Abstract
Glycosidases play an important role in a wide range of physiological and pathological conditions, and have become potential targets for the discovery and development of agents useful for the treatment of diseases such as diabetes, cancer, influenza, and even AIDS. In this study, several benzimidazole derivatives were prepared from o-phenylenediamine and aromatic and heteroaromatic carboxaldehydes in very good yields, using PdCl2(CH3CN)2 as the most efficient catalyst. Synthesized compounds were assayed for their activity on yeast and rat intestinal α-glucosidase inhibition and cytotoxic activity against colon carcinoma cell line HT-29. Compound 3e exhibited 95.6% and 75.3% inhibition of yeast and rat intestinal α-glucosidase enzyme, while showing 74.8% cytotoxic activity against the HT-29 cell line at primary screening concentrations of 2.1 mM for yeast and rat intestinal α-glucosidase enzyme and 0.2 mM for cytotoxic activity against the HT-29 cell line, respectively. Compound 3c displayed 76% and 34.4% inhibition of yeast and rat intestinal α-glucosidase enzyme, and 80.4% cytotoxic activity against the HT-29 cell line at similar primary screening concentrations. The IC50 value for the most potent intestinal α-glucosidase inhibitor compound 3e was found to be 99.4 μM. The IC50 values for the most active cytotoxic compounds 3c and 3e were 82 μM and 98.8 μM, respectively. Both compounds displayed significant antihyperglycemic activity in starch-induced postprandial hyperglycemia in rats. This is the first report assigning yeast and rat intestinal α-glucosidase enzyme inhibition, cytotoxic activity against the HT-29 cell line, and antihyperglycemic activity to benzimidazole compounds 3c and 3e.
Introduction
Benzimidazoles are heterocyclic compounds having various biological activities, among which albendazole, mebendazole, and thiabendazole are widely used antihelmintic drugsCitation1. Furthermore, benzimidazole compounds are also reported to possess biological activities such as inhibition of the angiopoietin receptor TIE-2 and tyrosine kinase receptor VEGFR-2 (vascular endothelial growth factor receptor-2)Citation2, antitumor activityCitation3, thrombopoietin receptor agonistic activityCitation4, γ-aminobutyric acid (GABA) agonistic acti- vityCitation5, antibacterial activityCitation6, antiprotozoalCitation7 and antimicrobial activityCitation8, topoisomerase inhibitionCitation9, neuropeptide Y1 receptor antagonistic activityCitation10, and inhibition of angiotensin IICitation11. Recently, benzimidazole derivatives have been found to be active on several targets useful for the development of antidiabetic therapeutics, e.g. inhibitors of dipeptidyl peptidase IVCitation12 and activators of peroxisome proliferator-activated receptor-γ (PPAR-γ)Citation13,Citation14.
In the course of our study to find α-glucosidase inhibitors from various sources, we observed benzimidazoles to be potent inhibitors of yeast as well as intestinal α-glucosidase. Intestinal α-glucosidase inhibitors have become an important therapeutic tool in mitigation of postprandial hyperglycemia (PPHG). PPHG has emerged as a prominent and early defect in type 2 diabetesCitation15 and as a predictor of cardiovascular or even all-cause mortality, as well as an independent risk factor for atherosclerosisCitation16. Several epidemiological studies have observed that subjects with type 2 diabetes have an increased risk for the development of colorectal cancerCitation17.
Therefore, the search for agents that mitigate postprandial hyperglycemia and possess cytotoxic activity against colon cancer cell lines may offer an opportunity for the development of novel therapeutics for diabetic patients. In this communication we report the synthesis of various benzimidazoles 3a–h by the condensation of o- phenylenediamines (1a–h) with aromatic and heteroaromatic carbonyl compounds (2a–h) in the presence of a catalytic amount of PdCl2(CH3CN)2 in dry MeOH at room temperature. Compounds 3a–h were evaluated for their in vitro α-glucosidase inhibitory activity and cytotoxic activity on colon carcinoma line HT-29, as well as antihyperglycemic property in starch-induced postprandial hyperglycemia in rats.
