
Abstract
Objectives. In the clinical setting, patients with slower resting heart rate are less prone to cardiovascular death compared with those with elevated heart rate. However, electrophysiological adaptations associated with reduced cardiac rhythm have not been thoroughly explored. In this study, relationships between intrinsic heart rate and arrhythmic susceptibility were examined by assessments of action potential duration (APD) rate adaptation and inducibility of repolarization alternans in sinoatrial node (SAN)-driven and atrioventricular (AV)-blocked guinea-pig hearts perfused with Langendorff apparatus. Design. Electrocardiograms, epicardial monophasic action potentials, and effective refractory periods (ERP) were assessed in normokalemic and hypokalemic conditions. Results. Slower basal heart rate in AV-blocked hearts was associated with prolonged ventricular repolarization during spontaneous beating, and with attenuated APD shortening at increased cardiac activation rates during dynamic pacing, when compared with SAN-driven hearts. During hypokalemic perfusion, the inducibility of repolarization alternans and tachyarrhythmia by rapid pacing was found to be lower in AV-blocked hearts. This difference was ascribed to prolonged ERP in the setting of reduced basal heart rate, which prevented ventricular capture at critically short pacing intervals required to induce arrhythmia. Conclusions. Reduced basal heart rate is associated with electrophysiological changes that prevent electrical instability upon an abrupt cardiac acceleration.
Introduction
There is considerable evidence that demonstrates a strong association between the resting heart rate and cardiovascular outcomes in human subjects. Epidemiological studies have shown that elevated resting heart rate is an independent predictor for cardiovascular mortality both in the general population (Citation1,Citation2), and in patients with preexisting ischemic heart disease and chronic heart failure (Citation3,Citation4). The contribution of increased heart rate to poor prognosis may be determined by associated clinical conditions including high sympathetic tone, increased insulin resistance, and poor physical fitness. Rapid resting heart rate also imposes a greater pulsatile stress on arterial vessels, which promotes endothelial injury (Citation5). Collectively, these factors accelerate progression of atherosclerosis, thus increasing cardiovascular mortality. In contrast, heart rate decrease reduces myocardial oxygen consumption, prolongs diastolic filling time, and improves coronary flow. During therapy with heart rate-lowering agents, such as beta-blockers and If channel inhibitors, a close direct relation was found to exist between the magnitude of heart rate decrease and subsequent reduction in risk of death from cardiovascular causes (Citation6,Citation7).
Much less attention was given to explore how reduced resting heart rate could affect myocardial electrophysiological properties and arrhythmic susceptibility. Ambulatory ECG monitoring in victims of sudden death suggests that fatal cardiac arrest is preceded by transient heart rate acceleration that may degenerate to ventricular fibrillation (VF) (Citation8,Citation9). VF is precipitated by critical shortening of action potential duration (APD), which promotes beat- to-beat APD oscillations (repolarization alternans) leading to spontaneous breakup and fragmentation of depolarization wave front (Citation10,Citation11). The present study was designed to test a hypothesis that slow resting heart rate may potentially prevent an excessive APD shortening following a sudden increase in cardiac activation rate, thus decreasing propensity to develop VF. This hypothesis offers a novel insight into the nature of mechanisms linking a reduced resting heart rate with improved clinical outcomes (Citation6,Citation7), and it is based on the following considerations. First, in the setting of slow resting heart rate, prolonged basal APD would likely limit APD shortening upon an abrupt cardiac acceleration via the mechanisms related to the short-term cardiac memory (Citation12,Citation13). Second, increased effective refractory period, which is an attribute of prolonged resting APD, is likely to prevent ventricular capture at rapid activation rates, at which APD becomes sufficiently short in order to precipitate VF. In order to address these possibilities, characteristics of ventricular APD rate adaptation and arrhythmic susceptibility upon rapid cardiac pacing were examined in isolated, perfused guinea-pig heart preparations from two study groups showing a large difference in intrinsic cardiac beating rate. Specifically, comparisons were made between sinoatrial node (SAN)-driven and atrioventricular (AV)-blocked heart preparations, by taking electrophysiological recordings both at baseline and in the setting of increased electrical instability produced by hypokalemic perfusion.
Methods
This study complies with the European Community Guidelines for the Care and Use of Experimental Animals, and was approved by the institutional Animal Ethics Screening Committee (clearance number: 2010/561-1799). Male Dunkin–Hartley guinea-pigs (supplied by Charles River, Sulzfeld, Germany) weighing 400–500 g were allowed to acclimate to the housing conditions, with free access to food and tap water, for at least 7 days before entry into the study.
