Abstract
Some new structural type inhibitors of urease, i.e. 2,5-disubstituted-1,3,4-oxadiazoles (4a–e) and 4,5-disubstituted-1,2,4-triazole-3-thiones (5a–e) were synthesized in two steps from mandelic acid hydrazides (2a–e) and aryl isothiocyantes. The hydrazides in turn were synthesized from mandelic acid via esterification. Compounds 4a–e and 5a–e were evaluated against jack bean urease. Compounds 4d, 5b, and 5d were found to be more potent, with IC50 values of 16.1 ± 0.12 µM, 18.9 ± 0.188 µM, and 16.7 ± 0.178 µM, respectively, when compared to the standard (thiourea; IC50 = 21.0 ± 0.011 µM). These compounds may be subjected to further investigations for the development of antiulcer drugs.
Introduction
Heterocycles, especially the bioisosteric 1,3,4-oxadiazoles, 1,2,4-triazoles, and 1,3,4-thiadiazoles, are important molecules known to interact with particular receptors on enzyme active sites. Among these, 1,3,4-oxadiazoles and 1,2,4-triazoles exhibit numerous biological activities such as antimicrobialCitation1–3, anticonvulsantCitation4, anti-human immunodeficiency virus (HIV)Citation5, anticancerCitation6,Citation7, and urease inhibitionCitation8,Citation9. Oxadiazoles and triazoles have been regarded as structural type inhibitors of ureaseCitation10. Serwar et al.Citation8 have attributed the urease inhibition activity of 1,2,4-triazole-3-thiones to the structural similarity of these compounds with the natural substrate of urease, i.e. urea.
As a result of our focus on the synthesis and biological evaluation of five-membered heterocyclesCitation11–13, in the present study, 2,5-disubstituted-1,3,4-oxadiazoles and 4,5-disubstituted-1,2,4-triazole-3-thiones were synthesized starting from mandelic acid () and their urease inhibition evaluated.
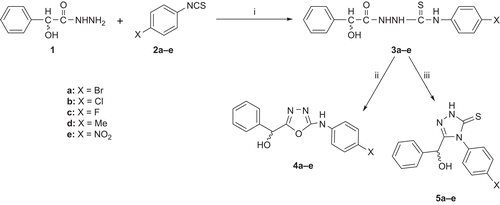
Scheme 1. Synthesis of 1,3,4-oxadiazoles and 1,2,4-triazole-3-thiones. Reagents and conditions: (i) MeOH, reflux; (ii) Hg(OAc)2; (iii) 5% NaOH.
Experimental
Materials
The synthesized compounds were characterized by their physical constants and spectroanalytical techniques. Thin layer chromatography (TLC) was carried out using precoated silica gel 60 HF254 aluminum sheets (Merck, Germany). The melting points were determined on a Sanyo Gallenkamp digital melting point apparatus (MPD 350) in open capillaries and are uncorrected. Infrared (IR) spectra were recorded on a Thermo Scientific Nicolet 6700 FT-IR spectrophotometer (USA) using the ATR (attenuated total reflectance) facility. 1H- and 13C-nuclear magnetic resonance (NMR) spectra were recorded on a Bruker Avance 300 MHz spectrometer and calibrated to residual solvent peaks. Mass spectra were recorded on a MAT-112-S spectrometer at 70 eV. Mandelic acid was purchased from Merck Chemicals; all the reagents were of high purity grade and the solvents were distilled before use.
General procedure for the synthesis of thiosemicarbazides (3a–e)
To a solution of carboxylic acid hydrazide (5.90 mmol) in absolute MeOH (30 mL) was added the respective isothiocyanate (5.9 mmol) separately dissolved in absolute MeOH (20 mL), with stirring. The reaction mixture was heated under reflux for 2–3 h (TLC). After cooling, the resulting solid was filtered and recrystallized from EtOH/water to give pure thiosemicarbazides (3a–e).
