Abstract
We investigated the molecular basis of specificity for the interaction between tumor necrosis factor-α converting enzyme (TACE) and peptidomimetic inhibitors. Four novel peptidomimetic TACE inhibitors (8a–d) were designed and synthesized by introducing a substituted sulfur group and a hydrophobic group to a novel matrix metalloprotease (MMP) inhibitor. Inhibition was determined by in vitro lipopolysaccharide (LPS) cytotoxicity tests in HL-60 cell lines and by measuring the expression of mTNF-α using FCM techniques and immunohistochemistry in vivo. We simulated the interaction of the inhibitors with the 3D structure of the TACE active site in the Brookhaven Protein Database (PDB). The four inhibitors (8a–d) inhibited activity by 9.1%, 54.5%, 27.3%, and 54.5%, respectively. 8b and 8d showed significant in vitro inhibition in cytotoxicity tests, which corresponded to the molecular docking results. 8d also showed inhibitory activity in vivo. We explored the interface between enzyme and substrate by combining bioinformatics with experimental observations to further the development of specific TACE inhibitors to reduce inflammatory responses.
Introduction
Tumor necrosis factor-α (TNF-α) is an important inflammatory cytokine secreted by monocytes/macrophages. Inflammatory diseases such as rheumatoid arthritis, septic shock, cachexia, multiple sclerosis, congestive heart failure, and regional ileitis involve over-expression of TNF-αCitation1–3.
TNF-α is converted from a membrane precursor (mTNF-α or Pro-TNF-α) into soluble TNF-α (sTNF-α) via TACE (tumor necrosis factor-α converting enzyme)Citation4. TACE is a zinc metalloprotease belonging to the ADAM (a disintegrin and a metalloprotease) family, which functions in proteolysis, cell adhesion, cell fusion, and cleavage of extracellular structures in transmembrane proteinsCitation5. TACE inhibitors block the secretion of soluble TNF-α and reduce inflammation, providing a novel target for anti-inflammatory compoundsCitation6–11.
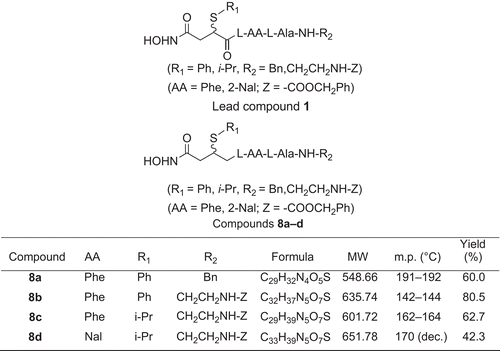
The active center of TACE shows remarkable homology with other zinc metalloproteases (MMPs), including a conserved amino acid sequence of -HexGHxxGxxH-Citation12–14. We chose a peptidomimetic MMP inhibitor compound 1 () as a lead compound for designing novel TACE inhibitors by modifying the center groups to include sulfur atoms and hydrophobic groups such as phenylCitation9–20. Accordingly, four novel compounds (8a–d) () were synthesized and their inhibitory activities against TACE were determined. 8b and 8d inhibited TACE activity in an in vitro model of inflammation, and 8d blocked inflammatory reactions in vivo. These results were consistent with molecular modeling.
Figure 1. Chemical structures of lead compound 1 and modified compounds 8a–d with molecular weight (MW), melting point (m.p.), and yield percentage. Ala, l-alanine; Phe, l-3-phenylalanine; Nal, l-3-(2’-naphthyl)alanine.
Materials and methods
Chemistry
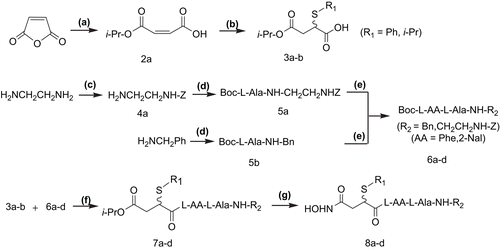
We used peptidomimetic inhibitor 2-(N-benzoxycarbonyl)aminoethyl-N-{4-hydroxyamino-4-oxo-2-(isopropyl)butanoyl}-l-AA-l-alanyl amide (compound 1), which inhibits MMP selectively, as a lead compoundCitation9 for TACE inhibitors (). Based on the scaffold of compound 1, our target compounds 8a–d contained a substitutive succinyl derivate and a small peptide fragment. We used amino acid residues with hydrophobic aryl side groups in the peptide segment and introduced alkyl/arylthio groups to position 2 of the succinyl core. The synthetic procedures () included: (a) synthesis of 2-alkyl/arylthio-succinic acid-4-isopropyl ester according to the method described in reference 20; (b) solid phase peptide synthesis (SPPS) of a small peptide fragment; (c) coupling reaction: 2-alkyl/arylthio-succinic acid-4- isopropyl ester linked with the small peptide fragment; (d) aminolysis reaction: the coupling product was aminolyzed to form four novel products.
Figure 2. Synthesis of 8a–d. Reagents and conditions: (a) i-PrOH; (b) R1SH/Et3N, benzene; (c) Z-Cl (PhCH2OCOCl); (d) Boc-l-AlaOH, HOBt/DCC, H2NCH2Ph; (e) TFA, Boc-l-AA-OH, HOBt/DCC/Et3N; (f) TFA, HOBt/DCC/Et3N; (g) HONH2/MeOH.
The reagents (chemicals) were purchased from Sigma-Aldrich (Beijing, China) and Beijing Chemical Reagent Company (Beijing, China), and were used with further purification. Melting points were measured in capillary tubes on a Yanako MP-500 (Kyoto, Japan) apparatus without correction. Nuclear magnetic resonance (NMR) spectra were obtained on a Varian-200 NMR spectrometer (Palo Alto, CA, USA). Mass spectra were obtained on a VG-ZAB-HS mass analyzer (Micromass, Manchester, UK).