Isolated, Langendorff-perfused heart preparations
The experiments on isolated, perfused hearts were performed as described previously (Citation14,Citation15). The guinea pigs were anesthetized with sodium pentobarbital (50 mg/kg i.p.), the chest was opened, and the hearts were immediately excised and mounted on a Langendorff perfusion setup (Hugo Sachs Elektronik-Harvard Apparatus GmbH, March-Hugstetten, Germany). The hearts were perfused via the aorta at a constant pressure (60–65 mmHg) with carefully filtered, warmed physiological saline solution saturated with 95%O2 and 5%CO2. The perfusion solution contained (in mM) NaCl, 118.0; KCl, 4.7; CaCl2, 2.5; NaHCO3, 25; KH2PO4, 1.2; MgSO4, 1.2; and glucose, 10.0; and had a pH of 7.4. The perfusion setup was equipped with two ISOTEC pressure transducers (Harvard Apparatus, Holliston, MA, USA) to measure aortic pressure and left ventricular (LV) developed pressure. The aortic pressure transducer was connected to the aorta block of the setup. The ventricular pressure transducer was coupled to the balloon-tipped catheter introduced into the LV cavity via an incision made in the left atrium. The volume of the LV balloon was adjusted to yield an end-diastolic pressure of 0–5 mm Hg. LV developed pressure was calculated as the difference between end-systolic and end-diastolic pressure. The coronary flow rate was determined using an ultrasonic flowmeter probe (Transonic Systems Inc., USA) placed just above the aortic cannula. The electrical activity of the heart preparations was assessed from the volume-conducted electrocardiogram (ECG) as well as monophasic action potential (MAP) recordings. Both ECG and MAP signals were amplified, filtered at high cutoff (1 kHz), and digitized at a sampling frequency of 5 kHz. Throughout the experiments, the heart preparations were kept immersed in the temperature-controlled, perfusate-filled chamber to minimize thermal loss. Aortic pressure, coronary flow rate, LV pressure, ECG, and ventricular MAPs were continuously monitored using a 16-channel PowerLab system (ADInstruments, Oxford, UK).
Study groups and assessments of ventricular repolarization and refractoriness
Electrophysiological recordings were obtained from intact (i.e., SAN-driven) and AV-blocked heart preparations. In the latter group, the right atrium was removed, and the AV node was crushed mechanically with forceps. The presence of complete AV block was verified by ECG recordings showing prolongation of the R–R interval above 500 ms (from its basal value of ∼300 ms), and the absence of ventricular responses during left atrial stimulation. In both AV-blocked and SAN-driven heart preparations, 30-min stabilization period was allowed before taking electrophysiological recordings. The heart preparations were used for the main part of the study, if they showed regular beating with no spontaneous arrhythmic events (e.g., frequent ectopic beats), and had appropriate coronary flow rates (10–14 ml/min) and contractile function (LV developed pressure, above 60 mm Hg) after the stabilization period. In addition, heart preparations with AV block were rejected if they showed no spontaneous beating or had abnormally slow activation rates (cardiac cycle longer than 800 ms).
The final electrophysiological assessments were made in 10 heart preparations from each group. In SAN-driven hearts, spontaneous beating rates ranged from 173 to 209 beats/min (Mean ± standard deviation [SD]: 190 ± 13 beats/min), and in AV-blocked hearts, they ranged from 94 to 115 beats/min (Mean ± SD: 110 ± 7 beats/min). In all heart preparations, ventricular repolarization, refractoriness, electrical restitution, and arrhythmic susceptibility were assessed both at baseline and in the setting of hypokalemia. Hypokalemic perfusion (2.5 mM K+ over 30 min) was used as a challenge, previously shown to markedly increase electrical instability in perfused guinea-pig heart model (Citation16).
MAPs were recorded using three LV and three right ventricular (RV) pressure contact electrodes applied in a circular manner to the basal epicardial surface. The MAP duration was measured at 90% repolarization (APD90) in arrhythmia-free conditions, and at 80% repolarization (APD80) during tachypacing-induced repolarization alternans. Transepicardial variability in repolarization was assessed by calculating the standard deviation of the mean APD90 value obtained from six ventricular recording sites used.
Electrical stimulation was applied at the LV epicardial base using 2-ms rectangular pulses of twice diastolic threshold current generated by a programmable stimulator (Hugo Sachs Electronik-Harvard Apparatus GmbH, March-Hugstetten, Germany). Effective refractory period (ERP) was measured using extrasystolic stimulation protocol, wherein a burst of 10 regular (S1) pulses was followed by a premature extrastimulus (S2). The S1 pulses in a basic drive train were applied at the longest interval producing no ventricular escaped beats (SAN-driven hearts: S1–S1 =270 ms; AV-blocked hearts: S1–S1 = 500 ms). The premature extrastimulus was initially applied at a coupling stimulation interval of 130 ms in SAN-driven hearts and 200 ms in AV-blocked preparations. Thereafter, the coupling stimulation interval was reduced in 5–10 ms steps until getting no capture. The ERP was defined as the longest S1–S2 interval producing no extrasystolic response.
Electrical restitution
Electrical restitution assessments were performed using dynamic pacing protocol as described previously (Citation17). A train of 30 regular pulses was applied at LV epicardium in progressively reducing pacing intervals. The stimulations were started with S1–S1 = 270 ms in SAN-driven hearts and S1–S1 = 500 ms in AV-blocked preparations. The pacing interval was then decreased in steps of 10 ms (SAN-driven hearts) or 20 ms (AV-blocked hearts) down to S1–S1 = 200 ms, followed by further 5–10 ms reductions until electrical stimulation produced either 2:1 conduction block or evoked arrhythmia. The APD90 values measured over a wide range of pacing cycle lengths were then plotted as a function of preceding diastolic interval (DI). The DI was measured between the APD90 time point in the previous beat and the beginning of the following action potential upstroke. The APD90 versus DI plot was fitted using double-exponential function: y = y0 + A1exp(−DI/τ1) + A2exp(− DI/τ2), where y represents APD90, y0 is a free-fitting variable, A1 and A2 are the amplitudes, and τ1 and τ2 are the time constants of the fast (A1 and τ1) and slow (A2 and τ2) exponential components obtained by a least-squares fit. The curve fitting was performed using Igor Pro 6.0 software (WaveMetrics, Inc., Portland, OR, USA). The APD90-to-DI plots were then differentiated to determine the maximum slope of electrical restitution.