1-[(2-Hydroxy-2-phenyl)acetyl]-4-(4-bromophenyl)thiosemicarbazide (3a)
Yield: 85%; m.p. 56°C; IR (ATR, cm−1): νmax 3485, 3227, 3119, 2952, 1690, 1642, 1590, 1511, 1484, 1170; 1H-NMR (300 Hz, DMSO-d6): δ 5.12 (1H, s, CHOH), 6.11 (1H, br s, OH), 7.33–7.48 (9H, m, Ar-H), 9.36 (1H, br s, NH), 9.80 (1H, br s, NH), 10.29 (1H, s, NH); EIMS: m/z 217, 215, 213, 173, 171, 148, 134, 107, 79, 77.
1-[(2-Hydroxy-2-phenyl)acetyl]-4-(4-chlorophenyl)thiosemicarbazide (3b)
Yield: 81%; m.p. 162–164°C; IR (ATR, cm−1): νmax 3479, 3227, 3125, 2946, 1689, 1643, 1591, 1510, 1488, 1170; 1H-NMR (300 Hz, DMSO-d6): δ 5.12 (1H, d, J = 2.4 Hz, CHOH), 6.12 (1H, br s, OH), 7.26–7.50 (9H, m, Ar-H), 9.39 (1H, br s, NH), 9.81 (1H, br s, NH), 10.31 (1H, s, NH); EIMS: m/z 208, 171, 170, 169, 148, 111, 107, 91, 79, 77.
1-[(2-Hydroxy-2-phenyl)acetyl]-4-(4-fluorophenyl)thiosemicarbazide (3c)
Yield: 87%; m.p. 178–180°C; IR (ATR, cm−1): νmax 3424, 3228, 3119, 2952, 1679, 1610, 1548, 1505, 1448, 1414, 1366, 1170; 1H-NMR (300 Hz, DMSO-d6): δ 5.11 (1H, d, J = 4.2 Hz, CHOH), 6.10 (1H, br s, OH), 7.16–7.50 (9H, m, Ar-H), 9.37 (1H, br s, NH), 9.73 (1H, br s, NH), 10.29 (1H, s, NH); EIMS: m/z 301, 212, 208, 194, 111, 110, 107, 105, 95, 79, 77.
1-[(2-Hydroxy-2-phenyl)acetyl]-4-(4-methylphenyl)thiosemicarbazide (3d)
Yield: 87%; m.p. 109–111°C; IR (ATR, cm−1): νmax 3479, 3244, 3130, 3033, 2952, 1692, 1644, 1594, 1505, 1449, 1414, 1366, 1170; 1H-NMR (300 Hz, DMSO-d6): δ 5.12 (1H, d, J = 2.4 Hz, CHOH), 6.12 (1H, br s, OH), 7.15 (2H, d, J = 8.1 Hz, Ar-H), 7.25–7.37 (5H, m, Ar-H), 7.48 (2H, d, J = 8.1 Hz, Ar-H), 9.24 (1H, br s, NH), 9.65 (1H, br s, NH), 10.28 (1H, s, NH); EIMS: m/z 297, 220, 208, 163, 148, 149, 121, 107, 91, 79, 77, 65.
1-[(2-Hydroxy-2-phenyl)acetyl]-4-(4-nitrophenyl)thiosemicarbazide (3e)
Yield: 79%; m.p. 174–175°C; IR (ATR, cm−1): νmax 3427, 3217, 3083, 2944, 1678, 1643, 1598, 1552, 1504, 1494, 1172; 1H-NMR (300 Hz, DMSO-d6): δ 5.13 (1H, s, CHOH), 6.18 (1H, br s, OH), 7.27–7.38 (3H, m, Ar-H), 7.49 (2H, d, J = 7.2 Hz, Ar-H), 7.87 (2H, d, J = 9.3 Hz, Ar-H), 8.23 (2H, dd, J = 9.0, 2.4 Hz, Ar-H), 9.69 (1H, br s, NH), 10.08 (1H, br s, NH), 10.38 (1H, s, NH); EIMS: m/z 208, 180, 148, 150, 134, 122, 107, 90, 79, 77.
