Abstract
Most non-steroidal anti-inflammatory drugs (NSAIDs) suffer from the deadlier gastrointestinal (GI) toxicities. The free -COOH group is responsible for the GI toxicity associated with all traditional NSAIDs. In the present research work, the main objective was to develop new chemical entities as potential anti-inflammatory agents with no GI toxicities. The results of synthesis and pharmacological screening of a series of hybrid molecules having general formula 2-(5-(5-(substituted phenyl)-2-oxo-ethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl)-2-oxoethyl nitrate are described. These compounds were tested in vivo for their anti-inflammatory, analgesic, and ulcerogenic properties, and subjected to histopathological studies. Compound 7c, 2-(5-(5-(3-hydroxyphenyl)-2-oxo-ethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl)-2-oxoethyl nitrate, was the most potent in this series. The compounds that showed significantly reduced GI ulcerogenicity also showed promising results in histopathological studies, and they were found to cause no mucosal injury. All the synthesized compounds were found to exhibit significant nitric oxide releasing activity in an in vitro method. In conclusion, the designed hybrid molecules were found to be significantly promising.
Introduction
Medical research is constantly seeking to improve the efficiency of drugs. One approach involves the use of so-called hybrid drugs, which comprises the incorporation of two drug pharmacophores in one single molecule with the intention to exert dual drug actionCitation1. For example, one of the hybrid parts may be incorporated to counterbalance the known side effects associated with the other hybrid part, or to amplify its effect through action on another biotarget.
Non-steroidal anti-inflammatory drugs (NSAIDs) are one of the most frequently used medications worldwide to treat pain, fever, and inflammation. NSAIDs exert their therapeutic activity by inhibiting cyclooxygenase derived prostaglandin synthesis, but this mechanism of action is inherently responsible for the gastrointestinal (GI)Citation2–6, renalCitation7–9, and hepaticCitation10 side effects observed in patients undergoing long term treatment. The most common side effects associated with NSAID therapy are upper GI irritation, ulceration, dyspepsia, bleeding, and in some cases deathCitation11. It is clear that NSAID induced toxicity is a serious public health problem, contributing significantly to morbidity and mortality.
Three different strategies have emerged to improve the safety profile of NSAIDs: (1) the development of selective cyclooxygenase-2 (COX-2) inhibitors; (2) the co-administration of a proton pump inhibitor with NSAIDs; and (3) the linkage of a nitrate based nitric oxide (NO) releasing moiety to classical NSAIDs (NO-NSAIDs).
Each strategy presents a different set of advantages and limitations. For example, despite the relatively safe profile of COX-2 inhibitors in the GI tract, their role with respect to the adverse cardiovascular effects reported in some patients undergoing chronic treatment has attracted considerable attentionCitation12. Different NO-NSAIDs have been developed, which are reported in the literature. Among the NO-NSAIDs that have come into clinical trials are nitroaspirin, nitronaproxene, nitroketoprofen, etc. Among the nitric oxide donors adopted to prove the validity of this principle are furoxanes, oximes, hydrazides, and organic nitratesCitation13–15. However, the long-term safety profile of such emerging classes of compounds is still to be determined.
Recent strategies adopted to minimize the side effects of NSAIDs include the use of hybrid molecules made up of non-selective or selective COX inhibitors together with a nitric oxide releasing functionCitation16–18.
Recent data have revealed serious cardiovascular side effects with selective COX-2 inhibitorsCitation17,Citation18. In addition, such drugs only minimize the development of new gastric ulcers, but do not affect existing onesCitation19.
The strategy involving the use of hybrid molecules made up of non-selective COX inhibitors together with a nitric oxide donating moiety constitutes one of the most promising approaches, because nitric oxide supports several endogenous GI tract defense mechanisms, including increase in mucus and bicarbonate secretions, increase in mucosal blood flow, and inhibition of the activation of proinflammatory cells. Moreover, because of the beneficial cardiovascular effects of NO, such drugs are expected to be devoid of the potential adverse cardiovascular effects associated with the use of selective COX-2 inhibitors.
Synthetic approaches based upon chemical modification of NSAIDs have been taken with aim of improving the safety profile and in turn the therapeutic window of NSAIDs. It has been reported in the literature that certain compounds bearing a 1,3,4-oxadiazole nucleus possess significant anti-inflammatory activityCitation20. In our attempt to discover new, safer, and potent agents for the treatment of inflammatory diseases, we functionalized the oxadiazole nucleus onto the indomethacin moiety.
In view of the above facts, herein we report the synthesis, anti-inflammatory, analgesic, and ulcerogenic properties, and in vitro nitric oxide releasing characteristic of a hybrid molecule which has an indomethacin like structure, oxadiazole moiety, and organic nitrate as the nitric oxide donating group.
In the present study, we modified indomethacin (non-selective COX-2 inhibitor) in three regions in an attempt both to increase the anti-inflammatory activity and to enhance drug safety by the covalent attachment of an organic nitrate moiety as a nitric oxide donor, as shown in .
Figure 1. Pharmacophore.
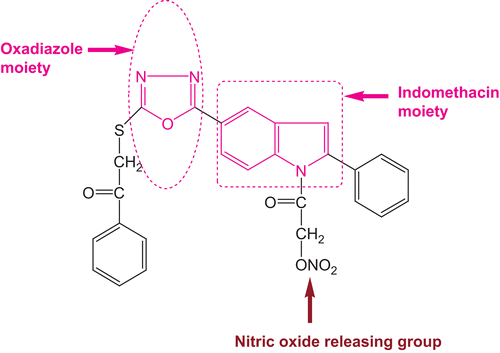
The compounds designed thus were found to possess a significant analgesic–anti-inflammatory profile with significant reduction in the potential for ulcerogenic toxicities.