Arrhythmic susceptibility
Arrhythmic propensity in SAN-driven and AV-blocked heart preparations was assessed by inducibility of repolarization alternans and ventricular tachyarrhythmias following rapid cardiac pacing (S1–S1 intervals ranging from 70 to 110 ms). The repolarization alternans preceding VT induction was defined as a regular beat-to-beat oscillation in APD, whereby the APD80 difference in each pair of long–short action potentials was ≥ 5 ms. T-wave alternans was determined by ECG when the periodic variations in T-wave amplitude in each pair of beats were ≥ 0.05 mV.
Data analysis
Data are expressed as means + standard errors of the mean. Unpaired t-tests were used to compare samples forming two data sets. The Fisher's exact test was used to assess the incidence of repolarization alternans and ventricular tachyarrhythmia at baseline and following hypokalemic perfusion. P values of less than 0.05 were considered to be significant.
Results
Basal electrophysiological and hemodynamic parameters determined upon spontaneous beating in SAN-driven and AV-blocked heart preparations
shows representative ECG and MAP recordings obtained in spontaneously beating heart preparations from two study groups. AV-blocked heart preparations exhibited slower cardiac beating rates (AV-blocked: 110 ± 4 beats/min; SAN-driven: 190 ± 5 beats/min; P < 0.0001), and had prolonged ventricular repolarization, as evidenced by increased QT intervals (AV-blocked: 203 ± 5 ms; SAN-driven: 157 ± 3 ms; P < 0.0001) and increased mean APD90 values determined in LV epicardium (AV-blocked: 168 ± 4 ms; SAN-driven: 131 ± 2 ms; P <0.0001) and RV epicardium (AV-blocked: 174 ± 4 ms; SAN-driven: 133 ± 2 ms; P < 0.0001). QRS complex duration was similar in both groups (AV-blocked: 24 ± 1 ms; SAN-driven: 22 ± 1 ms; P = 0.22). The effective refractory period determined at LV epicardium at the longest S1–S1 pacing intervals producing no escaped beats (AV-blocked: S1–S1 = 500 ms; SAN-driven: S1–S1 = 270 ms) was markedly longer in AV-blocked (ERP = 173 ± 3 ms) as compared with SAN-driven (ERP = 104 ± 3 ms; P < 0.0001) heart preparations.
Figure 1. Representative ECG and MAP recordings obtained from SAN-driven (panel A) and AV-blocked (panel B) heart preparations. Volume-conducted ECG and RV epicardial MAPs were recorded simultaneously in spontaneously beating heart preparations from two study groups. In panel B (middle trace) the double arrow shows diastolic interval.
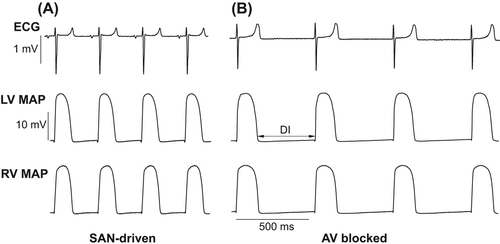
Similar aortic perfusion pressures (AV-blocked: 62 ± 1 mm Hg; SAN-driven: 61 ± 1 mm Hg; P = 0.85), coronary flow rates (AV-blocked: 13 ± 1 ml/min; SAN-driven: 12 ± 1 ml/min; P = 0.35), and LV developed pressures (AV-blocked: 67 ± 6 mm Hg; SAN-driven: 72 ± 7 mm Hg; P = 0.52) were found in heart preparations from two experimental groups.
Ventricular APD rate adaptation and “cardiac memory” effects
shows representative changes in monophasic APD produced by pacing at progressively reduced S1–S1 intervals in SAN-driven and AV-blocked heart preparations, and shows averaged electrical restitution curves along with APD90 restitution slope values determined at LV and RV epicardium. Owing to the greater spontaneous running cardiac cycle length (545 ± 12 ms and 316 ± 9 ms, respectively), dynamic pacing could have been started at longer S1–S1 intervals in AV-blocked as compared with SAN-driven hearts (, section 1 in A and B), which accounted for the longer plateau portion of the dynamic restitution curve in the former group (). In both study groups, a progressive reduction in pacing cycle length was associated with exponential shortening of APD90 (, sections 1–4 in A and B; ). The minimum pacing interval which allowed 1:1 ventricular capture was longer in AV-blocked (177 ± 3 ms) as compared with that in SAN-driven hearts (110 ± 4 ms; P < 0.0001), a difference ascribed to the longer basal ERP in AV-blocked heart preparations, as mentioned above. Electrical stimulations at shorter S1–S1 intervals produced 2:1 conduction block (, section 5 in A and B). No difference in the maximum electrical restitution slope values was found in AV-blocked versus SAN-driven heart preparations (, panels C–F).