Experimental
Chemistry
1H nuclear magnetic resonance (NMR) and 13C NMR spectra were recorded on a Varian Gemini 200 MHz and an Avance 300 MHz spectrometer in CDCl3 and DMSO-d6 using tetramethylsilane (TMS) as internal standard. All chemical shifts are reported as δ (ppm) values. Infrared (IR) spectra were recorded on a Nicollet 740 Fourier transform (FTIR) spectrometer. Mass spectra were obtained on an Agilent liquid chromatography (LCMS) instrument. Melting points were determined in open glass capillary tubes on a Metler FP 51 apparatus and are uncorrected. All reactions were monitored by thin layer chromatography (TLC) on pre-coated silica gel 60 F254 (mesh); spots were visualized under ultraviolet (UV) light. Merck silica gel (60–120; 100–200 mesh) was used for chromatography. All reactions were carried out using reagent-grade solvents, and the reagents were purchased from Sigma-Aldrich.
General procedure for the preparation of benzimidazoles 3a–h
To a stirred solution of o-phenylenediamine (1a, 1 mmol) and catalyst PdCl2(CH3CN)2 (20 mol%) in anhydrous methanol (5 mL) was added a solution of carbonyl compound (2a, 1 mmol) under a nitrogen atmosphere over a period of 5 min at room temperature, and the contents were stirred at the same temperature for 4 h. After completion of the reaction (TLC), the solvent was removed under reduced pressure, and the crude product was subjected to column chromatography purification to afford 2-(2- chloro-5-methyl-3-pyridyl)-1H-benzo[d]imidazole 3a as solid in 82% yield. Mp: 213–215°C; IR (KBr): 3443, 2922, 1569, 1436, 1071, 889 cm−1; 1H NMR (CDCl3, 300 MHz): δ 2.42 (s, 3H, CH3), 7.24–7.36 (m, 2H, aromatic), 7.62–7.72 (m, 2H, aromatic), 8.28 (s, 1H, heteroaromatic), 8.72 (s, 1H, heteroaromatic); 13C NMR (CDCl3, 75 MHz): δ 22.14, 120.71, 127.86, 130.93, 138.26, 143.92, 146.50, 150.31, 152.62, 155.82; LCMS: [M+ + 1] 243, 245.
Compounds 3b-h were synthesized similarly.
2-(2-chloro-5-methyl-3-pyridyl)-6-nitro-1H-benzo[d]imidazole.
3b 68% Yield; solid; Mp: 261–263°C; IR (KBr): 3363, 2924, 1619, 1439, 1339, 1189, 1064, 947 cm−1; 1H NMR (CDCl3 + DMSO-d6, 300 MHz): δ 2.42 (s, 3H, CH3), 7.78 (d, J = 10 Hz, 1H, aromatic), 8.22 (d, 1H, heteroaromatic), 8.32 (s, 1H, aromatic), 8.38 (s, 1H, heteroaromatic), 8.66 (s, 1H, heteroaromatic); LCMS: [M+ – 1] 287, 289.
4-(6-methyl-1H-benzo[d]imidazol-2-yl)-1,3- benzenediol.
3c 78% Yield; solid; Mp: 248–249°C; IR (KBr): 3559, 3356, 2922, 1740, 1616, 1494, 1426, 1250, 1040, 970 cm−1; 1H NMR (CDCl3 + DMSO-d6), 300 MHz): δ 2.42 (s, 3H, CH3), 6.40 (d, J = 8 Hz, 1H, aromatic), 6.50 (s, 1H, aromatic), 6.98 (s, 1H, aromatic), 7.30 (s, 1H, aromatic), 7.38 (d, J = 12 Hz, 1H, aromatic), 7.62 (d, J = 12 Hz, 1H, aromatic); 13C NMR (CDCl3 + DMSO-d6; 75 MHz): δ 21.19, 103.00, 104.55, 107.45, 123.69, 127.24, 131.62, 152.04, 159.78, 160.62; LCMS: [M+ – 1] 239.
6-Nitro-2-(3-pyridyl)-1H-benzo[d]imidazole.