General procedure for the synthesis of 1,3,4-oxadiazoles (4a–e)
The respective thiosemicarbazide (1.0 eq) was dissolved in methanol (10 mL) and mercuric acetate (1.1 eq) was added. The reaction mixture was refluxed for 2–3 h while completion of the reaction was monitored by TLC. The reaction mixture was filtered and the filtrate concentrated in vacuo. The resulting solid was recrystallized from aqueous ethanol.
5-(Hydroxybenzyl)-2-(4-bromophenyl)amino-1,3,4-oxadiazole (4a)
Yield: 79 %; m.p. 160–162°C; IR (ATR, cm−1): νmax 3425, 3061, 2926, 1581, 1493, 1486; 1H-NMR (300 Hz, CD3OD): δ 4.57 (1H, s, CHOH), 5.90 (1H, s, CHOH), 7.51–7.31 (9H, m, Ar-H); 13C-NMR (75 MHz, acetone-d6): δ 67.1, 113.8, 119.0, 126.3, 128.1, 128.4, 131.9, 138.2, 139.2, 160.1, 161.1; EIMS: m/z 345 [M+ •], 239, 196, 155, 133, 131, 107, 106, 105, 77.
5-(Hydroxybenzyl)-2-(4-chlorophenyl)amino-1,3,4-oxadiazole (4b)
Yield: 76 %; m.p. 166–168°C; IR (ATR, cm−1): νmax 3451, 3069, 3031, 2942, 1608, 1576, 1517, 1488, 1H-NMR (300 Hz, CD3OD): δ 4.56 (1H, s, CHOH), 5.89 (1H, s, OH), 7.51–7.26 (9H, m, Ar-H); 13C-NMR (75 MHz, acetone-d6): δ 67.1, 118.6, 126.3, 128.1, 128.2, 128.4, 128.9, 137.8, 139.5, 160.1, 161.1; EIMS: m/z 301 [M+ •], 195, 107, 106, 77, 69.
5-(Hydroxybenzyl)-2-(4-fluorophenyl)amino-1,3,4-oxadiazole (4c)
Yield: 79%; m.p. 175–177°C; IR (ATR, cm−1): νmax 3443, 3063, 3026, 2926, 1603, 1571, 1512, 1486, 1253; 1H-NMR (300 Hz, DMSO-d6): δ 4.73 (1H, d, J = 5.3 Hz, CHOH), 5.01 (1H, s, CHOH), 7.10–7.49 (9H, m, Ar-H), 9.62 (1H, s, NH); 13C-NMR (75 MHz, DMSO-d6): δ 72.8, 115.9 (d, J = 21.8 Hz), 120.6 (d, J = 7.5 Hz), 127.1, 128.1, 128.6, 132.1, 140.7, 158.1 (d, J = 237.0 Hz), 159.3, 161.7; EIMS: m/z 285 [M+ •], 272, 224, 194, 175, 149, 148, 136, 110, 105, 77.
5-(Hydroxybenzyl)-2-(4-methylphenyl)amino-1,3,4-oxadiazole (4d)
Yield: 81%; m.p. 172–173°C; IR (ATR, cm−1): νmax 3426, 3081, 3062, 2941, 1606, 1571,1508, 1489; 1H-NMR (300 Hz, CD3OD): δ 2.37 (3H, s, Ar-Me), 5.88 (1H, s, CHOH), 7.10 (2H, d, J = 8.3 Hz, Ar-H), 7.31-7.38 (5H, m, Ar-H) 7.49 (2H, d, J = 8.4 Hz, Ar-H); 13C-NMR (75 MHz, acetone-d6): δ 19.8, 67.1, 117.1, 126.3, 128.3, 128.3, 129.4, 131.3, 136.4, 139.7, 160.5, 160.7; EIMS: m/z 283 [M+ •], 281, 176, 148, 133, 132, 107, 106, 105, 91, 65.