Figure 2. Ventricular APD rate adaptation in SAN-driven (panel A) and AV-blocked (panel B) heart preparations. In panels A and B, sections 1–4 show LV epicardial MAPs obtained while progressively reducing S1–S1 pacing intervals from the maximum value that allowed no escaped beats (section 1) till the minimum value at which 1:1 ventricular capture was preserved (section 4). Further reduction in pacing cycle length resulted in 2:1 conduction block (section 5 in both panels). In all traces, the dashed vertical lines indicate the moments of pacing stimulus application. The S1–S1 pacing interval values are indicated in the first cardiac cycle, and the numbers above MAP traces indicate measured APD90 values. Note that cardiac pacing at S1–S1 =220 ms yields a greater APD90 value in AV-blocked (panel B, section 3) as compared with that in SAN-driven heart preparations (panel A, section 2). Also note a greater value of the minimum pacing interval in AV-blocked (180 ms) as compared with that in SAN-driven heart preparations (110 ms), as shown in section 4 (panels A and B).
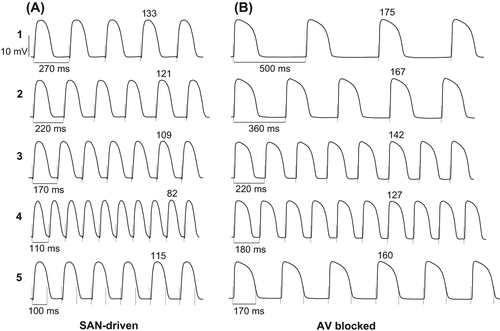
Figure 3. Electrical restitution plots obtained from SAN-driven (panels A, C and E) and AV-blocked (panels B, D, and F) heart preparations. APD90 values determined at three epicardial recording sites in each ventricular chamber were averaged and then plotted as a function of the preceding DI in panels A and B. Panels C and D show changes in APD90 restitution slope over a wide range of diastolic intervals used, and panels E and F show the maximum (Max) restitution slope values determined at the LV and RV epicardium. The dashed rectangular box in panels A and B is to highlight that over a range of diastolic intervals between 49 and 138 ms, the APD90 values determined at similar DIs were longer in AV-blocked as compared with those in SAN-driven heart preparations.
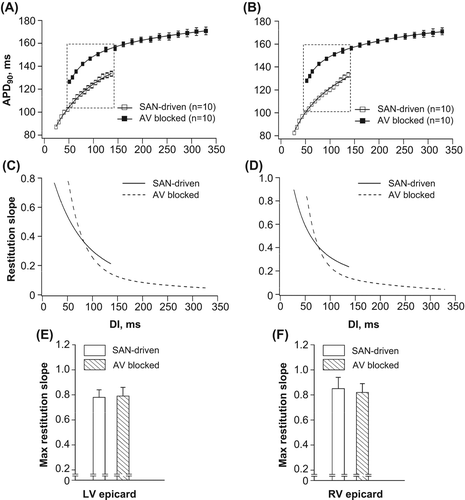
Pacing at the cycle lengths between 180 and 260 ms yielded a range of diastolic intervals (from 49 to 138 ms, as highlighted by rectangular box in ) over which the APD90 restitution curves obtained from AV-blocked and SAN-driven heart preparations overlapped. Importantly, over this range of S1–S1 stimulations, epicardial APD90 values obtained at the same diastolic intervals were longer in AV-blocked as compared with SAN-driven hearts, both in LV and RV chambers (). Thus, longer APD determined in spontaneously beating AV-blocked hearts was associated with less APD90 shortening in response to S1–S1 pacing at a given diastolic interval, indicating the presence of short-term cardiac memory effect.
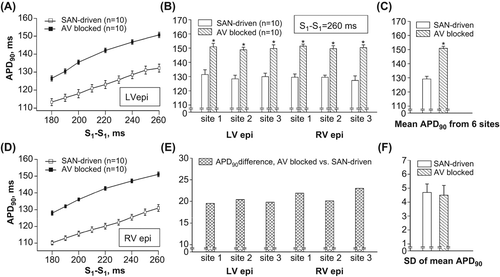
The cardiac memory effect could also be demonstrated by MAP recordings taken at an identical pacing interval in representative heart preparations from both groups. (section 2 in A and section 3 in B) shows that at S1–S1 =220 ms, APD90 value was significantly greater in AV-blocked as compared with SAN-driven preparation (142 and 121 ms, respectively). (panels A and D) shows composite data obtained from 10 preparations used in each group, which suggest that over a range of pacing intervals between 180 and 260 ms, APD90 values determined in AV-blocked hearts were 15–20 ms longer when compared with the APDs determined in SAN-driven hearts.
Figure 4. Epicardial APDs (APD90) obtained at identical pacing intervals in SAN-driven and AV-blocked heart preparations. APD90 values determined at three recording sites in LV epicardium (LV epi) and RV epicardium (RV epi) in SAN-driven and AV-blocked heart preparations were averaged and then plotted as a function of S1–S1 pacing interval in panels A and D. Panel B shows individual APD90 values determined at six epicardial recording sites at pacing interval of 260 ms, and panel E shows the relative difference in APD90 value (AV-blocked vs. SAN-driven heart preparations) obtained at each recording site used. The mean APD90 values from 6 recording sites and their SDs are shown in panels C and F, respectively. *P < 0.05 versus its corresponding value in SAN-driven heart preparations (in panels B and C).