Experimental
Synthesis studies
All the compounds were synthesized using reported literature procedures, and synthesis procedures were set and optimized as and where required. Melting points were determined by digital melting point apparatus (Veego), model VMP-D, and were uncorrected. Fourier transform infrared (FTIR) spectra of the powdered compounds were recorded using KBr on a Jasco FTIR V 430+ spectrometer, with diffuse reflectance attachment, and are reported in cm−1. 1H nuclear magnetic resonance (NMR) spectra were recorded on a Varian Mercury YH300 (300 MHz FT NMR) spectrophotometer using tetramethylsilane (TMS) as an internal reference (chemical shift represented in δ ppm). Mass spectra were recorded on a gas chromatography-mass spectrometry (GC-MS) QP5050A system (bench-top quadrupole mass spectrophotometer). NMR and MASS spectral studies were done at Department of Chemistry, University of Pune. Elemental analysis was recorded on a FlashEA112 series machine at the Shimadzu Analytical Center, University of Pune; the values were found to be within ±0.4% of the theoretical ones. The purity of the compounds was checked on thin layer chromatography (TLC) plates using silica gel G as the stationary phase and iodine vapor as the visualizing agent.
Synthesis of 4-hydrazino benzoic acid (1)
Compound (1) was synthesized as per the reported procedureCitation21. Yield: 82%, M.P. 108–110°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 3550 (O-H stretching in acid); 1715 (C=O stretching in acid); 3460 (primary Ar N-H stretching); 1560 (Ar C=C).
Synthesis of 4-(2-(1-phenylethylidene) hydrazinyl) benzoic acid (2)
Compound (2) was synthesized as per the reported procedureCitation21. Yield: 80%, M.P. 120–122°C. FTIR spectra (cm−1) of the compound showed bands at 3030 (Ar C-H); 3550 (O-H stretching in acid); 1715 (C=O stretching in acid); 1546 (C=N); 2941 (aliphatic C-H).
Synthesis of 2-phenyl-1-H-indole-5-carboxylic acid (3)
Compound (3) was synthesized as per the reported procedureCitation21. Yield: 75%, M.P. 192–194°C. FTIR spectra (cm−1) of the compound showed bands at 3030 (Ar C-H); 3540 (O-H stretching in acid); 1720 (C=O stretching in acid); 1260 (C-N stretching); 3350 (secondary Ar N-H stretching).
Synthesis of 2-phenyl-1-H-indole-5-carbohydrazide (4)
A mixture of 3 (4 g, 10 mmol) and excess of thionyl chloride was refluxed for 4 h; excess thionyl chloride was neutralized by using formic acid and the residue was refluxed with excess hydrazine hydrate in ethanol (20 mL) for 6 h. The mixture was left to cool and then evaporated under reduced pressure. The residue obtained was filtered, washed with water, dried, and crystallized from absolute ethanol. Yield: 73.49%, M.P. 200–202°C. FTIR spectra (cm−1) of the compound showed bands at 3020 (Ar C-H); 1650 (C=O stretching); 1275 (C-N stretching); 3520 (primary N-H stretching); 3320 (Ar N-H stretching).
Synthesis of 5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2-thiol (5)
Compound (5) was synthesized as per the reported procedureCitation22. Yield: 78.50%, M.P. 214–216°C. FTIR spectra (cm−1) of the compound showed bands at 3015 (Ar C-H); 1260 (C-N stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 3335 (Ar N-H stretching); 2560 (S-H stretching).
General procedure for the preparation of S-substituted phenacyl 1,3,4,-oxadaizole-2-thiol derivatives (5a–5f)
S-substituted phenacyl 1,3,4,-oxadaizole-2-thiol derivatives (5a-5f) were prepared as per the reported proceduresCitation23.
Synthesis of 2-(5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2ylthio)-1-(phenyl) ethanone(5a)
Yield: 72.70%, M.P. 228–230°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1635 (C=O stretching); 1250 (C-N stretching); 3300 (Ar N-H stretching); 1610 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2935 (aliphatic C-H). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.5 (s, 2H, -(CH2)); 6.9 (s, 1H, -CH-); 7.1–7.3 (m, 5H, -Ar-H); 7.4–7.5 (m, 5H, ArH); 10.1 (s, 1H, indole NH); 7.6–7.8 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 411 (M+), other important fragments were observed at 412 (M+ + 1), 413 (M+ + 2).
Synthesis of 2-(5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2ylthio)-1-(4-hydroxyphenyl) ethanone (5b)
Yield: 75.28%, M.P. 234–236°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1635 (C=O stretching); 1250 (C-N stretching); 3310 (Ar N-H stretching); 1610 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2920 (aliphatic C-H); 3293 (O-H stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.7 (s, 2H, -(CH2)); 6.4 (s, 1H, -CH-); 7.1–7.2 (m, 5H, -Ar-H); 7.3–7.5 (m, 5H, ArH); 9.6 (s, 1H, indole NH); 7.6–7.8 (m, 3H, 4, 6, 7 ArH); 8.23 (s, 1H, O-H). Mass spectra of compound exhibited molecular ion peak at m/z 427 (M+), other important fragments were observed at 428 (M+ + 1), 429 (M+ + 2).
Synthesis of 2-(5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2ylthio)-1-(3-hydroxyphenyl) ethanone (5c)
Yield: 74.88%, M.P. 232–234°C. FTIR spectra (cm−1) of the compound showed bands at 3035 (Ar C-H); 1675 (C=O stretching); 1250 (C-N stretching); 3300 (Ar N-H stretching); 1608 (C-N ring stretch of oxadiazole nucleus); 1040 (C-O stretch of oxadiazole nucleus); 2955 (aliphatic C-H); 3320 (O-H stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.7 (s, 2H, -(CH2)); 6.6 (s, 1H, -CH-); 7.1–7.21 (m, 5H, -Ar-H); 7.35–7.5 (m, 5H, ArH); 9.5 (s, 1H, indole NH); 7.6–7.82 (m, 3H, 4, 6, 7 ArH), 8.5 (s, 1H, O-H). Mass spectra of compound exhibited molecular ion peak at m/z 427 (M+), other important fragments were observed at 428 (M+ + 1), 429 (M+ + 2).