5-(Hydroxybenzyl)-2-(4-nitrophenyl)amino-1,3,4-oxadiazole (4e)
Yield: 66%; m.p. 179–180°C; IR (ATR, cm−1): νmax 3260, 3105, 2930, 1627, 1595, 1557, 1515, 1508, 1306; 1H-NMR (300 MHz, DMSO-d6): δ 5.96 (1H, d, J = 4.8 Hz, CHOH), 6.73 (1H, d, J = 4.8 Hz, CHOH), 7.33–7.50 (5H, m, Ar-H), 7.73 (2H, d, J = 9.0 Hz, Ar-H), 8.26 (2H, d, J = 9.0 Hz, Ar-H), 11.40 (1H, s, NH); 13C-NMR (75 MHz, DMSO-d6): δ 66.6, 117.2, 126.0, 126.8, 128.6, 128.9, 139.9, 141.6, 145.2, 159.7, 162.1; EIMS: m/z 312 [M+ •], 206, 165, 133, 119, 107, 79, 77.
General procedure for the synthesis of 1,2,4-triazoles (5a–e)
The respective thiosemicarbazide (3a–e; 1.10 mmol) was added portion-wise to a stirred solution of NaOH (5%, 30 mL) and the reaction mixture heated under reflux for 4 h. After cooling, the reaction mixture was filtered and the filtrate acidified with 6 N HCl to pH 2–3. The precipitated solid was filtered, washed thoroughly with water, and recrystallized from EtOH/water.
5-(Hydroxybenzyl)-4-(4-bromophenyl)-2H-1,2,4-triazole-3-thione (5a)
Yield: 64%; m.p. 188–190°C; IR (ATR, cm−1): νmax 3128, 3027, 2937, 1578, 1497, 1488; 1H-NMR (300 Hz, CD3OD): δ 5.68 (1H, s, CHOH), 6.92 (2H, d, J = 8.6 Hz, Ar-H), 7.08 (2H, dd, J = 7.4, 5.4 Hz, Ar-H), 7.51 (2H, d, J = 8.6 Hz, Ar-H), 7.27–7.19 (3H, m, Ar-H); 13C-NMR (75 MHz, acetone-d6): δ 67.8, 124.4, 126.5, 128.0, 128.3, 131.2, 133.4, 134.8, 139.2, 155.3, 171.5; EIMS: m/z 359/361 [M+ •], 256, 255, 228, 213, 155, 132, 131, 107, 106, 105.
5-(Hydroxybenzyl)-4-(4-chlorophenyl)-2H-1,2,4-triazole-3-thione (5b)
Yield: 67%; m.p. 178–180°C; IR (ATR, cm−1) νmax 3125, 3022, 2934, 1557, 1489, 1485; 1H-NMR (300 Hz, CD3OD): δ 5.68 (1H, s, CHOH), 6.99 (2H, d, J = 8.7 Hz, Ar-H), 7.08 (2H, dd, J = 7.4, 5.2 Hz, Ar-H), 7.24–7.19 (5H, m, Ar-H); 13C-NMR (75 MHz, acetone-d6): δ 67.5, 126.4, 127.8, 128.0, 128.9, 130.7, 133.1, 134.5, 139.0, 153.5, 170.4; EIMS: m/z 315/317 [M+ •], 245, 210, 183, 169, 132, 107, 106, 105, 99.
5-(Hydroxybenzyl)-4-(4-fluorophenyl)-2H-1,2,4-triazole-3-thione (5c)
Yield: 63%; m.p. 82–85°C; IR (ATR, cm−1): νmax 3453, 3069, 3026, 2930, 1599, 1562, 1508, 1487, 1216; 1H-NMR (300 Hz, DMSO-d6): δ 5.59 (1H, d, J = 5.4 Hz, CHOH), 6.37 (1H, d, J = 5.4 Hz, CHOH), 7.13–7.31 (9H, m, Ar-H), 13.91 (1H, s, NH); 13C-NMR (75 MHz, DMSO-d6): δ 66.9, 116.3 (d, J = 23.5 Hz), 126.8, 128.1, 128.3, 130.4 (d, J = 3 Hz), 131.4 (d, J = 9.8 Hz), 139.7, 154.0, 162.5 (d, J = 244.5 Hz), 169.2; EIMS: m/z 301/303 [M+ •], 300, 284, 272, 224, 194, 167, 153, 136, 122, 109, 105, 77.