Maleic acid monoisopropyl ester (2a)
Maleic anhydride (49 g, 0.5 mol) was heated to 75°C, and equal molar 2-propanol was added slowly. After the reaction was stirred for 12 h, the solvent was removed under vacuum. The crude monoester was purified by vacuum distillation and recrystallization to give 2a (6.18 g, 21%). m.p. 45–47°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 10.40 (br, 1H, -OH), 6.92 (d, 1H, J = 16, -CH=CH-), 6.84 (d, 1H, J = 16, -CH=CH-), 5.13 (m, 1H, -OCH(CH3)2), 1.33–1.22 (d, 6H, -OCH(CH3)2); 13C NMR (50 MHz, CDCl3): δ 170.0, 164.2, 136.2, 132.4, 69.30, 21.60.
General procedures for preparation of 2-alkyl/arylthio-succinic acid-4-isopropyl ester (3a–b)
Compound 2a (10 mmol) was dissolved in anhydrous benzene (CARE: CARCINOGENIC). Alkyl/arylthiol (20 mmol) and 0.14 mL Et3N were added with constant stirring at room temperature for 3d. After removing the solvent under vacuum, the crude product was separated by column chromatography on silica gel (H 60) to give 3a–b (Rf = 0.67, mobile phase: CHCl3/MeOH = 20:1 (v/v)).
2-Isopropylthio-succinic acid-4-isopropyl ester (3a) 3a was prepared as a yellow liquid (0.52 g), yield 25%. 1H NMR (200 MHz, CDCl3), δ (ppm): 9.38 (br, 1H, -COOH), 3.73 (dd, 1H, J = 6, J = 9.8, -CH(SPr-i)-), 3.72 (m, 1H, -OCH(CH3)2), 3.24 (m, 1H, -SCH(CH3)2), 2.98 (dd, 1H, J = 9.8, J = 17.2, -CH2CH(SPr-i)-), 2.67 (d, 6H, -OCH(CH3)2), 1.31 (d, 6H, -SCH(CH3)2).
2-Phenylthio-succinic acid-4-isopropyl ester (3b) 3b was prepared as a yellow liquid (0.84 g), yield 31%. 1H NMR (200 MHz, CDCl3), δ (ppm): 9.40 (br, 1H, -COOH), 7.52–7.27 (m, 5H, Ar-H), 4.98 (m, 1H, -OCH(CH3)2), 3.94 (dd, 1H, J = 5.6, J = 9.6, -CH(SPh)CO-), 3.00 (dd, 1H, J = 9.6, J = 17.6, -CH2CH(SPh)CO-), 2.77 (dd, 1H, J = 5.6, J = 17.6, -CH2CH(SPh)CO-), 1.22–1.14 (d, 6H, -OCH(CH3)2).
N-Benzoxycarbonyl-1,2-ethylenediamine (4a)
5 mL solution of Z-Cl (benzyl chloroformate) (4 M in CH2Cl2) was slowly added to anhydrous 1,2-ethylenediamine (13.4 g, 0.224 mol) for a period of 5 h, and was cooled in an ice bath. After addition, the reaction mixture was stirred overnight at room temperature. NaOH aq. (1 M, 15 mL) was added and stirred violently for 10 min. The organic layer was separated and washed with water, and condensed to afford a yellow oil. The crude product was separated by column chromatography on silica gel (H 60, saturated with Et3N) to give the product (Rf = 0.25, mobile phase: CHCl3/MeOH = 65:25 (v/v)). After recrystallization, 1.04 g (27%) of 4a as a white solid was obtained. m.p. 110–114°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.36 (s, 5H, Ar-H), 6.65 (br, 1H, -CONH), 5.11 (s, 2H, PhCH2-), 3.24 (t, 2H, J = 5.8, -NHCH2CH2NH2), 2.83 (t, 2H, J = 5.8, -NHCH2CH2NH2), 1.45 (t, 2H, -NH2).
General procedures for preparation of R2-N-Boc-l-alanyl amide (5a–b) are described as those for 2-(N-Benzoxycarbonyl)aminoethyl-N-Boc-l-alanyl amide (5a)
Boc-l-AlaOH (0.57 g, 3 mmol) was dissolved in 10 mL anhydrous CH2Cl2. HOBt (0.40 g, 3 mmol) and compound 4a (2.5 mmol) were added with stirring in an ice bath. After 15 min, a solution of dicyclohexylcarbodiimide (DCC; 0.62 g, 3 mmol) in 5 mL of CH2Cl2 was added slowly, and the reaction mixture was stirred overnight. The dicyclohexylurea (DCU) precipitate was filtered. After removing the solvent under vacuum, 60 mL of ethyl acetate was added and filtered again. The organic layer was washed respectively with 10% NaHCO3 solution, water, 5% citric acid solution, water, and saturated NaCl solution, and dried, condensed, and recrystallized from ethyl acetate to afford 5a (0.78 g, 85%). m.p. 100–102°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.34–7.24 (m, 5H, Ar-H), 6.65 (br, 1H, NH), 5.54 (br, 1H, NH), 5.09 (s, 2H, PhCH2O-), 4.98 (br, 1H, NH), 4.07 (m, 1H, -CH(CH3)-), 3.36 (m, 4H, -NHCH2CH2NH-), 1.43 (s, 9H, t-Bu), 1.31 (d, 3H, -CH(CH3)-).
Benzyl-N-Boc-L-alanyl amide (5b) 0.77g (97%) of 5b was obtained by recrystallization from ethyl acetate. m.p. 102–104°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.36–7.26 (m, 5H, Ar-H), 6.59 (br, 1H, NH), 5.01 (br, 1H, NH), 4.44 (d, 2H, PhCH2-), 4.17 (m, 1H, -CH(CH3)-), 1.41 (s, 9H, t-Bu), 1.38 (d, 3H, -CH(CH3)-).