Spatial repolarization gradients
APD90 variability throughout ventricular epicardium was assessed at S1–S1 = 260 ms, the longest pacing interval which was employed both in SAN-driven and AV-blocked heart preparations. shows individual APD90 values measured in six epicardial recording sites, and shows the relative APD90 difference between two study groups assessed at each recording site. In all recording sites used, APD determined in AV-blocked heart preparations was significantly longer compared with that in SAN-driven hearts (). However, this prolongation was highly uniform throughout ventricular epicardium, being over the range of values between 20 and 23 ms (). Consequently, although mean APD90 value from six epicardial recording sites was longer in AV-blocked compared with that in SAN-driven preparations (), its standard deviation, a measure of transepicardial APD90 variability, was similar in two groups ().
Arrhythmogenic responses upon rapid cardiac pacing
In basal conditions, rapid cardiac pacing elicited repolarization alternans in 3 out of 10 SAN-driven heart preparations. Following hypokalemic perfusion, repolarization alternans was induced in 9 out of 10 preparations, thus indicating an increase in its occurrence from 30% to 90% (P = 0.02). In six hypokalemic hearts, repolarization alternans precipitated ventricular tachyarrhythmia upon further reduction in S1–S1 pacing interval. These changes are illustrated by representative ECG and MAP recordings shown in . In this experiment, high amplitude beat-to-beat oscillations in T-wave and epicardial APD80 were promoted by shortening the S1–S1 pacing interval from 110 to 90 ms (), with subsequent induction of polymorphic ventricular tachyarrhythmia (VT) at S1–S1 = 80 ms ().
Figure 5. Arrhythmogenic responses during rapid cardiac pacing (panels A, B, and C), and changes in effective refractory periods (panel D) determined in hypokalemic heart preparations. Panels A–C show simultaneous recordings of volume-conducted ECG and LV and RV MAPs obtained upon tachypacing applied to SAN-driven heart preparations subjected to hypokalemic perfusion. In all MAP traces, the dashed vertical lines indicate the moments of pacing stimulus application. The S1–S1 pacing interval values are indicated in the first cardiac cycle (under the RV MAP traces), and the numbers above MAP recordings (panels A and B) indicate measured APD80 values. Panel A: low-amplitude T-wave alternans and regular beat-to-beat APD80 values obtained at S1–S1 = 110 ms. Panel B: shortening of S1–S1 interval till 90 ms provokes high amplitude beat-to-beat oscillations in T wave and APD80. Panel C: further shortening of S1-S1 interval till 80 ms promotes ventricular tachyarrhythmia. Panel D shows that reduced ERPs in hypokalemic setting allow 1:1 capture at S1–S1 intervals producing APD alternans in SAN-driven heart preparations, but not in AV-blocked heart preparations. The double arrows highlight the magnitude of ERP reduction which is required in both groups in order to allow pacing at arrhythmogenic S1–S1 intervals. *P < 0.05 versus its corresponding basal value (in panel D).
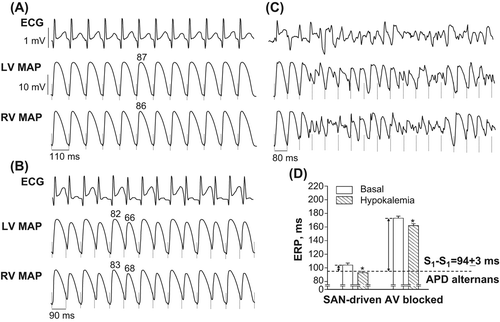
In hypokalemic heart preparations, repolarization alternans were invariably induced at pacing intervals of less than 100 ms (mean S1–S1 interval = 94 ± 3 ms). In this setting, beat-to-beat variability in ventricular repolarization has been manifested as alternations of the T-wave amplitude (Max: 0.35 ± 0.05 mV; Min: 0.11 ± 0.03 mV; Max-to-min change: 0.24 ± 0.05 mV), LV epicardial APD80 (Max: 86 ± 3 ms; Min: 74 ± 3 ms; Max-to-min change: 12 ± 2 ms), and RV epicardial APD80 (Max: 88 ± 3 ms; Min: 74 ± 2 ms; Max-to-min change: 14 ± 1 ms).
In contrast, no repolarization alternans or VT was induced by rapid cardiac pacing in AV-blocked heart preparations, either at baseline or following hypokalemic perfusion. Between-group comparisons using Fisher's exact test revealed that although inducibility of repolarization alternans in basal conditions was not different in SAN-driven versus AV-blocked hearts (30% vs. 0%, P = 0.21), in the setting of hypokalemia both repolarization alternans (inducibility in SAN-driven vs. AV-blocked hearts: 90% vs. 0%, P = 0.0001) and VT (inducibility in SAN-driven vs. AV-blocked hearts: 60% vs. 0%, P = 0.01) were elicited more frequently in SAN-driven preparations.