5-(Hydroxybenzyl)-4-(4-methylphenyl)-2H-1,2,4-triazole-3-thione (5d)
Yield: 63%; m.p. 82–85°C; IR (ATR, cm−1): νmax 3435, 3076, 3034, 2935, 1609, 1567, 1506, 1493, 1223; 1H-NMR (300 Hz, CD3OD): δ 2.37 (3H, s, Ar-Me), 5.59 (1H, s, CHOH), 6.88 (2H, d, J = 8.3 Hz, Ar-H), 7.07 (2H, dd, J = 7.6, 5.5 Hz), 7.17–7.22 (5H, m, Ar-H); 13C-NMR (75 MHz, acetone-d6): δ 20.3, 67.6, 122.0, 127.8, 128.5, 128.6, 129.4, 131.6, 139.2, 139.3, 153.9, 170.2; EIMS: m/z 297 [M+ •], 296, 295, 163, 150, 149, 132, 107, 106, 105, 91, 65.
5-(Hydroxybenzyl)-4-(4-nitrophenyl)-2H-1,2,4-triazole-3-thione (5e)
Yield: 64%; m.p. 122–123°C; IR (KBr, cm−1): νmax 3429, 3125, 3022, 2934, 1557, 1508, 1489, 1485, 1337; 1H-NMR (300 MHz, DMSO-d6): δ 5.93 (1H, d, J = 4.8 Hz, CHOH), 6.76 (1H, d, J = 4.8 Hz, CHOH), 7.33–7.50 (5H, m, Ar-H), 7.83 (2H, d, J = 9.0 Hz, Ar-H), 8.32 (2H, d, J = 9.0 Hz, Ar-H), 12.87 (1H, s, NH); 13C-NMR (75 MHz, DMSO-d6): δ 67.6, 116.4, 126.1, 126.7, 128.9, 129.1, 139.3, 141.9, 146.2, 162.7, 171.1; EIMS: m/z 328 [M+ •], 195, 180, 122, 107, 77.
Results and discussion
Synthesis
Mandelic acid was converted to its hydrazide via esterification. The hydrazide was treated with different isothiocyanates (2a–e) to afford thiosemicarbazides (3a–e). The 1,3,4-oxadiazoles (4a–e) and 1,2,4-triazoles (5a–e) were synthesized from thiosemicarbazides (3a–e) using different sets of reaction conditions. The 1,3,4-oxadiazoles were synthesized by cyclization of the thiosemicarbazides (3a–e) in the presence of mercuric acetateCitation14 and 1,2,4-triazoles by cyclodehydration under basic conditionsCitation15.
The IR spectra of thiosemicarbazides exhibited broad absorptions for NH groups in the region of 3485–3227 cm−1. A shift in carbonyl stretching was also observed, when compared to the parent hydrazide, along with the appearance of thiocarbonyl stretchings around 1170 cm−1.
In 1H-NMR spectra, formation of the product was confirmed by the appearance of two new doublets/multiplets in the aromatic region, corresponding to the para-disubstituted benzene ring in the molecule. The presence of three broad downfield singlets integrating to one proton each, assigned to the three NH protons, was strong evidence for the formation of thiosemicarbazides.
The syntheses of oxadiazoles (4a–e) and triazoles (5a–e) were indicated in the IR spectra by the appearance of weak bands in the region of 1620–1580 cm−1 for C=N stretchings, and also in that the strong carbonyl absorptions, observed for thiosemicarbazides, were absent. The disappearance of broad NH absorption bands, observed in the spectra of thiosemicarbazides in the region of 3480–3260 cm−1, also indicated cyclization into the respective nucleus.