Peptide segments Boc-l-AA-l-Ala-NH-R2 (6a–d)
Boc-l-Ala-NH-R2 (2 mmol) and CF3COOH (1.4 mL) were dissolved in 2 mL anhydrous CH2Cl2, respectively. The two solutions were mixed together under stirring in an ice bath for a period of 30 min. The reaction was continued for a further 1–2 h at room temperature. After removing CH2Cl2 under vacuum, 3 mL anhydrous CH2Cl2 was added and the pH value was adjusted to 8 by the addition of Et3N. Then, Boc-l-AA-OH (2 mmol) and HOBt (0.28 g, 2 mmol) were added with constant stirring in an ice bath. After 30 min, a solution of DCC (0.50 g, 2.4 mmol) in 8 mL of CH2Cl2 was added slowly to the reaction mixture and stirred overnight. After removing the solvent under vacuum, 60 mL ethyl acetate was added and the solution was filtered. The filtrate was washed respectively with 10% NaHCO3 solution, water, 5% citric acid solution, water, and saturated NaCl solution, and dried, condensed, and recrystallized from ethyl acetate to afford 6a–d.
Benzyl-N-Boc-L-3-phenylalanyl-L-alanyl amide (6a) Yield 85%: m.p. 177–179°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.36–7.26 (m, 10H, Ar-H), 6.57 (br, 1H, NH), 6.34 (d, 1H, J = 7, NH), 4.89 (br, 1H, NH), 4.39 (d, 2H, PhCH2NH-), 4.50–4.29 (m, 1H, -CH(CH3)-), 4.10 (m, 1H, -CH(CH2Ph)-), 3.03 (d, 2H, -CH(CH2Ph)-), 1.36 (s, 9H, t-Bu), 1.30 (d, 3H, J = 6 Hz, -CH(CH3)-).
Benzyl-N-Boc-L-3-(2’-naphthyl)alanyl-L-alanyl amide (6b) Yield 47%: m.p. 185–187°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.77 (d, 3H, Ar-H), 7.62 (s, 1H, Ar-H), 7.46 (m, 2H, Ar-H), 7.29–7.23 (m, 5H, Ar-H), 7.21 (s, 1H, Ar-H), 6.57 (br, 1H, NH), 6.42 (d, 1H, NH), 4.90 (br, 1H, NH), 4.28 (d, 2H, PhCH2NH-), 4.48 (m, 1H, -CH(CH2Nal)-), 4.39 (m, 1H, -CH(CH3)-), 3.21 (d, 2H, -CH(CH2Nal)-), 1.34–1.31 (m, 12H, t-Bu and -CH(CH3)-).
2-(N-Benzoxycarbonyl)aminoethyl-N-Boc-L-3-phenylalanyl-L-alanyl amide (6c) Yield 60.6%: m.p. 155–156°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.33–7.15 (m, 10H, Ar-H), 6.76 (br, 1H, NH), 6.56 (br, 1H, NH), 5.58 (br, 1H, NH), 5.08 (s, 2H, PhCH2O-), 5.01 (br, 1H, NH), 4.38 (m, 2H, -CH(CH2Ph)-, -CH(CH3)-), 3.30 (m, 4H, -NHCH2CH2NH-), 3.03 (d, 2H, -CH(CH2Ph)-), 1.38 (s, 9H, t-Bu), 1.28 (d, 3H, -CH(CH3)-).
2-(N-Benzoxycarbonyl)aminoethyl-N-Boc-L-3-(2’-naphthyl)alanyl-L-alanyl amide (6d) Yield 51%: m.p. 174–176°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.78 (m, 3H, Ar-H), 7.62 (s, 1H, Ar-H), 7.46–7.44 (m, 2H, Ar-H), 7.31–7.23 (m, 5H, Ar-H), 7.21 (s, 1H, Ar-H), 6.90 (d, 1H, NH), 6.82 (m, 1H, NH), 6.68 (m, 1H, NH), 5.69 (m, 1H, NH),5.07 (s, 2H, PhCH2O-), 4.50 (m, 1H, -CH(CH2-2-Nal)-), 4.39 (m, 1H, -CH(CH3)-), 3.40 (m, 4H, -NHCH2CH2NH-), 3.21 (d, 2H, -CH(CH2Nal)-), 1.35 (s, 9H, t-Bu), 1.26 (d, 3H, -CH(CH3)-).
General procedures for preparation of R2-N-{4-isopropoxy-4-oxo-2-(alkyl/arylthio)butanoyl}-l-AA-l-alanyl amide (7a–d)
Compounds 6a–d (1 mmol) and trifluoroacetic acid (TFA; 0.7 mL) were dissolved in CH2Cl2 (1 mL), respectively. The two solutions were mixed together under stirring in an ice bath for a period of 30 min. The reaction was continued for a further 1–2 h at room temperature. After removing CH2Cl2 under vacuum, 2 mL anhydrous CH2Cl2 was added and the pH value was adjusted to 8 by the addition of Et3N. Then, compounds 3a–b (1.5 mmol) and HOBt (1.1 mmol) were added with constant stirring in an ice bath. After 30 min, a solution of DCC (1.5 mmol) in 4 mL CH2Cl2 was added slowly to the reaction mixture and stirred overnight. After removing the precipitated DCU by filtration and then the solvent under vacuum, 20 mL ethyl acetate was added and the solution was filtered. The filtrate was washed respectively with 1 M HCl, water, 10% Na2CO3 solution, water, and saturated NaCl solution, and dried, condensed, and recrystallized from ethanol–petroleum ether to afford 7a–d.