Synthesis of 2-(5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2ylthio)-1-(4-nitrophenyl) ethanone (5d)
Yield: 71.50%, M.P. 238–240°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1230 (C-N stretching); 3320 (Ar N-H stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1045 (C-O stretch of oxadiazole nucleus); 2941 (aliphatic C-H); 1670 (N=O stretching); 1360 (C-N stretching in C-NO2). 1H NMR chemical shifts at (CDCl3, δ ppm): 4.2 (s, 2H, -(CH2)); 6.8 (s, 1H, -CH-); 7.01–7.1 (m, 5H, -Ar-H); 7.21–7.4 (m, 5H, Ar); 7.5–7.7 (m, 3H, 4, 6, 7 ArH); 8.1 (s, 1H, indole NH). Mass spectra of compound exhibited molecular ion peak at m/z 456 (M+), other important fragments were observed at 457 (M+ + 1), 458 (M+ + 2).
Synthesis of 2-(5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2ylthio)-1-(3-nitrophenyl) ethanone (5e)
Compound 5e was synthesized as per the reported procedure. Yield: 70.60%, M.P. 236–238°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1250 (C-N stretching); 3300 (Ar N-H stretching); 1615 (C-N ring stretch of oxadiazole nucleus); 1025 (C-O stretch of oxadiazole nucleus); 2935 (aliphatic C-H); 1620 (N=O stretching); 1340 (C-N stretching in C-NO2). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.5 (s, 2H, -(CH2)); 6.9 (s, 1H, -CH-); 7.01–7.1 (m, 5H, -Ar-H); 7.21–7.4 (m, 5H, ArH); 7.5–7.7 (m, 3H, 4, 6, 7 ArH); 8.3 (s, 1H, indole NH). Mass spectra of compound exhibited molecular ion peak at m/z 456 (M+), other important fragments were observed at 457 (M+ + 1), 458 (M+ + 2).
Synthesis of 2-(5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2ylthio)-1-(3,4-dimethoxyphenyl) ethanone (5f)
Yield: 76.70%, M.P. 244–246°C. FTIR spectra (cm−1) of the compound showed bands at 3040 (Ar C-H); 1675 (C=O stretching); 1230 (C-N stretching); 3310 (Ar N-H stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2930 (aliphatic C-H); 1150 (O-CH3 asymmetric stretch). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.4 (s, 2H, -(CH2)); 3.73 (s, 6H, (O-CH3)2); 6.4 (s, 1H, -CH-); 7.1–7.3 (m, 5H, -Ar-H); 7.4–7.6 (m, 5H, ArH); 7.5–7.7 (m, 3H, 4, 6, 7 ArH); 8.7 (s, 1H, indole NH). Mass spectra of compound exhibited molecular ion peak at m/z 471 (M+), other important fragments were observed at 472 (M+ + 1), 473 (M+ + 2).
General procedure for the preparation of N-substituted chloro acetyl chloride derivatives (6a–6f)
N-substituted chloro acetyl chloride derivatives (6a–6f) were prepared as per the reported proceduresCitation24.
2-Chloro-1-(5-(5-(2-oxo-2-phenylethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl) ethanone (6a)
Yield: 70.56%, M.P. 252–254°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1250 (C-N stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2941 (aliphatic C-H); 680 (C-Cl stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.6 (s, 2H, -(CH2)); 4.4 (s, 2H, -(CH2-Cl)). 6.5 (s, 1H, -CH-); 7.2–7.35 (m, 5H, -Ar-H); 7.4–7.54 (m, 5H, ArH); 7.6–7.8 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 487 (M+), other important fragments were observed at 488 (M+ + 1), 489 (M+ + 2).
2-Chloro-1-(5-(5-(2-(4-hydroxyphenyl)-2-oxo-2-phenylethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl) ethanone (6b)
Yield: 69.88%, M.P. 256–258°C. FTIR spectra (cm−1) of the compound showed bands at 3030 (Ar C-H); 1635 (C=O stretching); 1270 (C-N stretching); 1615 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2941 (aliphatic C-H); 3280 (O-H stretching); 695 (C-Cl stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.1 (s, 2H, -(CH2)); 4.2 (s, 2H, -(CH2-Cl)); 6.7 (s, 1H, -CH-); 7.3–7.4 (m, 5H, -Ar-H); 7.5–7.7 (m, 5H, ArH); 7.8–7.92 (m, 3H, 4, 6, 7 ArH); 8.3 (s, 1H, O-H). Mass spectra of compound exhibited molecular ion peak at m/z 503 (M+), other important fragments were observed at 504 (M+ + 1), 505 (M+ + 2).
2-Chloro-1-(5-(5-(2-(3-hydroxyphenyl)-2-oxo-2-phenylethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl) ethanone (6c)
Yield: 73.74%, M.P. 258–260°C. FTIR spectra (cm−1) of the compound showed bands at 3015 (Ar C-H); 1645 (C=O stretching); 1230 (C-N stretching); 1615 (C-N ring stretch of oxadiazole nucleus); 1035 (C-O stretch of oxadiazole nucleus); 2940 (aliphatic C-H); 3310 (O-H stretching); 670 (C-Cl stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.1 (s, 2H, -(CH2)); 4.2 (s, 2H, -(CH2-Cl)); 6.8 (s, 1H, -CH-); 7.1–7.32 (m, 5H, -Ar-H); 7.4–7.6 (m, 5H, ArH); 7.7–7.85 (m, 3H, 4, 6, 7 ArH); 8.6 (s, 1H, O-H). Mass spectra of compound exhibited molecular ion peak at m/z 503 (M+), other important fragments were observed at 504 (M+ + 1), 505 (M+ + 2).
2-Chloro-1-(5-(5-(2-(4-nitrophenyl)-2-oxo-2-phenlethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl) ethanone (6d)
Yield: 75.23%, M.P. 244–246°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1250 (C-N stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2941 (aliphatic C-H); 1670 (N=O stretching); 1360 (C-N stretching in C-NO2); 660 (C-Cl stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 4.1 (s, 2H, -(CH2)); 4.5 (s, 2H, -(CH2-Cl)) 6.3 (s, 1H, -CH-); 7.01–7.1 (m, 5H, -Ar-H); 7.21–7.4 (m, 5H, ArH); 7.5–7.7 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 532 (M+), other important fragments were observed at 533 (M+ + 1), 534 (M+ + 2).