In 1H-NMR spectra, the disappearance of three signals for NH protons confirmed the cyclization of thiosemicarbazides to the respective nucleus. A downfield singlet observed in the range of 5.90–5.59 ppm was assigned to the only CH proton in these compounds (4a–e, 5a–e). The signals for the aromatic protons were observed as three sets. The phenyl ring protons appeared as a multiplet integrating to five protons, and the signals for the p-disubstituted benzene ring were observed as two doublets. In 13C-NMR spectra, the carbon signal observed in the range of 63.6–67.6 ppm was assigned to the carbinol carbon (CHOH). The aromatic carbons were observed between 113 and 126 ppm. In the 13C-NMR spectra of triazoles (5a–e), the carbon signals in the range 169.7–170.4 ppm and 153.5–155.6 ppm were assigned to C-3 (C=S) and C-5 (C=N) of the triazole ring, respectively. The appearance of a carbon signal near 170 ppm for the thiocarbonyl group confirmed the existence of the triazole nucleus in the thione form. In the case of oxadiazoles (4a–e), the C-2 and C-5 (C=N) carbons were observed between 160.1 and 161.1 ppm. The mass spectra further confirmed formation of the desired nuclei. Molecular ion peaks were observed for all the compounds. In the case of chloro and bromo substituents, characteristic isotopic patterns were observed. Fragments corresponding to both aromatic rings were also observed along with fragments corresponding to cleavage of the oxadiazole and triazole moieties.
Urease inhibition studies
The synthesized compounds (4a–e, 5a–e) were tested for their in vitro urease inhibition activities at 0.2 mM concentration against jack bean urease. The urease inhibition activity was determined according to literature protocolsCitation8 using thiourea as the standard inhibitor, having an IC50 value of 21 ± 0.01 μM. The results are summarized in .
Table 1. Urease inhibition studies of oxadiazoles (4a–e) and triazoles (5a–e).
The tested compounds exhibited weak to moderate and excellent inhibition. The compounds 4d, 5b, and 5d were found to be more potent inhibitors than the standard, with IC50 = 16.1, 18.9, and 16.7 μM, respectively (compared to thiourea with IC50 = 21.0 μM). The most active compound was that with a methyl group in the aminophenyl part, in both 1,3,4-oxadiazole (4d) and 1,2,4-triazole (5d) nuclei. Compound 5b with a 4-chloro substituent in the aminophenyl part was also a stronger inhibitor than the standard, but its corresponding oxadiazole (4b) revealed no inhibition. The compounds with a p-fluoro substituent (4c, 5c) exhibited no observable activity.
It may be concluded from these results that triazolethiones (5a–e), in general, are more potent that their oxadiazole (4a–e) counterparts. This may be attributed to the substrate-like inhibition mechanismCitation10 where compounds 5a–e with N2–C(=S)–N4 have a structure similar to the natural substrate of the enzyme, i.e. ureaCitation8. It is supposed that these compounds (5a–e) bind to the enzyme active site. Thus, the enzyme fails to catalyze hydrolysis and the activity of the enzyme is retarded.
It may also be observed that compounds with an electron donating group, i.e. methyl (4d, 5d), are more potent inhibitors of urease, while those with strong electron withdrawing groups such as NO2 (4e, 5e) and F (4c, 5c) are either less potent or are not active. Compounds with bromo (5a) and chloro (5b) substituents are also strong inhibitors, while the corresponding fluoro substituted compound (5c) is devoid of any activity. The fluorine atom, being most electronegative, is a strong electron withdrawing group, while bromine and chlorine, being relatively less electronegative, may donate electrons mesomerically. This also augments the hypothesis that electron donor groups increase the inhibition activity, in contrast to strong electron withdrawing groups.
Conclusions
1,3,4-Oxadiazoles and 1,2,4-triazoles were synthesized from a common intermediate, i.e. thiosemicarbazides, and evaluated for urease inhibition. Three of the compounds, 4d, 5b, and 5d, exhibited excellent urease inhibition, with IC50 values lower than that of the standard. These compounds may be subjected to further structural modifications and investigations for the development of antiulcer drugs.