Benzyl-N-{4-isopropoxy-4-oxo-2-(phenylthio)butanoyl}-L-3-phenylalanyl-L-alanyl amide (7a) Yield 51%: m.p. 180–182°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.40–7.17 (m, 15H, ArH), 6.60 (br, 1H, NH), 6.57 (br, 1H, NH), 6.30 (br, 1H, NH), 4.90 (m, 1H, J = 6, -OCH(CH3)2), 4.60–4.51 (m, 1H, -CH(CH3)-), 4.39 (d, 2H, J = 6, PhCH2NH-), 3.95 (dd, 1H, J = 5.6, J = 9.6, -CH2CH(SPh)CO-), 3.48 (m, 1H, J = 7.2, -CH(CH2Ph)-), 3.05 (d, 2H, J = 6, -CH(CH2Ph)-), 2.72–2.58 (dd, 2H, J = 9.6, J = 17.6, -CH2CH(SPh)CO-), 1.39–1.22 (d, 3H, -CH(CH3)-), 1.19–1.07 (d, 6H, -OCH(CH3)2); (FAB-MS: m/z 576 (M + H)+; Anal. calcd for C32H37N3O5S: C, 66.76; H, 6.48; N, 7.30. Found: C, 66.50; H, 6.40; N, 7.39%).
2-(N-Benzoxycarbonyl)aminoethyl-N-{4-isopropoxy-4-oxo-2-(phenylthio)butanoyl}-L-3-phenylalanyl-L-alanyl amide (7b) Yield 69%: m.p. 170–172°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.37–7.18 (m, 15H, ArH), 6.78 (br, 1H, NH), 6.55 (br, 1H, NH), 6.17 (br, 1H, NH), 5.81 (br, 1H, NH), 5.06 (s, 2H, -OCH2Ph), 5.01 (m, 1H, J = 6, -OCH(CH3)2), 4.57 (m, 1H, J = 6, -CH(CH2Ph)-), 4.43 (m, 1H, -CH(CH3)-), 4.02 (dd, 1H, J = 7.4, J = 9.6, -CH(SPh)CO-), 3.31 (m, 4H, -NHCH2CH2NH-), 3.08 (d, 2H, J = 6, -CH(CH2Ph)-), 2.79 (dd, 1H, J = 9.6, J = 16.8, -CH2CH(SPh)CO-), 2.56 (dd, 1H, J = 7.4, J = 16.8, -CH2CH(SPh)CO-), 1.26 (d, 3H, -CH(CH3)-), 1.16–1.07 (d, 6H, -OCH(CH3)2); (FAB-MS: m/z 663 (M + H)+; Anal. calcd for C35H42N4O7S: C, 63.43; H, 6.39; N, 8.45. Found: C, 63.03; H, 6.43; N, 8.45%).
2-(N-Benzoxycarbonyl)aminoethyl-N-{4-isopropoxy-4-oxo-2-(isopropylthio)butanoyl}-L-3-phenylalanyl-L-alanyl amide (7c) Yield 72%: m.p. 158–160°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.34–7.15 (m, 10H, ArH), 6.76 (br, 1H, NH), 6.60 (br, 1H, NH), 6.28 (br, 1H, NH), 5.58 (br, 1H, NH), 5.09 (s, 2H, -OCH2Ph), 4.90 (m, 1H, -OCH(CH3)2), 4.50 (t, 1H, -CH(CH2Ph)-), 4.38 (m, 1H, -CH(CH3)-), 4.20 (t, 1H, -CH2CH(SPr-i)CO-), 3.48 (m, 1H, -S-CH(CH3)2), 3.30 (m, 4H, -NHCH2CH2NH-), 3.08 (d, 2H, -CH(CH2Ph)-), 2.98 (m, 1H, -CH2CH(SPr-i)CO-), 2.58 (m, 1H, -CH2CH(SPr-i)CO-), 1.25 (d, 6H, -OCH(CH3)2), 1.23 (d, 6H, -SCH(CH3)2); 1.18 (d, 3H, -CH(CH3)-); (FAB-MS: m/z 629 (M + H)+; Anal. calcd for C32H44N4O7S: C, 61.13; H, 7.05; N, 8.91. Found: C, 61.35; H, 7.06; N, 8.96%).
2-(N-Benzoxycarbonyl)aminoethyl-N-{4-isopropoxy-4-oxo-2-(isopropylthio)butanoyl}-L-3-(2’-naphthyl)alanyl-L-alanyl amide (7d) Yield 33%: m.p. 155–157°C; 1H NMR (200 MHz, CDCl3), δ (ppm): 7.73–7.16 (m, 12H, ArH), 7.63 (s, 1H, ArH), 7.46–7.42 (m, 2H, ArH), 7.31 (s, 5H, ArH), 7.16 (s, 1H, ArH), 6.95 (br, 1H, NH), 6.84 (br, 1H, NH), 5.99 (br, 1H, NH), 5.88 (br, 1H, NH), 5.07 (s, 2H, -OCH2Ph), 4.89 (m, 1H, -OCH(CH3)2), 4.74 (m, 1H, -CH(CH2-2-Nal)-), 4.47 (m, 1H, -CH(CH3)-), 3.16 (m, 6H, -NHCH2CH2NH-, -CH(CH2-2-Nal)-), 1.40–1.18 (m, 15H, -OCH(CH3)2), -SCH(CH3)2, -CH(CH3)-); (FAB-MS: m/z 679 (M + H)+; Anal. calcd for C36H46N4O7S: C, 63.64; H, 6.78; N, 8.25. Found: C, 63.60; H, 6.80; N, 8.27%).