3d 64% Yield; solid; Mp: 242–244°C; 1H NMR (CDCl3, 300 MHz): δ 7.42–7.54 (m, 1H, aromatic), 7.70 (d, J = 8 Hz, 1H, heteroaromatic), 8.18 (d, J = 10 Hz, 1H, heteroaromatic), 8.52–8.58 (m, 2H, heteroaromatic), 8.78 (s, 1H, aromatic), 9.46 (s, 1H, heteroaromatic). LCMS: [M+ + 1] 240.
6-Nitro-2-(4-pyridyl)-1H-benzo[d]imidazole.
3e 62% Yield; solid; Mp: 224–226°C; 1H NMR (CDCl3, 300 MHz): δ 7.26 (s, 1H, aromatic), 7.44–7.52 (m, 1H, aromatic), 7.74 (brs, 1H, NH), 7.92–8.02 (m, 1H, aromatic), 8.26 (d, J = 10 Hz, 1H, heteroaromatic), 8.54 (d, 1H, heteroaromatic), 8.68 (d, J = 8 Hz, 2H, heteroaromatic); LCMS: [M+ + 1] 241.
6-Methyl-2-(3-pyridyl)-1H-benzo[d]imidazole.
3f 76% Yield; solid; Mp: 244–246°C; IR (KBr): 3448, 3006, 2917, 1736, 1574, 1448, 1425, 1314, 1274, 1186, 962 cm−1; 1H NMR (CDCl3, 300 MHz): δ 2.46 (s, 3H, CH3), 6.98 (d, J = 10 Hz, 1H, aromatic), 7.28–7.52 (m, 3H, aromatic + heteroaromatic), 8.48 (d,d, J = 10 Hz, 1H, heteroaromatic), 8.66 (d,d, J = 8 Hz, 1H, aromatic), 9.34 (d, J = 8 Hz, 1H, heteroaromatic); 13C NMR (CDCl3 + DMSO-d6; 75 MHz): δ 21.26, 123.93, 126.24, 131.76, 133.56, 147.35, 148.45, 150.25; LCMS: [M+ + 1] 210.
6-Methyl-2-(4-pyridyl)-1H-benzo[d]imidazole.
3g 68% Yield; solid; Mp: 218–219°C; IR (KBr): 3448, 3038, 1683, 1606, 1483, 1373, 1235, 1000, 960 cm−1; 1H NMR (CDCl3, 300 MHz): δ 2.50, (s, 3H, CH3), 7.04 (d, J = 8 Hz, 1H, aromatic), 7.42 (s, 1H, aromatic), 7.58 (d, J = 8 Hz, 1H, aromatic), 8.04 (d, J = 10 Hz, 2H, heteroaromatic), 8.70 (d, J = 8 Hz, 2H, heteroaromatic); LCMS: [M+ + 1] 210.
6-Nitro-2-(2-pyridyl)-1-(2-pyridylmethyl)-1H-benzo[d]imidazole.
3h 64% Yield; solid; Mp: 173–175°C; IR (KBr): 3424, 3060, 1587, 1510, 1466, 1436, 1337, 1150, 992 cm−1; 1H NMR (CDCl3, 300 MHz): δ 6.34 (s, 2H, CH2), 7.06 (d, J = 10 Hz, 1H, aromatic), 7.14–7.22 (m, 1H, aromatic), 7.37–7.43 (m, 1H, heteroaromatic), 7.53–7.58 (m, 1H, heteroaromatic), 7.86–7.93 (m, 2H, heteroaromatic), 8.24 (d,d, J = 12 Hz, 1H, heteroaromatic), 8.44 (s, 1H, aromatic), 8.50 (d, J = 12 Hz, 1H, heteroaromatic), 8.58 (d, J = 6 Hz, 1H, heteroaromatic), 8.65 (d, J = 6 Hz, 1H, heteroaromatic); 13C NMR (CDCl3, 75 MHz): δ 51.49, 108.13, 119.27, 120.09, 121.19, 122.73, 125.14, 125.20, 136.39, 136.92, 137.19, 146.96, 148.80, 148.89, 149.44, 149.60, 154.20, 156.27; LCMS: [M+ + 1] 332.