2-Chloro-1-(5-(5-(2-(3-nitrophenyl)-2-oxo-2-phenlethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl) ethanone (6e)
Yield: 68.23%, M.P. 242–244°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1250 (C-N stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2941 (aliphatic C-H); 1630 (N=O stretching); 1340 (C-N stretching in C-NO2); 675 (C-Cl stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 4.2 (s, 2H, -(CH2)); 4.6 (s, 2H, -(CH2-Cl)); 6.3 (s, 1H, -CH-); 7.21–7.4 (m, 5H, -Ar-H); 7.4–7.6 (m, 5H, ArH); 7.7–7.85 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 532 (M+), other important fragments were observed at 533 (M+ + 1), 534 (M+ + 2).
2-Chloro-1-(5-(5-(2-(3,4-dimethoxyphenyl)-2-oxo-2-phenylethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl) ethanone (6f)
Yield: 74.55%, M.P. 250–252°C. FTIR spectra (cm−1) of the compound showed bands at 3017 (Ar C-H); 1675 (C=O stretching); 1220 (C-N stretching); 1630 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2945 (aliphatic C-H); 1140 (O-CH3 asymmetric stretch); 680 (C-Cl stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.2 (s, 2H, -(CH2)); 3.45 (s, 6H, (O-CH3)2); 4.4 (s, 2H, -(CH2-Cl)); 6.4 (s, 1H, -CH-); 7.01–7.1 (m, 5H, -Ar-H); 7.21–7.4 ( m, 5H, ArH ); 7.5–7.7 (m, 3H, 4, 6, 7 ArH).. Mass spectra of compound exhibited molecular ion peak at m/z 547 (M+), other important fragments were observed at 548 (M+ + 1), 549 (M+ + 2).
General procedure for the preparation of nitrate derivatives (7a–7f)
Nitrate derivatives (7a–7f) were prepared as per the reported proceduresCitation25.
2-Oxo-2-(5-(5-(2-oxo-2-phenylethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl) ethyl nitrate (7a)
Yield: 75.63%, M.P. 260–262°C. FTIR spectra (cm−1) of the compound showed bands at 3015 (Ar C-H); 1665 (C=O stretching); 1255 (C-N stretching); 1604 (C-N ring stretch of oxadiazole nucleus); 1025 (C-O stretch of oxadiazole nucleus); 2965 (aliphatic C-H); 1444 (CH2-O stretching); 1632 (-N=O stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.7 (s, 2H, -(CH2)); 4.6 (s, 2H, -(CH2–ONO2)); 6.4 (s, 1H, -CH-); 6.9–7.1 (m, 5H, -Ar-H); 7.21–7.4 (m, 5H, ArH); 7.5–7.7 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 514 (M+), other important fragments were observed at 515 (M+ + 1), 516 (M+ + 2). Elemental analysis was observed as C (60.64%), H (3.56%), N (10.92%).
2-(5-(5-(4-Hydroxyphenyl)-2-oxo-ethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl)-2-oxoethyl nitrate (7b)
Yield: 52.50%, M.P. 266–268°C. FTIR spectra (cm−1) of the compound showed bands at 3035 (Ar C-H); 1675 (C=O stretching); 1240 (C-N stretching); 1608 (C-N ring stretch of oxadiazole nucleus); 1046 (C-O stretch of oxadiazole nucleus); 3010 (aliphatic C-H); 3293 (O-H stretching); 1430 (CH2-O stretching); 1635 (-N=O stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.6 (s, 2H, -(CH2)); 4.3 (s, 2H, -(CH2–ONO2)); 6.8 (s, 1H, -CH-); 7.1–7.26 (m, 5H, -Ar-H); 7.3–7.4 (m, 5H, ArH); 7.5–7.7 (m, 3H, 4, 6, 7 ArH); 8.12 (s, 1H, O-H). Mass spectra of compound exhibited molecular ion peak at m/z 530 (M+), other important fragments were observed at 531 (M+ + 1), 532 (M+ + 2). Elemental analysis was observed as C (58.90%), H (3.45%), N (10.52%).
2-(5-(5-(3-Hydroxyphenyl)-2-oxo-ethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl)-2-oxoethyl nitrate (7c)
Yield: 64.23%, M.P. 254–256°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1270 (C-N stretching); 1630 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2920 (aliphatic C-H); 1460 (CH2-O stretching); 1650 (-N=O stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.5 (s, 2H, -(CH2)); 4.4 (s, 2H, -(CH2–ONO2)); 6.9 (s, 1H, -CH-); 7.01–7.1 (m, 5H, -Ar-H); 7.21–7.4 (m, 5H, ArH); 7.5–7.7 (m, 3H, 4, 6, 7 ArH); 8.23 (s, 1H, O-H). Mass spectra of compound exhibited molecular ion peak at m/z 530 (M+), other important fragments were observed at 531 (M+ + 1), 532 (M+ + 2). Elemental analysis was observed as C (58.82%), H (3.46%), N (10.52%).
2-(5-(5-(4-Nitrophenyl)-2-oxo-ethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl)-2-oxoethyl nitrate (7d)
Yield: 59.52%, M.P. 274–276°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1250 (C-N stretching); 3350 (Ar N-H stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2941 (aliphatic C-H); 1670 (N=O stretching); 1360 (C-N stretching in C-NO2); 1450 (CH2-O stretching); 1630 (-N=O stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.6 (s, 2H, -(CH2)); 4.3 (s, 2H, -(CH2–ONO2)) 6.4 (s, 1H, -CH-); 6.9–7.1 (m, 5H, -Ar-H); 7.3–7.4 (m, 5H, ArH); 7.6–7.8 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 559 (M+), other important fragments were observed at 560 (M+ + 1), 561 (M+ + 2). Elemental analysis was observed as C (55.84%), H (3.10%), N (12.56%).