General procedures for preparation of R2-N-{4-hydroxyamino-4-oxo-(2-alkyl/arylthio)butanoyl}-l-AA-l-alanyl amide (8a–d)
Hydroxylamine hydrochloride (0.14 g, 2 mmol) and KOH (0.18 g, 3.2 mmol) were dissolved in 2 mL anhydrous methanol, respectively. The two solutions were mixed together under stirring and cooled in an ice bath for 1 h. After the mixture was filtered, the filtrate was added slowly to a solution of 7a–b (0.56 mmol, in 2 mL methanol). The reaction was monitored by thin layer chromatography (TLC; CHCl3/MeOH = 20:1 (v/v)). About 15 min later, the reaction was stopped and the solvent was removed under vacuum. 2 mL water was added to dissolve the condensate and the pH value was adjusted to 5 with 1 mol/L HCl and neutralized with saturated NaHCO3 aq. The crude product shown as a white precipitate was washed and recrystallized from ethanol–petroleum ether to obtain 8a–d as a white solid.
Benzyl-N-{4-hydroxyamino-4-oxo-2-(phenylthio)butanoyl}-L-3-phenylalanyl-L-alanyl amide (8a) Yield 60%: m.p. 191–192°C; 1H NMR (200 MHz, DMSO-d6), δ (ppm): 10.57 (s, 1H, -OH), 8.84–8.14 (m, 4H, NH), 7.29–7.19 (m, 15H, ArH), 4.60–4.40 (m, 2H, -CH(CH2Ph)-, -CH(CH3)-), 4.25 (d, 2H, J = 6.4, PhCH2NH-), 4.12 (dd, 1H, J = 6, J = 9.4, -CH2CH(SPh)CO-), 3.05 (d, 2H, J = 14, -CH(CH2Ph)-), 2.73 (dd, 1H, J = 9.4, J = 14.2, -CH2CH(SPh)CO-), 2.24 (dd, 1H, J = 6, J = 14.2, -CH2CH(SPh)CO-), 1.27 (d, 3H, -CH(CH3)-); (FAB-MS: m/z 549 (M + H)+).
2-(N-Benzoxycarbonyl)aminoethyl-N-{4-hydroxyamino-4-oxo-2-(phenylthio)butanoyl}-L-3-phenylalanyl-L-alanyl amide (8b) Yield 81%: m.p. 142–144°C; 1H NMR (200 MHz, DMSO-d6), δ (ppm): 10.59 (s, 1H, -OH), 9.05–7.97 (m, 5H, NH), 7.29–7.19 (s, 15H, ArH), 4.97 (s, 2H, -COOCH2Ph), 4.40 (m, 1H, -CH(CH2Ph)-), 4.16 (m, 1H, -CH(CH3)-), 4.12 (dd, 1H, J = 6, J = 9.4, -CH2CH(SPh)CO-), 3.04 (m, 6H, -NHCH2CH2NH-, -CH2Ph), 2.45 (m, 1H, -CH2CH(SPh)CO-), 2.30 (m, 1H, -CH2CH(SPh)CO-), 1.18 (d, 3H, J = 6.8, -CH(CH3)-); (FAB-MS: m/z 636 (M + H)+, 658 (M + Na)+, 674 (M + K)+).
2-(N-Benzoxycarbonyl)aminoethyl-N-{4-hydroxyamino-4-oxo-2-(isopropylthio)butanoyl}-L-3-phenylalanyl-L-alanyl amide (8c) Yield 63%: m.p. 162–164°C; 1H NMR (200 MHz, DMSO-d6), δ (ppm): 10.65 (br, 1H, -OH), 8.95–8.17 (m, 5H, NH), 7.58–7.47 (s, 10H, ArH), 5.25 (s, 2H, -COOCH2Ph), 4.50–4.40 (m, 2H, -CH(CH2Ph)-, -CH(CH3)-), 4.18 (t, 1H, -CH2CH(SPr-i)CO-), 3.38 (m, 1H, -SCH(CH3)2), 3.06 (m, 6H, -NHCH2CH2NH-, -CH2Ph), 2.78 (m, 1H, -CH2CH(SPr-i)CO-), 2.58 (m, 1H, -CH2CH(SPr-i)CO-), 1.25 (d, 6H, -SCH(CH3)2), 1.10 (d, 3H, -CH(CH3)-); (FAB-MS: m/z 602 (M + H)+).
2-(N-Benzoxycarbonyl)aminoethyl-N-{4-hydroxyamino-4-oxo-2-(isopropylthio)butanoyl}-L-3-(2’-naphthyl)alanyl-L-alanyl amide (8d) Yield 42%: m.p. 170°C (dec.); 1H NMR (200 MHz, DMSO-d6), δ (ppm): 10.61 (br, 1H, -OH), 8.83–8.02 (m, 5H, NH), 7.77–7.30 (m, 12H, ArH), 4.97 (s, 2H, -COOCH2Ph), 4.56 (m, 1H, -CH(CH2-2-Nal)-), 4.17 (m, 1H, -CH(CH3)-), 4.10 (t, 1H, J = 9.6, -CH2CH(SPr-i)CO-), 3.28 (m, 1H, -SCH(CH3)2), 3.10 (d, 2H, -CH(CH2-2-Nal)-), 3.01 (m, 4H, -NHCH2CH2NH-), 2.78 (m, 1H, -CH2CH(SPr-i)CO-), 2.40 (m, 1H, -CH2CH(SPr-i)CO-), 1.17–1.12 (d, 6H, -SCH(CH3)2), 1.03–0.88 (d, 3H, -CH(CH3)-); (FAB-MS: m/z 679 (M + H)+).
