2O2 (DAB-kit, Vector Laboratories). All slides were counterstained with hematoxylin, dehydrated and mounted with Pertex (Histolab, Gothenburg, Sweden). As negative control, the primary antibody was omitted and the sections were incubated only with the solvent. Murine brain tissue served as a positive control. Analyses of ERα and ERβ expression in different tissues were performed blinded.
Capillary density. Capillary density was analysed in the sections that were, after fixation in formaldehyde, incubated with Griffonia Badeiraea Simplicifolia Isolectin B4 (GSL-I-B4, Vector Laboratories), which was followed by a second incubation with ABC Complex. Finally, capillaries were visualized using DAB with 0.03% hydrogen peroxide. Capillaries were counted with a 20 × objective using a LCD camera (Olympus, Japan) connected to a microscope. Ten fields of pictures from the periinfarct area from each specimen were taken and the capillary count was analyzed with an image analysis program (Micro Image, Olympus) (Citation26). Quantification of capillaries was performed blinded.
Arteriolar density. Frozen sections of myocardium were fixed in 2% formaldehyde diluted in 1 × PBS for 10 min and then incubated with normal rabbit serum as blocking solution for 30 min. Sections were incubated with primary antibody α-actin (Sigma) overnight in a 1:250 dilution. Finally, rabbit anti-mouse secondary antibody (TRITC, Dako) was used to visualize the blood vessels. Blood vessels were blindly analyzed at 20 × magnification under a fluorescence microscope and counted.
Inflammatory response. The inflammatory response was evaluated by counting CD3- and CD11b- positive cells in the periinfarct area (Citation27). After fixation with 4% formaldehyde (Histolab Products Ltd, Gothenburg, Sweden) the sections were blocked with 5% rabbit serum (code No. X0902; Dako Cytomation) and 5% mouse serum (code no. X0910; Dako Cytomation) in TBS (stock solution 10 × concentration: 87.66 g NaCl, 60.55 g Tris diluted to 1000 ml in distilled water, pH: 7.4). The sections were subsequently incubated overnight in a humidified chamber with the primary antibody. These included rat anti-mouse CD3 (clone KT3) and rat anti-mouse CD11b (clone 5C6) (Serotec, Oslo, Norway). The slides were then incubated with the secondary antibody rabbit anti-rat-IgG (FITC code No. F0234; Dako Cytomation) for two hours in a humidified chamber and mounted with an anti-fading reagent containing 4,6-diamidino-2-phenylindole (DAPI) before visualization in the fluorescence microscope (Olympus BX60). Spleen from C57BL/6 mouse was used as positive control.
Statistical analysis
Values are expressed as mean ± SEM. Statistical non-homogeneity was tested with Student's unpaired t-test or with one way Analysis of Variance (ANOVA). A p-value of <0.05 was considered significant.
Results
Real time PCR
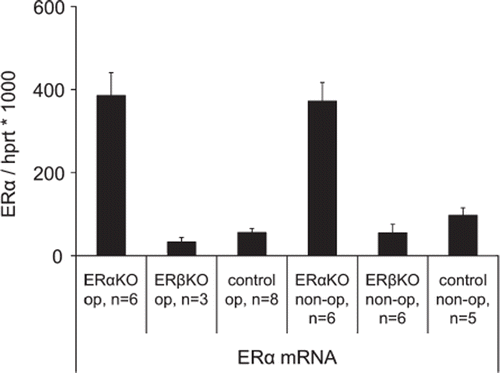
ERαKO Disrupted mRNA coding for ERα was higher (p < 0.005) in ERαKO than native ERα expression in ERβKO and wild type mouse (). The expression of ERα did not change after MI and there was no compensatory increase of ERβ mRNA.
Figure 2. ERα expression in ERαKO, ERβKO and littermate wild type control mice 12 days after induction of myocardial infarction and in non-operated groups. In the infarcted specimens periinfarct tissue was used. Values are mean with SEM.
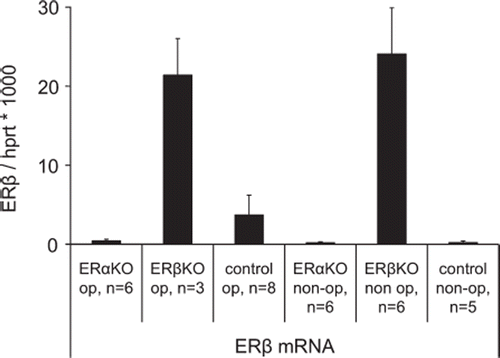
ERbKO. Disrupted mRNA coding for ERβ was higher (p < 0.005) in ERβKO than native ERβ expression in ERαKO and wild type mouse (). The expression of ERβ did not change after MI and there was no compensatory increase of ERα mRNA.