2-(5-(5-(3-Nitrophenyl)-2-oxo-ethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl)-2-oxoethyl nitrate (7e)
Yield: 67.62%, M.P. 286–288°C. FTIR spectra (cm−1) of the compound showed bands at 3035 (Ar C-H); 1675 (C=O stretching); 1255 (C-N stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1040 (C-O stretch of oxadiazole nucleus); 2931 (aliphatic C-H); 1620 (N=O stretching); 1344 (C-N stretching in C-NO2); 1430 (CH2-O stretching); 1630 (-N=O stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.3 (s, 2H, -(CH2)); 4.4 (s, 2H, -(CH2–ONO2)); 6.9 (s, 1H, -CH-); 7.1–7.2 (m, 5H, -Ar-H); 7.31–7.5 (m, 5H, ArH); 7.6–7.86 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 559 (M+), other important fragments were observed at 560 (M+ + 1), 561 (M+ + 2). Elemental analysis was observed as C (55.81%), H (3.02%), N (12.56%).
2-(5-(5-(3,4-Dimethoxyphenyl)-2-oxo-ethylthio)-1,3,4-oxadiazole-2-yl)-2-phenyl-1H-indol-1-yl)-2-oxoethyl nitrate (7f)
Yield: 61.34%, M.P. 292–294°C. FTIR spectra (cm−1) of the compound showed bands at 3025 (Ar C-H); 1675 (C=O stretching); 1250 (C-N stretching); 1602 (C-N ring stretch of oxadiazole nucleus); 1055 (C-O stretch of oxadiazole nucleus); 2941 (aliphatic C-H); 1150 (O-CH3 asymmetric stretch); 1450 (CH2-O stretching); 1630 (-N=O stretching). 1H NMR chemical shifts at (CDCl3, δ ppm): 3.5 (s, 2H, -(CH2)); 3.73 (s, 6H, (O-CH3)2); 4.4 (s, 2H, -(CH2–ONO2)); 6.62 (s, 1H, -CH-); 7.1–7.21 (m, 5H, -Ar-H); 7.2–7.38 (m, 5H, ArH); 7.62–7.7 (m, 3H, 4, 6, 7 ArH). Mass spectra of compound exhibited molecular ion peak at m/z 574 (M+), other important fragments were observed at 575 (M+ + 1), 576 (M+ + 2). Elemental analysis was observed as C (58.51%), H (3.83%), N (9.70%).
Pharmacology
Animals
Swiss albino mice of either sex weighing 20–25 g and Wistar rats weighing in the range 100–120 g were obtained from Yash Farm, Pune, India. All the animals were housed under standard ambient conditions of temperature (25 ± 2°C) and relative humidity of 50 ± 5%. A 12:12 h light/dark cycle was maintained. All the animals were allowed to have free access to water, and standard pelletized laboratory animal diet until 24 h prior to pharmacological studies. All the experimental procedures and protocols used in this study were reviewed and approved by the Institutional Animal Ethical Committee (IAEC) of the AISSMS College of Pharmacy, Pune, constituted for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA/257, CPCSEA/300), Government of India.
Preparation of test compounds
After suspending the test compounds in a 1.0% aqueous solution of sodium carboxymethyl cellulose (CMC), test samples were administered to the test animals orally. The negative and positive control group animals received the same experimental handling as those of the test groups except that the control group animals received only appropriate volumes of vehicle and of the reference drug, indomethacin or diclofenac acid, respectively.
Anti-inflammatory activity
Anti-inflammatory activity was evaluated using the well known carrageenan induced rat paw edema model of Winter et al.Citation26, using groups of six animals each. A freshly prepared aqueous suspension of carrageenan (1.0%, w/v, 0.1 mL) was injected in the subplantar region of the right hind paw of each rat. One group was kept as control, and the animals of the other group were pretreated with the test compounds, 1 h before the carrageenan treatment. The volume was measured before and after carrageenan treatment at 60 min intervals with the help of a digital plethysmometer (Panlab LE 7500, USA).
Analgesic activity
The analgesic activity was evaluated using the acetic acid induced writhing methodCitation27.
Acute ulcerogenicity studies
Acute ulcerogenicity screening was done according to method reported by Cioli et al.Citation28. The mucosal damage was examined by means of an electron microscope. For each stomach specimen, the mucosal damage was assessed according to the following scoring system:
0.0, normal (no injury, bleeding, and latent injury);
0.5, latent injury or widespread bleeding (>2 mm);
1.0, slight injury (2–3 dotted lines);
2.0, severe injury (continuous lined injury or 5–6 dotted injuries);
3.0, very severe injury (several continuous lined injuries);
4.0, widespread lined injury or widened injury.
The mean score for each treated group minus the mean score for the control group was regarded as the severity index of gastric mucosal damage. Data are expressed as mean ulcer score ± SEM, data analyzed by one way analysis of variance (ANOVA) followed by Dunnett’s t test to determine the significance of the difference between the standard group and rats treated with the test compounds. The differences in results were considered significant when p was found to be <0.01.
Histopathological studies20,29,30
For the histopathological study, rats were sacrificed 4 h after cold stress and their stomach specimens removed and put into 10% formalin solution. A longitudinal section of the stomach along the greater curvature, which included the ulcer base and both sides of the ulcer margin, was taken and fixed in 10% formalin for 24 h at 4°C and embedded in white solid paraffin. Morphological examination was performed using hematoxylin and eosin staining to analyze histological changes and done under an electron microscope.
Detection of nitrite25
A solution of the appropriate compound (20 µL) in dimethylsulfoxide (DMSO) was added to a 2 mL of 1:1, v/v, mixture either of 50 mM phosphate buffer (pH 7.4) or of HCl solution (pH 1) with MeOH, containing 5 × 10−4 mM l-cysteine. The final concentration of test compound was 10−4 M. After 1 h at 37°C, 1 mL of the reaction mixture was treated with 250 µL of Griess reagent (sulfanilamide (4 g), N-naphthylethylenediamine dihydrochloride (0.2 g), 85% phosphoric acid (10 mL) in distilled water (final volume: 100 mL)). After 10 min at room temperature, the absorbance was measured at 540 nm. Sodium nitrite standard solutions (10–80 µmol/mL) were used to construct the calibration curve. The results are expressed as the percentage of NO released (n = 2) relative to a theoretical maximum release of 1 mol NO/mol of test compound.