Biology assay
Cytotoxicity assay by MTT
Human promyelocytic leukemia cells (HL-60) and high TNF-susceptible mouse L929 cells, purchased from ATCC (stored by WTCC) (Wuhan, China), were used for the cytotoxicity assay. Cells were cultivated in RPMI-1640 (Gibco, USA) supplemented with 100 mL/L fetal calf serum (FCS; Gibco, USA), penicillin (100 μg/mL), and streptomycin (100 μg/mL) in a humidified atmosphere of 5% CO2 at 37°C. LPS was obtained from Sigma-Aldrich (Beijing, China). Cytotoxicity was evaluated by modified MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide) assayCitation21,Citation22. Briefly, HL-60 cells were seeded at 1 × 10Citation6 cells/well in six-well plates and L929 cells in 96-well plates (Falcon, USA), and incubated for 12 h in 100 mL culture media with 10% FCS. Media were then replaced with serum-free medium. After a 6 h treatment of HL-60 with lipopolysaccharide (LPS; 200 ng/mL), cells were incubated with 8a (2 mg/mL), 8b (1 mg/mL), 8c (4 mg/mL), and 8d (2 mg/mL) dissolved in 1% absolute alcohol/phosphate buffered saline (PBS) at 37°C for 2 h. The supernatant was then added to L929 cell culture medium (200 μL/well), followed by incubation at 37°C for 12 h. Then the L929 cells were treated with MTT (Sigma, USA) 10 μL (5 g/L) for 4 h. After removal of the supernatant, the purple-blue sediment was dissolved in 100 μL/well dimethylsulfoxide (DMSO), and the optical densities were read on a multi-well spectrophotometer (Labsystems Dragon, Finland) at 570 nm (OD570). The inhibitory activity of 8a–d against TACE was then determined based on the growth inhibitory rate (GIR) of sTNF-α in L929 cells. The GIR value was calculated by Equation (1) as follows:
where LPS(–) and LPS(+) indicate the supernatants of HL-60 culture media in the absence or presence of LPS, respectively.
mTNF-α content in the macrophage cell surface by FCM
We tested the effect of inhibitors on the amount of mTNF-α on the macrophage surface using the following procedure. KM mice (weight about 20 g) were sacrificed by cervical dislocation and the abdominal macrophages were isolated and cultivated. Transmembrane TNF-α on the macrophage surface was labeled with primary goat anti-mouse TNF-α polyclonal antibody followed by a secondary antibody of FITC (fluorescein isothiocyanate)-labeled rabbit anti-goat immunoglobulin G (IgG) antibody. Finally, mTNF-α content was determined with flow cytometry (FCM). The specific procedures are described in reference 23.
m-TNFα content after LPS-endotoxemia using FCM and histology
A mouse model of LPS endotoxemia was prepared according to the method of our project team. Briefly, 0.1 mL of a solution containing 10 mg d-GalN (d-galactosamine) and 0.4–2 μg LPS was intra-caudally injected into clean KM mice weighing about 20 g. Systemic reactions such as shivering and piloerection were observed 20 min after injection, and 40 min later, some mice showed cyanosis and drowsiness. Mice were sacrificed by cervical dislocation 6 h later.
Grouping methods were: intraperitoneal injection (IP injection) of 0.1 mL PBS (group A, blank control), PBS + Tween-80 (group B, solvent control), Tween-80 containing 1.5 mg of compound 8d (group C), and Tween-80 containing 2.5 mg of compound 8d (group D). 0.5 h after injection of the four solutions, 0.1 mL of the solution containing 10 mg d-GalN and 0.4–2 μg LPS was intra-caudally injected. A positive control injected with only 0.1 mL of 10 mg d-GalN and 0.4–2 μg LPS was considered as group E. Each group contained five mice. Mice were sacrificed 6 h later by cervical dislocation and macrophage cells were isolated and cultivated from the abdominal cavity. mTNF-α content was determined by FCM.
Livers, kidneys, and lungs were collected, fixed with 10% formalin, dehydrated, cleared, embedded, stained with hematoxylin and eosin (HE), and mounted with neutral gum for histological examination.
Molecular modeling
The crystal structures of the TACE catalytic domain were acquired from the Brookhaven Protein Database (PDB) (http://www.pdb.org). To find the binding mode of compounds 8a–d with TACE, we used the advanced Insight II 2003.3L molecular modeling software, Catalyst 4.3 pharmacophore modeling software, and MLD chemical information system to automatically dock the inhibitors to the TACE binding site on an SCI Octane 2 workstation.
We used flexible docking methods for TACE and the inhibitors. First, we obtained three-dimensional (3D) conformations of 8a–d by energy minimization; second, the crystal structures (INN, 1BKC were the PDB codes for inhibitor and TACE complex, respectively) of the 1BKC complex of the catalytic domain of TACE and INN of the inhibitor were obtained by a database search in PDB. The detailed docking procedures were: open 1BKC file in PDB and display the 3D structure of 1BKC; determine the initial position of inhibitor in the TACE binding site by molecular alignment of 8a–d with INN; delete INN and molecularly dock the catalytic domain of TACE with 8a–d.
Molecular docking should be conducted in a cvff (consistent valence force field)Citation19,Citation24. However, zinc could not be charged because zinc force field parameters do not exist in a cvff. Therefore, we changed zinc in the active center to copper to complete the charge distribution for each atom in the 1BKC complex. This substitution of copper for zinc should only introduce a minor error since copper is close to zinc in the periodic table and has a similar structure.
Amino acid residues within 1 nm around the inhibitor were designated as active regions, and the conformations of these regions could freely change during molecular docking: nitrogen (of 405, 409, and 415 histidine of TACE) linked with zinc; oxygen (of the inhibitor) linked with zinc; and zinc itself. Conformations outside of these regions remained unchanged. Calculation following docking was done in two steps: determine 10 lower energy conformations with a Monte Carlo algorithm, and obtain a minimum energy conformation by simulated annealing dynamics using a cell multipole method for nonbonding interactions.