Figure 3. ERβ expression in ERαKO, ERβKO and littermate wild type control mice 12 days after induction of myocardial infarction and in non-operated groups. In the infarcted specimens periinfarct tissue was used. Values are mean with SEM.
Immunohistochemistry
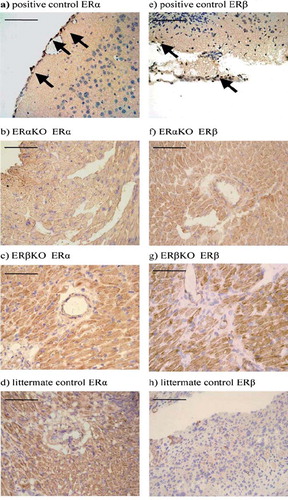
ERa and ERb- staining. In this blinded assay and compared to positive controls (murine brain), ERαKO, ERβKO and wild type mice did not show any expression of ERα and ERβ in periinfarct myocardial tissue sections containing cardiomyocytes, capillaries and arterioles (). Negative results were obtained for all of the antibodies tested (ERα n = 3 and ERβ n = 2).
Figure 4. Rabbit polyclonal antibody to mouse was used for detection of ERα in a) positive control, murine brain, b) ERαKO mouse myocardium and c) ERβKO mouse myocardium, (d) littermate control mouse myocardium; and for detection of ERβ in e) positive control, murine brain, f) ERαKO mouse myocardium and g) ERβKO mouse myocardium, (h) littermate control, mouse myocardium. The marker represents 100 μm. Arrows indicate positive cells.
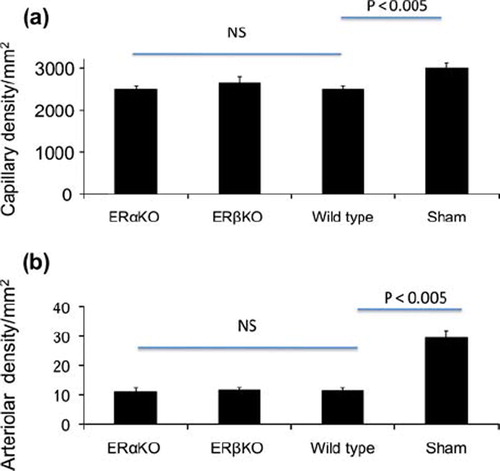
Capillary and arteriolar densities. Capillaries in the periinfarct areas were in the ERαKO-, ERβKO-, wild type and sham operated control groups 2503 ± 241, 2667 ± 385, 2513 ± 382, and / mm2, and arterioles were 11 ± 2, 11 ± 1, 11 ± 1 and 29.5 ± 2.5 mm2, respectively. While the infarcted groups had lower capillary and arteriolar densities than sham controls (p < 0.005), there were no differences in periinfarct vascular densities between ERαKO-, ERβKO- and wild type mice ().
Figure 5. Periinfarct (a) capillary and (b) arteriolar densities 12 days after induction of myocardial infarction in ERαKO, ERβKO, wild type and sham operated mice. While after myocardial infarction vascular densities were lower than in sham operated controls, there were no differences between ERαKO, ERβKO and wild type mice. The assessment was made blinded. Values are mean with SEM.
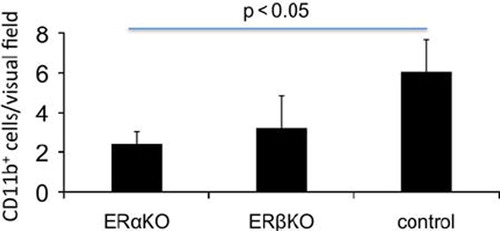
Inflammatory response. The inflammatory response was assessed by counting CD3 + and CD 11b + cells in the periinfarct areas. In the ERαKO group there was less macrophages (CD11b +, p < 0.05) than in the control group (), but there was no difference between the groups in T-cell response (CD3).
Figure 6. The number of macrophages in the periinfarct area was assessed by counting anti CD11b+ cells. In the periinfarct area of ERαKO mice the number of macrophages was lower compared to control (p < 0.05).