Statistical analysis
Data obtained for each set of anti-inflammatory models are expressed as the mean of change in paw volume ± SD and were analyzed by one-way ANOVA followed by Dunnett’s t test. Data from the acetic acid induced writhing model are expressed as the mean number of writhes ± SEM and were analyzed by one-way ANOVA followed by Dunnett’s t test. The level of significance was set at p < 0.05. All statistical calculations were performed using the evaluation version of GraphPad Prism 3.0 (USA) statistical software.
Results and discussion
Chemistry
The synthetic route used to synthesize the title compounds is outlined in . The compound 4-hydrazinobenzoic acid (1) was prepared according to the method reported in the literature, using PABA (para-aminobenzoic acid). 4-(2-(1-Phenylethylidene) hydrazinyl) benzoic acid (2) was prepared by reacting 1 with acetophenone in ethanol. 2-Phenyl-1-H-indole-5-carboxylic acid (3) was prepared by reacting 2 with H3PO4. The reaction of 3 with thionyl chloride followed by hydrazine hydrate yielded 2-phenyl-1-H-indole-5-carbohydrazide (4). The reaction of 4 with carbon disulfide in an alkaline medium afforded 5-(2-phenyl-1-H-indol-5-yl)-1,3,4-oxadiazole-2-thiol (5). The compounds 5a–5f were prepared by reacting 5 with differently substituted phenacyl bromide in the presence of pyridine. The reaction of these (5a–5f) with the substituted chloro acetyl chloride gave the corresponding haloalkyl derivatives (6a–6f). The terminal chloro function was then substituted with nitric oxide releasing moieties (AgNO3 using acetonitrile as solvent) to get the nitrate derivatives (7a–7f). The structures of the various synthesized compounds were assigned on the basis of results of different chromatographic and spectral studies. The physical data, FTIR, 1H-NMR, and mass spectral data, and elemental analysis data for all the synthesized compounds are given in the above experimental protocols.
Scheme 1. Synthesis route for designed NCEs, 7a–7f.
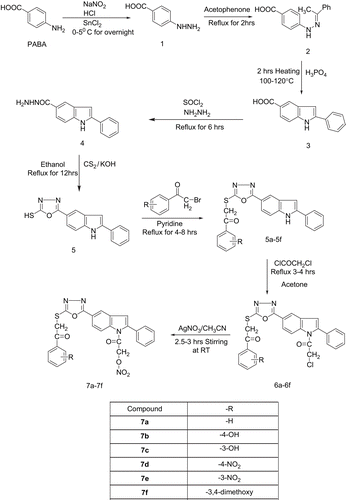
The FTIR spectra of the title compounds (7a–7f) exhibited very similar features and showed the expected bands for the characteristic groups present in the compounds, such as C=O stretching and another specific band for -NO2 vibrations. In the NMR spectral data, all protons were seen according to the expected chemical shift and integration values. The aromatic protons appeared as multiplet peaks within the range 7.01–7.86 δ ppm.
Pharmacology
We modified indomethacin (non-selective COX-2 inhibitors) in three regions in an attempt both to increase the anti-inflammatory activity and to enhance drug safety by the covalent attachment of an organic nitrate moiety as a nitric oxide donor. We investigated the anti-inflammatory, analgesic, and ulcerogenic properties as well as the nitric oxide releasing characteristic of the hybrid molecules in which indomethacin was substituted with the oxadiazole moiety and organic nitrate was the nitric oxide donating group. Indomethacin and diclofenac acid were used as reference standards. The experiments were performed using Wistar rats of either sex, weighing 100–120 g, and Swiss albino mice of either sex, weighing 20–25 g. The animals were maintained at 25 ± 2°C, 50 ± 5% relative humidity, and in a 12 h light/dark cycle. The animals were fasted for 24 h prior to the experiments and water was provided ad libitum. The test compounds were suspended in 1% aqueous carboxymethyl cellulose (CMC) solution and administered orally to experimental animals for all the studies.
Anti-inflammatory activity
Anti-inflammatory activity of the synthesized compounds was evaluated by the carrageenan induced rat paw edema model. Equimolar dose, that is, equivalent to indomethacin subplantar injection, of 0.1 mL, 1% carrageenan in the rat paw produced an increase in paw volume (edema) of all the animals of the various groups. The onset of action was evident from 1 h in the various test groups. A significant (p < 0.01) reduction of rat paw edema was observed for most of the test compounds at 3 h, compared to the control group; indomethacin was used as standard (). Compounds with significant anti-inflammatory profile were subjected to GI-ulcerogenicity potential studies at 12 times the therapeutic doses with additional physical (cold) stress. A thorough examination of the results of histopathological studies indicated the absence of disruption of morphology of gastric epithelial cells and absence of ulcers/erosion in test (test compound treated) group animals compared to the reference standard, indomethacin, and control group animals. The results of the ulcerogenicity studies are presented in and results of the histopathological studies are depicted in . Of the synthesized new chemical entities (NCEs), 7a, 7b, and 7c (55.27%, 62.35%, and 70.65%, respectively) exhibited very significant anti-inflammatory activity, compared to compounds 5 and 6c. The haloalkyl derivatives were less potent than the nitrooxy derivatives. S-substituted phenacyl 1,3,4,-oxadaizole-2-thiol derivatives (6a–6f) were more potent than compound 5. This indicates that substituted phenacyl bromide may increase the activity. Derivatives 7a–7f were more potent than the corresponding haloalkyl derivatives 6a–6f. This indicates that the nitrate moiety may contribute to the anti-inflammatory action of the final compounds. Based on the findings of these preclinical results, further studies need to be carried out to investigate the other specifications, such as in vitro assays, chronic ulcerogenecity studies, and toxicological studies, and the mechanism by which these drugs exhibit potential analgesic and anti-inflammatory activities ().
Table 1. Results for anti-inflammatory activity of synthesized compounds against carrageenan induced rat paw edema model in rats.