Results and discussion
Biological activity
We first determined the effects of peptidomimetic inhibitors on LPS-medicated sTNF-α production in a cytotoxicity test (). The sTNF-α production of HL-60 cells induced by the same amount of LPS caused different GIR in L929 cells. 8b and 8d inhibited TACE better than 8a and 8c.
Table 1. Effects of inhibitors on LPS-induced sTNF-α secretion by HL-60.
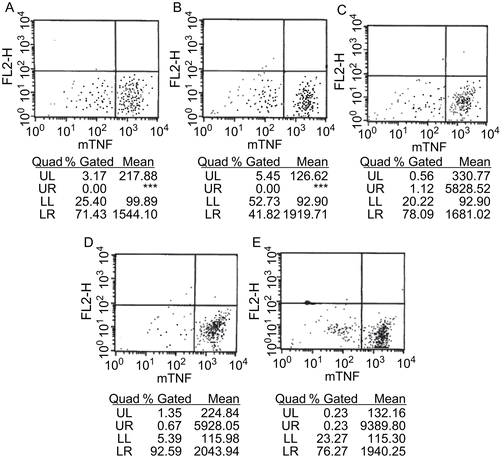
sTNF-α secretion from macrophages was then assayed by FCM (). mTNF-α fluorescence on peritoneal macrophage surfaces was low because of high sTNF-α secretion after LPS treatment. Inhibitor injection increased the fluorescence intensity of mTNF-α on peritoneal macrophages, suggesting that LPS-mediated conversion of mTNF-α into sTNF-α was inhibited.
Figure 3. Content assay of mTNF-α on peritoneal macrophage surfaces of LPS-treated mice by FCM. (A) 0.1 mL PBS (blank control); (B) PBS + Tween-80 (solvent control); (C) Tween-80 containing 1.5 mg of compound 8d; (D) Tween-80 containing 2.5 mg of compound 8d; (A–D) i.p. injection of the above solutions, respectively, followed by intracaudate injection of 0.1 mL solution containing 10 mg d-GalN and 0.4–2 μg LPS; (E) intracaudate injection of 0.1 mL solution containing 10 mg d-GalN and 0.4–2 μg LPS only (positive control).
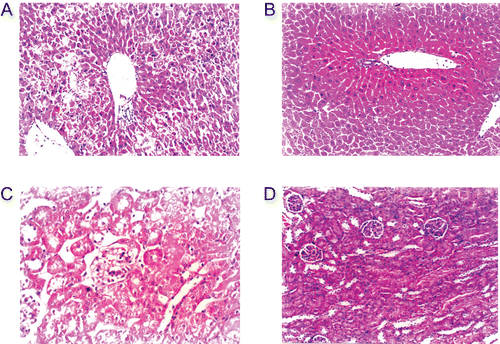
LPS treatment produced swelling, degeneration, and necrosis of mouse liver and kidney in HE staining, and 8d could reverse this inflammatory reaction, and alleviate degeneration and necrosis of the liver and kidney (). LPS did not affect the lungs.
Figure 4. Hematoxylin and eosin (HE) staining of liver and kidney before and after injection of LPS, inhibitor 8d (×200). (A) LPS in liver; (B) LPS and 8d (2.5 mg) in liver; (C) LPS in kidney; (D) LPS and 8d (2.5 mg) in kidney.
Molecular modeling
The conformations between 8c and TACE in the molecular docking are shown as protein surfaces expressed as a pseudo-Connolly solvent accessible surface, and the radius of the probe atom (solvent atom) was 0.05 nm (). Stronger nonbonding interactions (larger negative value) between the inhibitors and TACE indicated that the inhibitor had a larger binding constant with TACE and higher inhibitory activity ().
Figure 5. Resultant conformation after molecular docking between inhibitor 8c and TACE on SCI Octane 2 graph workstation using advanced Insight II 2003.3L molecular modeling software.
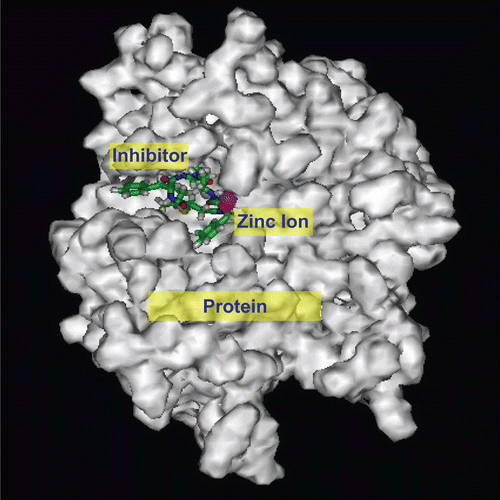
Table 2. Comparison of nonbonding interaction energies between four inhibitors (8a–d) and TACE with results of cell experiments.
The electrostatic interaction energy between 8a and TACE was significantly lower than for the other three inhibitors, which might be attributed to the few charged groups of 8aCitation24. Molecular docking and cell experiments showed similar results, indicating that molecular docking could simulate the interaction between molecules.
Conclusions
We tried to model the molecular interactions of a small molecular peptidomimetic inhibitor and TACE using 3D modeling and biological experiments for developing specific TACE inhibitors. By introducing a sulfur atom at position 2 of the succinyl core of compound 1, and using l-3-phenylalanine and l-3-(2’-naphthyl)alanine with hydrophobic aryl side groups in the peptide segment, we successfully produced four novel TACE inhibitors, and cell experiments confirmed that two of the four inhibitors inhibited TACE. The inhibitors could also alleviate inflammatory reactions and inhibit the conversion of membrane TNF-α on the macrophage surface into soluble TNF-α. We established a method for flexible molecular docking between TACE and inhibitors that showed good agreement with biological experiments. Appropriate molecular modifications of these potent compounds might improve their ability to inhibit TACE.