Biological evaluation
Chemicals
Yeast α-glucosidase and rat intestinal acetone powder as a source of intestinal α-glucosidae, p-nitrophenyl α-d-glucopyranoside (pNPG), soluble potato starch, DMEM (Dulbecco’s modified Eagle’s medium), MTT (3-(4, 5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide), trypsin, and EDTA (ethylenediaminetetraacetic acid) were purchased from Sigma Chemical Co., St. Louis, MO, USA. Fetal bovine serum (FBS) was purchased from Gibco. Other chemicals of analytical grade were procured from indigenous manufacturers. The HT-29 (colon cancer) cell line was obtained from the National Center for Cell Science (NCCS), Pune, India.
In vitro α-glucosidase inhibitory assay
α-Glucosidase inhibitory activities were determined as per earlier reported methodsCitation18,Citation19. Rat intestinal acetone powder in normal saline (100: 1; w/v) was sonicated thoroughly and the supernatant was used as a source of crude intestinal α-glucosidase after centrifugation. In brief, 10 μL of test sample (5 mg/mL DMSO solution) was reconstituted in 100 μL of 100 mM phosphate buffer (pH 6.8) in a 96-well microplate and incubated with 50 μL of crude intestinal α-glucosidase for 5 min before 50 μL substrate (5 mM pNPG prepared in same buffer) was added. Release of p-nitrophenol was measured at 405 nm spectrophotometrically (SpectraMax Plus384; Molecular Devices Corp., Sunnyvale, CA, USA) 5 min after incubation with substrate. Individual blanks for test samples were prepared to correct background absorbance where substrate was replaced with 50 μL of buffer. The control sample contained 10 μL of dimethylsulfoxide (DMSO) in place of test sample. The percentage of enzyme inhibition was calculated as (1 – B/A) × 100 where A represents the absorbance of control without test sample, and B represents the absorbance in the presence of test sample. The IC50 values were determined by applying logarithmic regression analysis to the data for at least five concentration dilutions of the compound.
In vitro assay for cytotoxic activity
HT-29 (colon cancer) cells were plated at a density of 1 × 103 cells per well in 100 μL of DMEM supplemented with 10% FBS medium in a 96-well plate and grown for 24 h. The cells were then exposed to a series of concentrations of test compounds for 24 h and the viability of cells was measured with the MTT method as reported earlierCitation20. Briefly, the above media were replaced with 90 μL of fresh serum-free DMEM and 10 μL of MTT reagent (5 mg/mL) and plates were incubated at 37°C for 4 h; thereafter the above media were replaced with 200 μL of DMSO and incubated for 15 min. The absorbance at 570 nm was measured on a spectrophotometer (SpectraMax; Molecular Devices). The values for each point were calculated from triplicate wells. IC50 values were determined applying logarithmic regression analysis to the data for at least three dilutions.
Animal experiment
The study of antihyperglycemic activity was done according to a method reported earlierCitation21. Male Wistar rats weighing between 195 and 215 g were obtained from the National Institute of Nutrition (CPCSEA Reg. No. 154, Government of India), Hyderabad. The animals were housed in standard polyvinyl cages. The room temperature was maintained at 22 ± 1°C with an alternating 12 h light/dark cycle. Food and water were provided ad libitum. Experiments were performed as per the Institutional Animal Ethics Committee norms. The rats were divided into three groups, viz. control, and 3c and 3e groups, containing six rats in each group. All animals were subjected to overnight fasting. The next day, forenoon blood was collected from the retro-orbital plexus in EDTA-containing tubes, and basal (0 h) plasma glucose levels were measured by the glucose-oxidase test method using a blood autoanalysis instrument (Bayer Express Plus). Test compounds were suspended in normal saline and administered orally in a dose of 50 mg/kg body weight. The control group of animals was given only normal saline. Fifteen minutes after test sample treatment, animals were fed with soluble starch dissolved in normal saline at a dose of 2 g/kg body weight. Thereafter, blood was collected at intervals of 30, 60, 90, and 120 min post-starch feeding. Plasma was separated out for glucose measurement as described above.