Effective refractory periods and minimum pacing intervals
Differential arrhythmic susceptibility in SAN-driven versus AV-blocked heart preparations was ascribed to the basal difference in the amount of ERP reduction which was required for inducing repolarization alternans in both study groups. These relations are illustrated in . The basal LV epicardial ERP values in SAN-driven and AV-blocked heart preparations were found to be 104 ± 3 ms and 173 ± 3 ms, and the S1–S1 pacing interval to induce repolarization alternans (as assessed in hypokalemic SAN-driven hearts) was 94 ± 3 ms. This means that in order to allow 1:1 pacing at arrhythmogenic stimulation intervals, the ERP should have been reduced by ∼ 10 ms in SAN-driven hearts, and by ∼79 ms in AV-blocked preparations (as shown by double arrows in ). Hypokalemia evoked a proportional ERP reduction in both study groups (SAN-driven hearts: − 11 ± 1 ms; AV-blocked hearts: − 10 ± 1 ms; P = 0.91). In SAN-driven hearts, the magnitude of this change was sufficient in order to bring the reduced ERP (93 ± 2 ms) toward the range of values which allowed ventricular capture at arrhythmogenic S1–S1 intervals (). In contrast, in hypokalemic AV-blocked heart preparations, the effective refractory period (162 ± 3 ms) and the associated minimum pacing interval (156 ± 4 ms) remained much greater compared with the threshold value of S1–S1 interval for inducing repolarization alternans (94 ± 3 ms), thereby preventing electrical instability.
Discussion
Main findings
Although clinical trials reported on direct relationships between the magnitude of resting heart rate reduction and improved outcomes (Citation6,Citation7), the contributing electrophysiological mechanisms remain incompletely understood. The present study is the first to show that reduced intrinsic heart rate is associated with attenuated APD shortening upon sudden heart rate acceleration, which contributes to reduced propensity to develop tachyarrhythmia. APD reduction at fast activation rates is limited owing to cardiac memory attributed to the past “slow cardiac beating” history associated with prolonged repolarization. Furthermore, in hearts with slower intrinsic beating rate, prolonged basal ERP may prevent ventricular capture at rapid cardiac activation rates, which would otherwise lead to the critical APD shortening required to promote arrhythmia. In this regard, this study suggests that a relative difference between the basal ERP and the threshold pacing interval value for inducing repolarization alternans may represent a novel parameter suitable to quantify pro-arrhythmic tendency, especially upon interventions which abbreviate ventricular refractoriness, such as hypokalemia.
Ventricular APD, arrhythmic susceptibility, and short-term cardiac memory
The excessive shortening in APD at fast cardiac activation rates is thought to contribute to conversion of ventricular tachycardia to VF. A segment of the wave front with critically short APD carries insufficient current strength to depolarize downstream cardiac cells; this translates to the source-to-sink mismatch which causes local conduction failure (Citation10,Citation11). The localized conduction block provokes fragmentation of the propagating wave front, thus leading to highly disorganized electrical activation and precipitating VF.
The shortening of ventricular APD in tachycardia is attributed to reduction of the preceding diastolic interval (the electrical restitution effect). During cardiac acceleration, reduced diastolic interval contributes to incomplete recovery of outward K+ currents that govern ventricular repolarization, thus leading to decreased APD. Rate adaptation of ventricular repolarization, however, may include more complexities, when considering that APD in a given cardiac cycle is determined not only by the immediately preceding diastolic interval, but also by the entire activation history (Citation12,Citation13,Citation18). In general, ventricular APD tends to be longer than expected from preceding diastolic interval if APD in previous cardiac beats was long, and vice versa; this kind of regulation was called the short-term cardiac memory (Citation12,Citation13). Cardiac memory effect may be revealed, for example, when measuring APD in extrasystolic beat evoked immediately after a train of regular pulses applied at different S1–S1 pacing intervals (Citation12,Citation17). At a given S1–S2 coupling stimulation interval, APD in a premature beat tends to be longer upon increasing the S1–S1 interval (and hence increasing APD) in a series of regular beats that precede extrastimulus application. Memory effect is thought to be an important determinant of repolarization variability in cardiac arrhythmia. Indeed, in the setting of VF, activation history was found to be equally important or even predominating over the electrical restitution in explaining beat-to-beat APD variability (Citation18).
The mechanisms involved in cardiac memory are thought to be related to slowly developing alterations in ionic concentrations on both sides of the cardiac myocyte membrane, which result from changes in intrinsic beating rate (Citation19). Fast beating rates are associated with K+ accumulation in extracellular clefts, which causes APD shortening owing to increased conductance of IK, the delayed rectifier K+ current (Citation19). APD shortening in rapid cardiac rates could also be attributed to increased intracellular Na+ levels, which stimulate the reversed mode of the Na+–Ca2+ exchange and Na+–K+ pump, or to elevated intracellular Ca2+ concentrations, which contribute to faster inactivation of ICa (Citation19). Collectively, these changes lead to an enhanced net repolarizing current in ventricular cells with faster intrinsic activation rates; this was likely to contribute to the APD difference found at similar diastolic intervals (ranged from 49 to 138 ms) in paced SAN-driven versus AV-blocked heart preparations used in this study.
Importantly, over this range of overlapping diastolic intervals, the relative APD lengthening in AV-blocked hearts was found to be uniform throughout epicardium, thus translating to no change in spatial repolarization gradients. Such an APD prolongation is thought to be anti-arrhythmic (Citation20). This notion is further supported by outcomes of simulation studies (Citation21) and experimental works (Citation22), which suggest that cardiac memory (i.e., longer APD than expected from the preceding diastolic interval) can suppress arrhythmogenic repolarization alternans evoked by sudden cardiac acceleration.