Analgesic activity
The analgesic activity of the compounds was studied using the acetic acid induced writhing test in mice. The compounds that exhibited significant anti-inflammatory activity, comparable to that of indomethacin, were screened for analgesic activity. The analgesic activity was evaluated at equimolar dose, equivalent to 10 mg/kg (indomethacin) body weight. These compounds exhibited an important analgesic profile measured by the classical acetic acid induced writhing model. From the results, it was noted that 7b (50.48%) and 7c (68.4%) possessed significant analgesic activity ().
Table 2. Results for analgesic activity of synthesized compounds against acetic acid induced writhing test in mice.
Acute ulcerogenecity studies
The ulcerogenic effect of derivatives 7a, 7b, and 7c with the best overall profile in the animal efficacy model was evaluated for gastric ulcerogenic potential in the rat stress model at 12 times the therapeutic dose of diclofenac acid (). When compared with diclofenac acid, these three compounds did not cause any gastric ulceration and disruption of gastric epithelial cells at the abovementioned oral dose. The results are shown in .
Table 3. Ulcerogenic effects of synthesized compounds in comparison with diclofenac sodium.
Histopathological studies
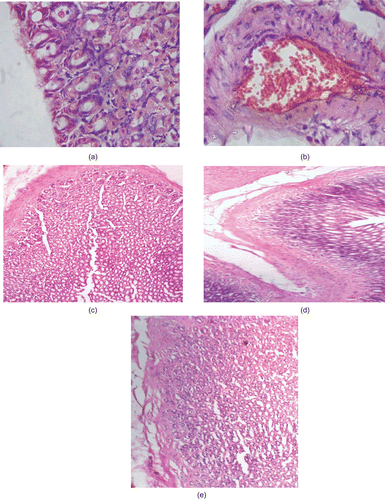
Stomach specimens of diclofenac acid treated rats were characterized by complete disruption of the protective mucosal layer (). Histopathological analysis also showed characteristic features of ulceration in the diclofenac acid treated group of animals. The tissue of diclofenac acid treated rats showed that some epithelial cells in the ulcer margin had proliferated and migrated into the ulcer crater, which indicated complete disruption of the gastric epithelial layer. Scanning of stomach specimens using an electron microscope revealed that in the rats treated with synthesized derivatives 7a, 7b, and 7c there was no injury in the stomach mucosa, as illustrated in , which was identical to that of the control ().
Figure 2. Hematoxylin and eosin immunohistochemical staining of gastric ulcers after ulcer induction in rats. (a) Intact mucous membrane in control treated rat showing granular tissues composed of macrophages, fibroblasts, and endothelial cells forming microvessels. (b) Congestion of mucosal blood vessels observed in diclofenac treated group. (c–e) No damage to mucosa of rats treated with test compounds 7a, 7b, and 7c, respectively. These specimens were identical to that of the control (a). Original magnification ×200.
Nitric oxide release study
The ability of the test compounds to release NO was studied by performing an in vitro nitric oxide release study. This study involved evaluation of nitric oxide releasing properties of the synthesized compounds in phosphate buffer, pH 7.4, in the presence of l-cysteine, relative to nitric oxide released from standard sodium nitrite solution ().
Table 4. Nitric oxide releasing property of compounds 7a–7f.
Conclusions
Six hybrid derivatives were synthesized and screened for analgesic, anti-inflammatory, and ulcerogenic potential, and subjected to nitric oxide releasing studies. Compounds 7a, 7b, and 7c exhibited significant analgesic and anti-inflammatory activity. Compounds 7b and 7c showed strong analgesia in acetic acid induced writhing tests. Among all the synthesized compounds, compound 7c exhibited the most prominent and consistent anti-inflammatory activity. From detailed analysis of the results of histopathological studies, we conclude that the synthesized compounds have not only retained but show an enhanced anti-inflammatory profile, and are devoid of the deadlier gastrointestinal toxicities; also, all the synthesized derivatives exhibit a significant nitric oxide releasing property. Therefore, it can be concluded that the rationale on which the design of these NCEs was based has proved to be superior, compared to currently used NSAIDs. The outcome of the present research work is very promising. The only concern to be addressed now is the pharmacokinetics profile of the designed NCEs, since the molecular weight of all the designed compounds is more than 500 Da.
The most potent derivatives are in the process of chronic toxicity and pharmacokinetics studies, and, based on those results, a further action plan will be finalized.
Scope of further research
The most potent compounds will be subjected to following studies:
Acute and chronic toxicity studies.
Mechanism of release of NO.
Pharmacokinetics profile.
Mechanism of action.
Acknowledgements
The authors are thankful to the Principal, AISSMS College of Pharmacy, Pune, for providing laboratory facilities. The authors are also thankful to Professor N. S. Vyawahare, Head, Department of Pharmacology, for his continuous support and guidance throughout the project. The authors also wish to thank the Shimadzu Analytical Center and Department of Chemistry, University of Pune, for providing facilities for spectral studies of compounds. The authors are also thankful to Satav’s Pathology Laboratory, Pune, for providing facilities for histopathological studies.
Declaration of interest
The authors report no conflict of interest. The authors alone are responsible for the content of this paper.
References
- Morphy R, Kay C, Rankovic Z. From magic bullets to designed multiple ligands. Drug Discov Today 2004;9:641–51.
- Cryer B. NSAID-associated deaths: the rise and fall of NSAID-associated GI mortality. Am J Gastroenterol 2005;100:1694–5.
- Go MF. Drug injury in the upper gastrointestinal tract: nonsteroidal anti-inflammatory drugs. Gastrointest Endosc Clin N Am 2006;16:83–97.
- James MW, Hawkey CJ. Assessment of non-steroidal anti-inflammatory drug (NSAID) damage in human gastrointestinal tract. Br J Clin Pharmacol 2003;56:146–55.
- Lozzaroni M, Porro GB. Gastrointestinal side-effects of traditional anti-inflammatory drugs and new formulations. Aliment Pharmacol Ther 2004;20(2):48–58.
- Naesdal J, Brown K. NSAID-associated adverse effects and acid control aids to prevent them: a review of current treatment options. Drug Saf 2006;29:119–32.