Results and discussion
Chemistry
Due to the importance of benzimidazoles, a few methods have been reported in the literature, which include reaction of o-aryldiamines with carboxylic acids or their derivatives under strong acidic conditions at high temperaturesCitation22,Citation23. Cyclodehydration of o-aryldiamines with aldehydes in the presence of oxidative reagents is another method to synthesize benzimidazolesCitation24. Single electron transformation (SET) reactions of 2-nitroaniline with benzaldehyde in the presence of indium/BNPCitation25 as well as the reductive cyclization of 2-nitroaniline in the presence of Na2S2O4 are also reported for the synthesis of benzimidazoles. However, all of these methods have drawbacks in terms of using expensive reagents, oxidation processes, severe conditions, long reaction times, and tedious work-up procedures. It is also known that catalysts based on palladium are very important in organic synthesis, particularly in the total synthesis of natural products for carbon–carbon bond formationCitation26. PdCl2(CH3CN)2 is considered an important catalyst among all palladium catalysts. We carried out the synthesis of benzimidazoles by the condensation of o-phenylenediamine and substituted o-phenylenediamines (1a–h, 1 mmol) with aryl and heteroaryl carbonyl compounds (2a–h, 1 mmol) in the presence of a catalytic amount of PdCl2(CH3CN)2 in anhydrous methanol at room temperatureCitation27. The mode of reaction is the formation of a Schiff base (1H NMR, δ 8.45 singlet, 1H), followed by intramolecular cyclization resulting in benzimidazoles (3a–h; and ) in 62–82% yield. Compounds such as 2-(2-chloro-5-methyl-3-pyridyl)-1H-benzo[d]imidazole (3a) and 2-(2-chloro-5-methyl-3-pyridyl)-6-nitro-1H-benzo[d]imidazole (3b) were prepared by the condensation of o-phenylenediamine (1a) and nitro o-phenylenediamine (1b) with 2-chloro-5-methylpyridine-3-carboxaldehyde (2a) in very good yields. The compound 2-chloro-5-methylpyridine-3-carboxaldehyde (2a) was prepared by the Vilsmeier reaction of an enamide as per our earlier reported methodCitation28. 6-Nitro-2-(2-pyridyl)-1-(2-pyridylmethyl)-1H-benzo[d]imidazole (3h) was prepared by the condensation of nitro o-phenylenediamine (1b) with 2 mol of 2-pyridinecarboxaldehyde (2h). The synthesized compounds were characterized by spectral data (1H NMR, IR, and MS).
Scheme 1. Reaction route for the synthesis of benzimidazoles and their structures.
Table 1. Synthesis of benzimidazoles.
Inhibition of glycosidases has recently become important not only due to their interesting role in delineating enzyme mechanisms and the control of postprandial hyperglycemic excursion in diabetes mellitus, but also in development of newer therapeutics targeted at, for example, cancer, viral infections including human immunodeficiency virus (HIV) and influenza, and lysosomal storage diseases, with a number of drugs in current clinical useCitation29. All the compounds were tested in vitro first, for α-glucosidase inhibitory activity. Compounds were screened using two models of α-glucosidase enzyme sources: one from yeast that represents α-glucosidase type-ICitation30 that has been extensively used as a model for screening potential inhibitors against viral diseases and cancerCitation31, and another from rat intestine that represents α-glucosidase type-IICitation30 that serves as a potential target for screening compounds that may be developed as antihyperglycemic agents against carbohydrate-induced postprandial hyperglycemic excursionCitation32. It is evident from that compounds 3c, 3e, and 3g displayed potent inhibitory activity for yeast α-glucosidase.
Figure 1. Percentage of activity of benzimidazole derivatives on various targets. The primary concentration at which compounds were screened was 500 μg/mL for yeast [A] and intestinal [B] α-glucosidase inhibition assay, and 50 μg/mL for cytotoxic activity [C]. Data represent mean ± SD, n = 3.