Tachypacing-induced electrical instability
Rapid cardiac rates are often associated with repolarization alternans, which is manifested in regular beat-to-beat variations in APD and T-wave amplitude (). Repolarization alternans is clinically recognized as a strong marker of increased vulnerability to malignant ventricular tachyarrhythmia (Citation23). In line with this notion, in the present study, repolarization alternans was found to precede VF development in 6 out of 10 SAN-driven heart preparations exposed to hypokalemia. Importantly, repolarization alternans is inducible in conditions wherein ventricular capture is preserved at a certain critical level of increased cardiac pacing rate. For instance, in the guinea-pig hearts used in this study, repolarization alternans invariably occurred when 1:1 capture was maintained at S1–S1 stimulation intervals less than 100 ms. The importance of a given critical degree of heart rate acceleration for promoting repolarization alternans is likely to be accounted for by abnormalities in intracellular Ca2+ cycling associated with tachypacing (Citation23). Increased systolic Ca2+ fluxes at rapid stimulation rates may perturb the activity of Ca2+-dependent sarcolemmal ion channels and exchangers, as well as impair sarcoplasmic reticulum (SR) Ca2+ handling via effects on SR Ca2+-ATPase (SERCA) and Ca2+/calmodulin-dependent kinase II (CaMKII). Collectively, these changes translate to abnormalities in SR Ca2+ uptake and release, thereby leading to beat-to-beat oscillations in cytosolic Ca2+ levels (i.e., Ca2+ transient alternans). Fluctuations in intracellular Ca2+ in turn promote beat-to-beat APD changes secondary to Ca2+-induced effects on ICa inactivation, or the electrogenic Na+–Ca2+ exchanger, or both (Citation23).
Basal LV effective refractory period is the major determinant of the minimum 1:1 pacing interval that could be achieved upon progressive increase in cardiac stimulation rate. Consequently, in heart preparations with short ERP, ventricular capture could be maintained at faster stimulation rates, thus increasing propensity for repolarization alternans. In contrast, prolonged basal ERP would contribute to 2:1 conduction block at those levels of cardiac acceleration, which are required to induce arrhythmia. These considerations suggest that the difference between the basal ERP and the threshold pacing interval value required for promoting repolarization alternans is a critical determinant of arrhythmic susceptibility. For instance, in SAN-driven heart preparations used in this study, hypokalemia was found to reduce epicardial ERP to such an extent that the difference between the basal ERP and the arrhythmogenic pacing interval value was totally eliminated (); this contributed to an increase in occurrence of repolarization alternans during tachypacing from 30% to 90%. Conversely, although ERP was reduced by the same amount in hypokalemic AV-blocked heart preparations, it nevertheless remained much greater compared with the value of arrhythmogenic stimulation interval (), thus eliminating arrhythmic substrate.
Limitations
The present findings should not be overinterpreted in a sense that AV block may be clinically viewed as an anti-arrhythmic condition. In fact, in this study, an acute AV block was used just as a methodological instrument in order to produce two study groups with markedly different levels of intrinsic heart rate (i.e., AV-blocked vs. SAN-driven preparations). Chronic AV block seen in cardiac patients is most certain to be arrhythmogenic, because a sustained increase in diastolic filling time (resulting from prolonged cardiac cycle) would eventually lead to volume overload, eccentric LV remodeling, and systolic dysfunction, thus creating arrhythmic substrate.
Reduction in the resting heart rate may provoke different changes in arrhythmic substrate depending on whether cardiac repolarization reserve in a given patient is impaired or not. In the setting of long QT syndrome caused by inherited (ion channel mutation) or acquired (e.g., drug-induced) defects in ventricular repolarization, heart rate decrease may impose an additional hit on reduced repolarization reserve, thus precipitating arrhythmia. In contrast, in subjects with preserved repolarization mechanisms, a reduction in resting heart rate by pharmacological therapy and lifestyle changes was found to be associated with improved cardiovascular outcomes (Citation6,Citation7). The present work examined relationships between the intrinsic heart rate and arrhythmic susceptibility in normal guinea-pig hearts. Further studies are warranted in order to determine how these relationships are modified in heart preparations with impaired repolarization mechanisms (e.g., upon IK blocker administration).
This study reports on the difference in arrhythmic susceptibility in AV-blocked versus SAN-driven heart preparations determined in the setting of hypokalemia. The effects produced by other arrhythmogenic challenges, for example, by beta-adrenergic agonist infusion (Citation24,Citation25), on inducibility of repolarization alternans in both study groups, remain to be determined.
Conclusions
In perfused guinea-pig hearts, prolonged baseline action potential duration and effective refractory period in the setting of reduced intrinsic heart rate attenuate arrhythmogenic APD shortening upon cardiac acceleration, and prevent ventricular capture at rapid pacing rates that promote repolarization alternans and VT.
Declaration of interest: The author reports no conflicts of interest. The author alone is responsible for the content and writing of the paper.
This study was supported by the Novo Nordisk Foundation and the Obel Family Foundation.
References
- Kannel WB, Cupples LA. Heart rate and cardiovascular mortality: the Framingham study. Am Heart J. 1987;113: 1489–94.
- Johansen CD, Olsen RH, Pedersen LR, Kumarathurai P, Mouridsen MR, Binici Z, et al. Resting, night-time, and 24 h heart rate as markers of cardiovascular risk in middle-aged and elderly men and women with no apparent heart disease. Eur Heart J. 2013;34:1732–9.