- Mounier G, Guy C, Berthoux F, Beyens MN, Ratrema M, Ollagnier M. Severe renal adverse events with arylcarboxylic non-steroidal anti-inflammatory drugs: results of an eight-year French national survey. Therapie 2006;61:255–66.
- Schneider V, Levesque LE, Zhang B, Hutchinson T, Brophy JM. Association of selective and conventional nonsteroidal anti-inflammatory drugs with acute renal failure: a population-based nested case-control analysis. Am J Epidemiol 2006;164:881–9.
- Zadrazil J. Nonsteroidal antiinflammatory drugs and kidney. Vnitr Lek 2006;52:686–90.
- Adebayo D, Bjarnason I. Is non-steroidal anti-inflammatory drug (NSAID) enteropathy clinically more important than NSAID gastropathy? Postgrad Med J 2006;82:186–91.
- Thomsen RW, Riis A, Munk EM, Norgaard M, Christensen S, Sorensen HT. 30-Day mortality after peptic ulcer perforation among users of newer selective COX-2 inhibitors and traditional NSAIDs: a population based study. Am J Gastroenterol 2006;101:2704–10.
- Dogne JM, Supuran CT, Pratico D. Adverse cardiovascular effects of the coxibs. J Med Chem 2005;48:2251–7.
- Mann NS. NO-aspirin. Gastroenterology 2003;125:1918–19.
- Keeble JE, Moore PK. Pharmacology and potential therapeutic applications of nitric oxide-releasing non-steroidalanti-inflammatory and related nitric oxide-donating drugs. Br J Pharmacol 2002;137:295–310.
- Rehse K, Brehme F.New NO donors with antithrombotic and vasodilating activities, Part 27. Azide oximes and 1-hydroxytetrazoles. Arch Pharm Pharm Med Chem 2000;333:157–61.
- Bias P, Buchner A, Klesser B, Laufer S. The gastrointestinal tolerability of the LOX/COX inhibitor, licofelone, is similar to placebo and superior to naproxen therapy in healthy volunteers: results from a randomized, controlled trial. Am J Gastroenterol 2004;99:611–18.
- Doggrell SA. The safety of lumiracoxib when used in the treatment of arthritis. Expert Opin Pharmacother 2005;6:347–50.
- Velazquez C, Rao PN, McDonald R, Knaus EE. Synthesis and biological evaluation of 3,4-diphenyl-1,2,5-oxadiazole-2-oxides and 3,4-diphenyl-1,2,5-oxadiazoles as potential hybrid COX-2 inhibitor/nitric oxide donor agents. Bioorg Med Chem 2005;13:2749–57.
- Hunt RH, Barkun AN, Baron D, Bombardier C, Bursey FR, Marshall JR, et al. Recommendations for the appropriate use of anti-inflammatory drugs in the era of the coxibs: defining the role of gastroprotective agents. Can J Gastroenterol 2002;16:231–40.
- Omar FA, Mahfouz NM, Rahman MA. Design, synthesis and anti-inflammatory activity of some 1,3,4-oxadiazole derivatives. Eur J Med Chem 1996;31:819–25.
- Vogel AI. Text Book of Practical Organic Chemistry. New York: Longman Scientific & Technical, 1994:458, 1161.
- Mohd Amir, Kumar S. Synthesis and evaluation of anti-inflammatory, analgesic, ulcerogenic and lipid peroxidation properties of ibuprofen derivatives. Acta Pharm 2007;57:31–45.
- Bhandari SV, Bothara KG, Raut MK. Design, synthesis, and evaluation of anti-inflammatory, analgesic and ulcerogenicity studies of novel S-substituted phenacyl-1,3,4-oxadiazole-2-thiol and Schiff bases of diclofinac acid as nonulcerogenic derivatives. Bioorg Med Chem 2008;16:1822–31.
- Shrivastava SK, Nema A, Shrivastava SD. Conventional as well as microwave assisted synthesis of some new N9-[hydrazinoacetyl-(2-oxo-3-chloro-4-substituted aryl azetidine)]-carbazoles: antifungal and antibacterial studies. Indian J Chem 2008;47B:606–12.
- Abadi HA, Gehan HH, Asmaa A. Synthesis of novel 4-substituted-7-trifluoro methylquinoline derivatives with nitric oxide releasing properties and their evaluation as analgesic and anti-inflammatory agents. Bioorg Med Chem 2005;13:5759–65.
- Winter CA, Risley EA, Nuss GN. Carrageenan induced edema in hind paw of the rats as an assay for anti-inflammatory drugs. Proc Soc Exp Biol 1962;111:544–7.
- Koster R, Anderson M, De Beer EJ. Acetic acid for analgesic screening. Fed Proc 1959;18:412.
- Cioli V, Putzolu S, Rossi V, Sorza Barcellona P, Corradino C. The role of direct tissue contact in the production of gastrointestinal ulcers by anti-inflammatory drugs in rats. Toxicol Appl Pharmacol 1979;50:283–9.
- Fidalgo SS, Lacave MI, Illanes M, Motilva V. Angiogenesis, cell proliferation and apoptosis in gastric ulcer healing. Effect of a selective cox-2 inhibitor <http://www.sciencedirect.com/science?_ob=ArticleURL&_udi=B6T1J-4DNRXR4-2&_user=8570523&_coverDate=11%2F28%2F2004&_rdoc=23&_fmt=high&_orig=browse&_srch=doc-info%28%23toc%234892%232004%23994949998%23529595%23FLA%23display%23Volume%29&_cdi=4892&_sort=d&_docanchor=&_ct=33&_acct=C000107336&_version=1&_urlVersion=0&_userid=8570523&md5=ced063171e366758dc95cf5a7f921ef0>. Eur J Pharmacol 2004;505:187–94.
- Palaska E, Sahin G, Kelicen P, Durlu N, Altinok G. Synthesis and anti-inflammatory activity of 1-acylthiosemicarbazides, 1,3,4-oxadiazoles, 1,3,4-thiadiazoles and 1,2,4-triazole-3-thiones. Farmaco 2002;57:101–7.