Discussion
The aim of this study was to explore whether ERα and ERβ are expressed and functional in the myocardium, if they are upregulated in the periinfarct region following MI and if they stimulate angiogenesis. Such stimulation would counteract the decreased density of capillaries and arterioles that is an adaptation to the ischemia/hibernation that occurs in the periinfarct region. The model used is relevant and sensitive as in previous studies therapeutic angiogenesis with administration of proteins or genes of angiogenic growth factors such as VEGF, FGF, PDGF-BB or angiopoietin indeed induced normalization of vessel densities in the periinfarct region.
Although the disrupted mRNA in both ERαKO and ERβKO mice was overexpressed in the myocardium suggesting responsive promoters, no change occurred following MI. Further, native ER mRNAs in wild type or in ERαKO and ERβKO mice did not change. No ERs were observed with immunohistochemistry in spite of tests with positive controls for several antibodies. Further, no change in capillary or arteriolar densities were observed in this blinded investigation. These observations are in keeping with Förster et al. who reported that no ER could be demonstrated in the mouse heart (Citation21).
There is ample evidence on the vascular protective effects of estrogen in premenopausal women. Estrogen modifies systemic hemodynamics. For example vascular tone is modified by effects on NO mediated mechanisms (Citation28,Citation29). Such systemic effects may indirectly modify the load on the damaged heart, and consequently remodeling and prognosis.
Macrophage density was depressed in the periinfarct area in ERαKO mice suggesting a decreased postinfarct inflammatory response. This effect may be viewed as an indirect ER effect on the myocardium. Although this anti-inflammatory response did not affect periinfarct angiogenesis or expression of VEGF mRNA (unpublished results) it is in keeping with estrogen-dependent proinflammatory effects and macrophage activation (Citation30) and that this effect is related to ERα.
Conclusions
In ERβKO mice the absence of functional ERβ and in ERαKO the absence of functional ERα do not influence myocardial angiogenesis or arteriogenesis after myocardial infarction.
Acknowledgements
This study was supported by Karolinska Institutets Fonder. We thank Margaret Warner for generous supply of antibody. The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Grodstein F, Stampfer M. The epidemiology of coronary heart disease and estrogen replacement in postmenopausal women. Prog Cardiovasc Dis. 1995;38:199–210.
- Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, . Randomized trial of estrogen plus progestin for secondary prevention of coronary heart disease in postmenopausal women. JAMA. 1998;280:605–13.
- Hulley S, Grady D, Bush T, Furberg C, Herrington D, Riggs B, . Postmenopausal hormone treatment. JAMA. 2009;301:2493–5.
- Rossouw JE, Anderson GL, Prentice RL, LaCroix AZ, Kooperberg C, Stefanick ML, . Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results From the Women's Health Initiative randomized controlled trial. JAMA. 2002;288:321–33.
- Chen D-B, Bird IM, Zheng J, Magness RR. Membrane estrogen receptor – dependent extracellular signal-regulated kinase pathway mediates acute activation of endothelial nitric oxide synthase by estrogen in uterine artery endothelial cells. Endocrinology. 2004;145:113–25.
- Traupe T, Stettler CD, Li H, Haas E, Bhattacharya I, Minotti R, . Distinct roles of estrogen receptor α and β mediating acute vasodilation of epicardial coronary arteries. Hypertension. 2007;49:1364–70.
- Taylor AH, Al-Azzawi F. Immunolocalisation of oestrogen receptor beta in human tissues. J Mole Endocrin. 2000;24: 145–55.
- Grohé C, Kahlert S, Löbbert K, Stimpel M, Karas RH, Vetter H, . Cardiomyocytes and fibroblast contain functional estrogen receptors. FEBS Lett. 1997;416:107–12.
- Karas RH, Baur WE, van Eickels M, Mendelsohn ME. Human vascular smooth muscle cells express an estrogen receptor isoform. FEBS Lett. 1995;377:103–8.
- Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, . 17-beta estradiol reduces cardiomyocyte apopotosis in vivo and in vitro via activation of phospho-inositide-3 kinase/Akt signaling. Circ Res. 2004;95:692–9.
- Babiker FA, Lips D, Meyer R, Delvaux E, Zandberg P, Janssen B, . Estrogen receptor β protects the murine heart against left ventricular hypertrophy. Arterioscler Thromb Vasc Biol. 2006;26:1524–30.
- Pedram A, Razandi M, Aitkenhead M, Levin ER. Estrogen inhibits cardiomyocyte hypertrophy in vitro. J Biol Chem. 2005;280:26339–48.
- Groten T, Pierce AA, Huen AC, Schnaper HW. 17β-estradiol transiently disrupts adherens junctions in endothelial cells. FASEB J. 2005;19:1368–70.