Acknowledgments
We thank Professor Xu Jiaxi for his assistance with the synthesis work. This study was supported by the National Natural Sciences Foundation of China (30371309, 30500085; Y.-Y.Z.).
Declaration of interest
We declare all support given has been mentioned above; there is no conflict of other interest.
References
- Newton R, Decicco C. Therapeutical potential and strategies for inhibiting tumor necrosis factor α. J Med Chem 1999;42:2295–314.
- Sekut L, Connolly KM. Pathophysiology and regulation of TNF-α in inflammation. Drug News Perspect 1996;9:261–7.
- Rink L, Kirchner H. Recent progress in the tumour necrosis factor-α field. Int Arch Allergy Immunol 1996;111:199–209.
- Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, et al. A metalloproteinase disintegrin that releases tumor necrosis factor-α from cells. Nature 1997;385:729–33.
- Black RA, Fitzner JN, Sleath PR. Inhibitors of TNF-α secretion. United States Patent 5,594,106.
- Blobel CP. Functional and biochemical characterization of ADAMs and their predicted role in protein ectodomain shedding. Inflamm Res 2002;51:83–4.
- Xue C-B, He X, Roderick J, Corbett RL, Duan James JW, Liu R-O, et al. Rational design, synthesis and structure–activity relationships of a cyclic succinate series of TNF-α converting enzyme inhibitors. Part 2: Lead optimization. Bioorg Med Chem Lett 2003;13:4299–304.
- Kamei N, Tanaka T, Kawai K, Miyawaki K, Okuyama A, Murakami Y, et al. Reverse hydroxamate-based selective TACE inhibitors. Bioorg Med Chem Lett 2004;14:2897–900.
- Zask A, Gu Y, Albright JD, Du X, Hogan M, Levin JI, et al. Synthesis and SAR of bicyclic heteroaryl hydroxamic acid MMP and TACE inhibitors. Bioorg Med Chem Lett 2003;13:1487–90.
- Zhang Y, Hegen M, Xu J, Keith JC Jr, Jin G, Du X, et al. Characterization of (2R, 3S)-2-({[4-(2-butynyloxy) phenyl] sulfonyl}amino)-N,3-dihydroxybutanamide, a potent and selective inhibitor of TNF-α converting enzyme. Int Immunopharmacol 2004;4:1845–57.
- Xue C-B, Chen X-T, He X, Roderick J, Corbett RL, Ghavimi B, et al. Synthesis and structure–activity relationship of a novel sulfone series of TNF-α converting enzyme inhibitors. Bioorg Med Chem Lett 2004;14:4453–9.
- Venkatesan AM, Davis JM, Grosu GT, Baker J, Zask A, Levin JI, et al. Synthesis and structure-activity relationships of 4-alkynyloxy phenyl sulfanyl, sulfinyl, and sulfonyl alkyl hydroxamates as tumor necrosis factor-α converting enzyme and matrix metalloproteinase inhibitors. J Med Chem 2004;47:6255–69.
- Maskos K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie 2005;87:249–63.
- Lukacova V, Yufen Z, Kroll DM, Raha S, Comez D, Balaz S. A comparison of the binding sites of matrix metalloproteinases and tumor necrosis factor-α converting enzyme: implications for selectivity. J Med Chem 2005;48:2361–70.
- Maskos K, Fernandez-Catalan C, Huber R, Bourenkov GP, Bartunik H, Ellestad GA, et al. Crystal structure of the catalytic domain of human tumor necrosis factor-α-converting enzyme. Proc Natl Acad Sci USA 1998;95:3408–12.
- Lovejoy B, Welch AR, Carr S, Luong C, Broka C, Hendricks RT, et al. Crystal structures of MMP-1 and -13 reveal the structural basis for selectivity of collagenase inhibitors. Nat Struct Biol 1999;6:217–21.
- Feng Y, Likos J, Zhu L, Woodward H, McDonald J, Stevens A, et al. 1H, 13C and 15N resonance assignments for a truncated and inhibited catalytic domain of matrix metalloproteinase-2. J Biomol NMR 2000;17:85–6.
- Pavlovsky AG, Williams MG, Ye QZ, Ortwine DF, Purchase CF 2nd, White AD, et al. X-ray structure of human stromelysin catalytic domain complexed with nonpeptide inhibitors: implications for inhibitor selectivity. Protein Sci 1999;8:1455–62.
- Rowsell S, Hawtin P, Minshull CA, Jepson H, Brockbank SM, Barratt DG, et al. Crystal structure of human MMP9 in complex with a reverse hydroxamate inhibitor. J Mol Biol 2002;319:173–81.
- Levy DE, Lapierre F, Liang W, Ye W, Lange CW, Li X, et al. Matrix metalloproteinase inhibitors: a structure-activity study. J Med Chem 1998;41:199–206.
- Yin B, Li Z, Yu S, Jiang X, Gong F, Xu Y, et al. Relationship between the increase of secretion of sTNF-α induced by lipopolysaccharides and the enhanced expression of TACE mRNA in HL-60 cells and adhesive cells from human spleen. J Tongji Med Univ 2001;21:265–7.
- Wang Z, Wang Y, Zhu K-L, Guo L-J, Yang Y-Z. Mechanism of three inhibitors of TACE in blocking the converting of pro-TNFα into sTNFα. J Huazhong Univ Sci Technol (Med Sci) 2003;23:458–61.
- Shen G, Zhou R. Modern Experimental Techniques in Immunology.Wuhan, China: Hubei Science and Technology Publishing Company, 1998:226–4.
- Zhao Y, Feng W-F, Yang Y-Z, Ling L-J, Chen R-S. Comparison of property of tumor necrosis factor-α converting enzyme (TACE) and some matrix metalloproteases (MMPs) in catalytic domain. J Huazhong Univ Sci Technol (Med Sci) 2006;26:637–9.