![Figure 1. Percentage of activity of benzimidazole derivatives on various targets. The primary concentration at which compounds were screened was 500 μg/mL for yeast [A] and intestinal [B] α-glucosidase inhibition assay, and 50 μg/mL for cytotoxic activity [C]. Data represent mean ± SD, n = 3.](/cms/asset/da52bfdf-08c2-47b1-a351-a160a83d0b12/ienz_a_401885_f0002_b.gif)
Despite the fact that amino acid sequences in the catalytic site of the α-glucosidase family are highly conserved, the aglycon specificity is differentCitation33. It has been advised, therefore, to use enzymes of target tissues or organs for screening of agents for viral diseases, cancer, or diabetesCitation34. Excessive intake of a high-calorie, quickly digestible high- carbohydrate diet results in abnormal surges in the blood glucose level, referred to as PPGHCitation35. Slowing the digestion and absorption of digestible dietary carbohydrates by intestinal α-glucosidase inhibitors in particular, which reduce the rate of enzymatic digestion of starch and delay the release of glucose molecules for absorption, has shown promise in reducing PPHG, hyperinsulinemia, and unwanted consequences on pancreatic function along with the development of hemodynamic disturbancesCitation36–37. When compounds under study were screened for their rat intestinal α-glucosidase inhibitory activity, only 3e could display strong inhibitory potential for enzyme inhibition also (, IC50 = 99.4 μM).
Evaluation of the cytotoxic activity of these compounds was done on colon caner cell line HT-29. It was observed that at a primary screening concentration of 0.2 μM, compounds 3c and 3e displayed 80.4% and 74.8% cytotoxic activity (). The IC50 values for compounds 3c and 3e were calculated to be 82 μM and 98.75 μM, respectively.
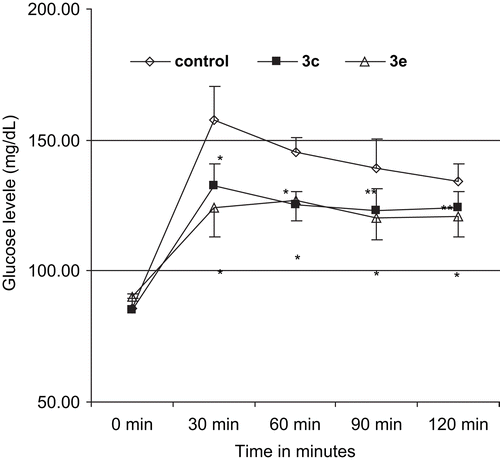
Because compounds 3c and 3e possessed both α-glucosidase inhibitory as well as cytotoxic activity, they were selected for evaluation of antihyperglycemic activity in vivo on starch-induced hyperglycemia in rats. Both compounds significantly reduced starch-induced postprandial hyperglycemic excursion in rats (), 3e being slightly (insignificantly) superior to 3c.
Figure 2. Antihyperglycemic activity of the compounds on starch tolerance test. For statistical analysis ANOVA followed by Dunnett’s multiple comparison test was applied to compare the difference between the means. *p < 0.01, **p < 0.05. Values represent mean ± SD, n = 6.
Conclusion
Synthesis of benzimidazole compounds 3a–h was achieved by the condensation of aromatic and heteroaromatic aldehydes with o-phenylenediamine and substituted o- phenylenediamine in the presence of PdCl2(CH3CN)2 with very good yields. Compounds 3c, 3e, and 3g exhibited potent inhibition of yeast α-glucosidase enzyme. However, only 3e could display potent intestinal α-glucosidase enzyme inhibition. Compounds 3c and 3e displayed cytotoxic potential also against colon cancer cell line HT-29. Furthermore, both compounds (3c and 3e) displayed significant antihyperglycemic activity in starch-induced postprandial hyperglycemia in rats. These compounds, therefore, open new avenues for the development of antihyperglycemic anticancer therapeutics targeted toward the treatment of type 2 diabetes.
Acknowledgements
The authors are thankful to the Head, Organic Chemistry Division-1, and Director-IICT for constant encouragement. One of the authors (J.A.K.) is thankful to UGC (New Delhi) for the award of a Fellowship.
Declaration of interest: The authors report no conflicts of interest.
References
- Kohler P. The biochemical basis of anthelmintic action and resistance. Int J Parasitol 2001;31:336–45.
- Hasegawa M, Nishigaki N, Washio Y, Kano K, Harris PA, Sato H, et al. Discovery of novel benzimidazoles as potent inhibitors of TIE-2 and VEGFR-2 tyrosine kinase receptor. J Med Chem 2007;50:4453–70.