- Diaz A, Bourassa MG, Guertin MC, Tardif JC. Long-term prognostic value of resting heart rate in patients with suspected or proven coronary artery disease. Eur Heart J. 2005;26:967–74.
- Castagno D, Skali H, Takeuchi M, Swedberg K, Yusuf S, Granger CB, et al. Association of heart rate and outcomes in a broad spectrum of patients with chronic heart failure: results from the CHARM (Candesartan in Heart Failure: Assessment of Reduction in Mortality and morbidity) program. J Am Coll Cardiol. 2012;59:1785–95.
- Whelton SP, Blankstein R, Al-Mallah MH, Lima JA, Bluemke DA, Hundley WG, et al. Association of resting heart rate with carotid and aortic arterial stiffness: multi-ethnic study of atherosclerosis. Hypertension. 2013;62:477–84.
- Flannery G, Gehrig-Mills R, Billah B, Krum H. Analysis of randomized controlled trials on the effect of magnitude of heart rate reduction on clinical outcomes in patients with systolic chronic heart failure receiving beta-blockers. Am J Cardiol. 2008;101:865–9.
- Fox K, Komajda M, Ford I, Robertson M, Bohm M, Borer JS, et al. Effect of ivabradine in patients with left ventricular systolic dysfunction: a pooled analysis of individual patient data from the BEAUTIFUL and SHIFT trials. Eur Heart J. 2013;34:2263–70.
- Panidis IP, Morganroth J. Sudden death in hospitalized patients: cardiac rhythm disturbances detected by ambulatory electrocardiographic monitoring. J Am Coll Cardiol. 1983;2:798–805.
- Milner PG, Platia EV, Reid PR, Griffith LSC. Ambulatory electrocardiographic recordings at the time of fatal cardiac arrest. Am J Cardiol. 1985;56:588–92.
- Kim YH, Yashima M, Wu TJ, Doshi R, Chen PS, Karagueuzian HS. Mechanism of procainamide-induced prevention of spontaneous wave break during ventricular fibrillation. Circulation. 1999;100:666–74.
- Harada M, Tsuji Y, Ishiguro YS, Takanari H, Okuno Y, Inden Y, et al. Rate-dependent shortening of action potential duration increases ventricular vulnerability in failing rabbit heart. Am J Physiol Heart Circ Physiol. 2011;300: H565–73.
- Gilmour RF, Otani NF, Watanabe MA. Memory and complex dynamics in cardiac Purkinje fibers. Am J Physiol Heart Circ Physiol. 1997;272:H1826–32.
- Watanabe MA, Koller ML. Mathematical analysis of dynamics of cardiac memory and accommodation: theory and experiment. Am J Physiol Heart Circ Physiol. 2002;282: H1534–47.
- Osadchii O, Norton G, Deftereos D, Woodiwiss A. Rat strain-related differences in myocardial adrenergic tone and the impact on cardiac fibrosis, adrenergic responsiveness, and myocardial structure and function. Pharmacol Res. 2007;55:287–94.
- Osadchii OE, Soltysinska E, Olesen SP. Na+ channel distribution and electrophysiological heterogeneities in guinea-pig ventricular wall. Am J Physiol Heart Circ Physiol. 2011; 300:H989–1002.
- Osadchii OE, Olesen SP. Electrophysiological determinants of hypokalemia-induced arrhythmogenicity in the guinea-pig heart. Acta Physiol (Oxf). 2009;197:273–87.
- Osadchii OE. Effects of ventricular pacing protocol on electrical restitution assessments in guinea-pig heart. Exp Physiol. 2012;97:807–21.
- Toal SC, Farid TA, Selvaraj R, Chauhan VS, Masse S, Ivanov J, et al. Short-term memory and restitution during ventricular fibrillation in human hearts. Circ Arrhythm Electrophysiol. 2009;2:562–70.
- Eisner DA, Dibb KM, Trafford AW. The mechanism and significance of the slow changes of ventricular action potential duration following a change of heart rate. Exp Physiol. 2009;94:520–8.
- Hondeghem LM, Carlsson L, Duker G. Instability and triangulation of the action potential predict serious proarrhythmia, but action potential duration prolongation is antiarrhythmic. Circulation. 2001;103:2004–13.
- Cherry EM, Fenton FH. Suppression of alternans and conduction blocks despite steep APD restitution: electrotonic, memory, and conduction velocity restitution effects. Am J Physiol Heart Circ Physiol. 2004;286: H2332–41.
- Benville I, Chattipakorn N, Gray R. Restitution dynamics during pacing and arrhythmias in isolated pig hearts. J Cardiovasc Electrophysiol. 2004;15:455–63.
- Laurita KR, Rosenbaum DS. Cellular mechanisms of arrhythmogenic cardiac alternans. Prog Biophys Mol Biol. 2008;97:332–47.
- Gibbs M, Veliotes DG, Anamourlis C, Badenhorst D, Osadchii O, Norton GR, Woodiwiss AJ. Chronic beta-adrenoreceptor activation increases cardiac cavity size through chamber remodeling and not via modifications in myocardial material properties. Am J Physiol Heart Circ Physiol. 2004;287:H2762–7.
- Soltysinska E, Thiele S, Olesen SP, Osadchii OE. Chronic sympathetic activation promotes downregulation of β-adrenoceptor-mediated effects in the guinea-pig heart independently of structural remodeling and systolic dysfunction. Pflugers Arch. 2011;462:529–43.