Declaration of interest
This work was financially supported by the Higher Education Commission (HEC) of Pakistan through project no. 20-674/R&D/06/1764, under the National Research Program for Universities.
References
- Al-Masoudi IA, Al-Soud YA, Al-Salihi NJ, Al-Masoudi NA. 1,2,4-Triazoles: synthetic approaches and pharmacological importance. Chem Heterocycl Compd 2006;42:1377–403.
- Torres HA, Hachem RY, Chemaly RF, Kontoyiannis DP, Raad II. Posaconazole: a broad spectrum triazole antifungal. Lancet Infect Dis 2005;5:775–85.
- Macaev F, Rusu G, Pogrebnoi S, Gudima A, Stingaci E, Vlad L, et al. Synthesis of novel 5-aryl-2-thio-1,3,4-oxadiazoles and the study of their structure–anti-mycobacterial activities. Bioorg Med Chem 2005;13:4842–50.
- Zarghi A, Tabatabai SA, Faizi M, Ahadian A, Navabi P, Zanganeh V, et al. Synthesis and anticonvulsant activity of new 2-substituted-5-(2-benzyloxyphenyl)-1,3,4-oxadiazoles. Bioorg Med Chem Lett 2005;15:1863–5.
- Rosa MDL, Kim HW, Gunic E, Jenket C, Boyle U, Koh Y, et al. Tri-substituted triazoles as potent non-nucleoside inhibitors of the HIV-1 reverse transcriptase. Bioorg Med Chem Lett 2006;16: 4444–9.
- Al-Soud YA, Al-Masoudi NA, Ferwanah Ael-R. Synthesis and properties of new substituted 1,2,4-triazoles: potential antitumor agents. Bioorg Med Chem 2003;11:1701–8.
- Lin R, Connolly PJ, Huang S, Wetter SK, Lu Y, Murray WV, et al. 1-Acyl-1H-(1,2,4)triazole-3,5-diamine analogues as novel and potent anticancer cyclin-dependent kinase inhibitors: synthesis and evaluation of biological activities. J Med Chem 2005;48:4208–11.
- Serwar M, Akhtar T, Hameed S, Khan KM. Synthesis, urease inhibition and antimicrobial activities of some chiral 5-aryl-4-phenylpropyl-1,2,4-triazoles. ARKIVOC2009;(vii):210–21.
- Akhtar T, Hameed S, Khan KM, Choudhary MI. Syntheses, urease inhibition, and antimicrobial studies of some chiral 3-substituted-4-amino-5-thioxo-1H,4H-1,2,4-triazoles. Med Chem 2008;4: 539–43.
- Amtul Z, Rasheed M, Choudhary MI, Rosanna S, Khan KM, Rahman A. Kinetics of novel competitive inhibitors of urease enzymes by a focused library of oxadiazoles/thiadiazoles and triazoles. Biochem Biophys Res Commun 2004;319:1053–63.
- Akhtar T, Hameed S, Al-Masoudi NA, Khan KM. Synthesis, and anti-HIV activity of new chiral 1,2,4-triazoles and 1,3,4-thiadiazoles. Heteroatom Chem 2007;18:316–22.
- Akhtar T, Hameed S, Al-Masoudi NA, Loddo R, La Colla P. In vitro antitumor and antiviral activities of new benzothiazole and 1,3,4-oxadiazole-2-thione derivatives. Acta Pharm 2008;58:135–49.
- Zahid M, Yasin KA, Akhtar T, Hameed S, Rama NH, Al-Masoudi NA, et al.Synthesis and in vitro antiproliferative activity of new adamantylthiazolyl-1,3,4-oxadiazoles. ARKIVOC 2009;(xi):85–93.
- Kidwai M, Bhushan KR. Microwave induced novel synthetic route to organomercurials. J Agric Food Chem 1999;52:114–117.
- Zamani K, Faghihi K, Sangi MR, Zolgharnein J. Synthesis of some new substituted 1,2,4-triazole and 1,3,4-thiadiazole and their derivatives. Turk J Chem 2003;27:119–125.