- Hranjec M, Kralji M, Piantanida I, Sedic M, Suman L, Pvelic K, et al. Novel cyno and amidino-substituted derivatives of styryl-2-benzimidazoles and benzimidazo[1,2-a]quinolines. Synthesis, photochemical synthesis, DNA binding, and antitumor evaluation, Part 3. J Med Chem 2007;50:5696–711.
- Safonov IG, Heerding DA, Keenan RM, Price AT, Ericson-Miller CL, Hopson CB, et al. New benzimidazoles as thrombopoietin receptor agonists. Bioorg Med Chem Lett 2006;16:1212–16.
- Falco JL, Pique M, Gonzalez M, Buira I, Mendez E, Terencio J, et al. Synthesis, pharmacology and molecular modeling of N-substituted 2-phenyl-indoles and benzimidazoles as potent GABAA agonists. Eur J Med Chem 2006;41:985–90.
- Siva Kumar BV, Vaidya SD, Vinod Kumra R, Bhirud SB, Mane RB. Synthesis and anti-bacterial activity of some novel 2-(6-fluorochroman-2-yl)-1-alkyl/acyl/aroyl-1H-benzimidazoles. Eur J Med Chem 2006;41:599–604.
- Navarette-Vazquez N, Rojano-Vilchis MM, Yepez-Mulia L, Melendez V, Gerena L, Hernandez-Campos A, et al. Synthesis and antiprotozoal activity of some 2-(trifluoromethyl)-1H-benzimidazole bioisosteres. Eur J Med Chem 2006;41:135–41.
- Kazimierczuk Z, Rzejewska M, Kaustova J, Klimesova V. Synthesis and antimycobacterial activity of 2-substituted halogenobenzimidazoles. Eur J Med Chem 2005;40:203–8.
- Kim JS, Sun Q, Gatto B, Yu C, Liu A, Liu LF, et al. Structure-activity relationship of benzimidazoles and related heterocycles as topoisomerase I inhibitors. Bioorg Med Chem 1996;4:621–30.
- Zarrinmayeh H, Zimmerman D M, Cantrell B E, Schober D A, Bruns R F. Structure-activity relationship of series of diaminoalkyl substituted benzimidazole as neuropeptide YY1 receptor antagonists. Bioorg Med Chem Lett 1999;9:647–52.
- Kohara Y, Kubo K, Imamiya E, Wada T, Inada Y, Naka T. Synthesis and angiotensin II receptor antagonist activities of benzimidazole derivatives bearing acidic heterocycles as novel tetrazole bioisosters. J Med Chem 1996;39:5228–35.
- Wallace MB, Feng J, Zhang Z, Skene RJ, Shi L, Caster CL, et al. Structure based design and synthesis of benzimidazole derivatives as dipeptidyl peptidase IV inhibitors. Bioorg Med Chem Lett 2008;18:2362–7.
- Minoura H, Takeshita S, Ita M, Hirosumi J, Mbuchi M, Kawamura I, et al. Pharmacological characteristics of a novel nonthiazolidinedione insulin sensitizer FK 614. Eur J Pharmacol 2004;494:273–81.
- Minoura H, Takeshita S, Kimura C, Hirosumi J, Takakura S, Kawamura I, et al. Mechanism by which a novel nonthiazolidinedione peroxisome proliferator-activated receptor gamma agonist, FK 614, ameliorates insulin resistance in Zucker fatty rats. Diabetes Obes Metab 2007;9:369–78.
- Carroll MF, Gutierrez A, Castro M, Tsewang D, Schade DS. Targeting postprandial hyperglycemia: a comparative study of insulin tropic agents in type 2 diabetes. J Clin Endocrinol Metab 2003;88:5248–54.
- Boutati EI, Raptis SA. Postprandial hyperglycemia in type 2 diabetes: pathophysiological aspects, teleological notions and flags for clinical practice. Diabetes Metab Res Rev 2004;20:S13–23.
- Berseter JM, Goke B. Type 2 diabetes mellitus as risk factor for colorectal cancer. Arch Physiol Biochem 2008;114:84–98.