- Mahmoodzadeh S, Eder S, Nordmeyer J, Ehler E, Huber O, Martus P, . Estrogen receptor alpha up-regulation and redistribution in human heart failure. FASEB J. 2006;20:926–34.
- Mancino A, Mancino MG, Glaser SS, Alpini G, Bolognese L, Izzo A, . Estrogens stimulate the proliferation of human cholangiocarcinoma by inducing the expression and secretion of vascular endothelial growth factor. Dig Liver Dis. 2009;41:156–63.
- Gargett CE, Zaitseva M, Bucak K, Chu S, Fuller PJ, Rogers PA. 17Beta-estradiol up-regulates vascular endothelial growth factor receptor-2 expression in human myometrial microvascular endothelial cells: Role of estrogen receptor-alpha and -beta.J Clin Endocrinol Metab. 2002;87:4341–9.
- Lizotte E, Grandy SA, Tremblay A, Allen BG, Fiset C. Expression, distribution and regulation of sex steroid hormone receptors in mouse heart. Cell Physiol Biochem. 2009;23:75–86.
- Wang M, Wang Y, Weil B, Abarbanell A, Herrmann J, Tan J, . Estrogen receptor β mediates increased activation of PI3K/Akt signalling and improved myocardial function in female hearts following acute ischemia. Am J Physiol Regul Integr Comp Physiol. 2009;296:R972–8.
- Smith PJW, Ornatsky O, Stewart DJ, Picard P, Dawood F, Wen WH, . Effects of estrogen replacement on infarct size, cardiac remodeling, and the endothelin system after myocardial infarction in ovariectomized rats. Circulation. 2000;102:2983–9.
- van Eickels M, Patten RD, Aronovitz MJ, Alsheikh-Ali A, Gostyla K, Celestin F, . 17-beta-estradiol increases cardiac remodelling and mortality in mice with myocardial infarction. J Am Coll Cardiol. 2003;41:2048–92; (published correction in J AM Coll Cardiol. 2003;42:143.
- Förster C, Kietz S, Hultenby K, Warner M, Gustafsson JÅ. Characterization of the ERβ-/- mouse heart. PNAS. 2004; 101:14234–9.
- Lubahn DB, Moyer JS, Golding TS, Couse JF, Korach KS, Smithies O. Alteration of reproductive function but not prenatal sexual development after insertional disruption of the mouse estrogen receptor gene. PNAS. 1993;90:11162–6.
- Krege JH, Hodgin JB, Couse JF, Enmark E, Warner M, Mahler JF, . Generation and reproductive phenotypes of mice lacking estrogen receptor β. PNAS. 1998;95:15677–82.
- Winer J, Jung CKS, Shackel I, Williams PM. Development and validation of real-time quantitative reverse transcriptase-polymerase chain reaction for monitoring gene expression in cardiac myocytes in vitro. Analyt Biochem. 1999;270: 41–9.
- Cheng G, Weihua Z, Warner M, Gustafsson JÅ. Estrogen receptors ER alpha and ER beta in proliferation in the rodent mammary gland. PNAS. 2004;101:3739–46.
- Siddiqui AJ, Mansson-Broberg A, Gusafsson T, Grinnemo KH, Dellgren G, Hao X, . Antagonism of the renin-angiotensin system can counteract cardiac angiogenic vascular endothelial growth factor gene therapy and myocardial angiogenesis in the normal heart. AJH. 2005;18:1347–52.
- Grinnemo KH, Genead R, Kumagai-Braesh M, Andersson A, Danielsson C, Månsson-Broberg A, . Costimulation blockade induces tolerance to HESC transplanted to the testis and induces regulatory T-cells to HESC transplanted into the heart. Stem Cells. 2008;26:1850–7.
- Arias-Loza PA, Jazbutyte V, Pelzer T. Genetic and pharmacologic strategies to determine the function of estrogen receptor α and estrogen receptor β in the cardiovascular system. Gender Med. 2008;5(Suppl A):34–45.
- Byers MJ, Zangl A, Phernetton TM, Lopez G, Chen D-B, Magness RR. Endothelial vasodilator production by ovine uterine and systemic arteries: Ovarian steroid and pregnancy control of ERα and ERβ levels. J Physiol. 2005;565:85–99.
- Cutolo M, Sulli A, Capellino S, Villaggio B, Montagna P, Seriolo B, . Sex hormones influence on the immune system: Basic and clinical aspects in autoimmunity. Lupus. 2004;13:635–8.