- Reddy SV, Tiwari AK, Kumar US, Rao JR, Rao JM. Free radical scavenging, enzyme inhibitory constituents from antidiabetic Ayurvedic medicinal plant Hydnocarpus wigtiana Blume. Phytother Res 2005;19:277–81.
- Gowri PM, Tiwari AK, Ali AZ, Rao JM. Inhibition of α-glucosidase and amylase by bartogenic acid isolated from Barringtonia racemosa Roxb. seeds. Phytother Res 2007;21:796–9.
- Kamal A, Tekumalla V, Raju P, Naidu VGM, Diwan PV, Sistala R. Pyrrolo[2,1-c][1,4]benzodiazepine-β-glucuronide prodrugs with a potential for selective therapy of solid tumors by PMT and ADEPT strategies. Bioorg Med Chem Lett 2008;18:3769–73.
- Tiwari AK, Kumbhare RM, Agawane SB, Ali AZ, Kumar KV. Reduction in postprandial hyperglycemic excursion through α-glucosidase inhibition by β-acetamido carbonyl compounds. Bioorg Med Chem Lett 2008;18:4130–2.
- Phillips MA. The formation of 2-substituted benziminazoles. J Chem Soc 1928:2393–9.
- Preston PN. Benzimidazoles and congeneric trycyclic compounds. In: Weissberger A, Tayler EC eds. The Chemistry of Heterocyclic Compounds. New York: Wiley, 1981;40:6–60.
- Dubey PK, Ratnam CV. Synthesis and spectra of 2-styryl-1H-imidazole[4,5-b]pyridines. Indian J Chem B 1979;18:428–31.
- Kim BH, Han R, Kim JS, Jun YM, Baik W, Lee BM. Indium-mediated reductive intermolecular coupling reaction of 2-nitroaniline with aromatic aldehydes to benzimidazoles. Heterocycles 2004;63:41–54.
- Yin L, Liebscher J. Carbon-carbon coupling reactions catalyzed by heterogeneous palladium catalysts. Chem Rev 2007;107:133–73.
- Ashok Kumar J, China Raju B. Manuscript under consideration in European Journal of Medicinal Chemistry. An efficient strategy for the synthesis of benzimidazoles using Pd (II) catalyst. 2008
- Gangadasu B, Narender P, Kumar SB, Ravinder M, Rao BA, Ramesh C, et al. Facile and selective synthesis of chloronicotinaldehydes by the Vilsmeier reaction. Tetrahedron 2006;62:8398–403.
- Gloster TM, Meloncelli P, Stick RV, Zechel D, Vasella A, Davies GJ. Glycosidase inhibition: an assessment of the binding of 18 putative transition state mimics. J Am Chem Soc 2007;129:2345–54.
- Chiba S. Molecular mechanism in α-glucosidase and glucoamylase. Biosci Biotechnol Biochem 1997;61:1233–9.
- Asano N. Glycosidase inhibitors: update and perspectives on practical use. Glycobiology 2003;13:93R–104R.
- Matsui T, Ogunwande IA, Abesundara KJM, Matsumoto K. Anti-hyperglycemic potential of natural products. Mini-Rev Med Chem 2006;6:109–20.
- Kimura A. Molecular anatomy of α-glucosidase. Trends Glycosci Glycotechnol 2000;12:373–80.
- Hakamata W, Muroi M, Kadokura K, Nishio T, Oku T, Kimur A, et al. Aglycon specificity profiling of α-glucosidases using synthetic probes. Bioorg Med Chem Lett 2005;15:1489–92.
- O’Keefe JH, Bell DSH. Postprandial hyperglycemia/hyperlipidemia (postprandial dysmetabolism) is a cardiovascular risk factor. Am J Cardiol 2007;100:899–904.
- Van de Laar FA, Lucassen PL, Akkermans RP, Van de Lisdonk EH, Rutter GE, Van Weel C. Alpha-glucosidase inhibitors for patients with type 2 diabetes. Diabetes Care 2005;28:154–62.
- Maki KC, Carlson ML, Miller MP, Turowski M, Bell M, Wilder DM, et al. High viscosity hydroxypropylmethyl cellulose blunts post- prandial glucose and insulin responses. Diabetes Care 2007;30:1039–43.