Abstract
A linear quantitative structure–activity relationship (QSAR) model is presented for the modelling and prediction for the interleukin-1 receptor associated kinase 4 (IRAK-4) inhibition activity of amides and imidazo[1,2-α] pyridines. The model was produced using the multiple linear regression (MLR) technique on a database that consisted of 65 recently discovered amides and imidazo[1,2- α] pyridines. Among the different constitutional, topological, geometrical, electrostatic and quantum-chemical descriptors that were considered as inputs to the model, seven variables were selected using the genetic algorithm subset selection method (GA). The accuracy of the proposed MLR model was illustrated using the following evaluation techniques: cross-validation, validation through an external test set, and Y-randomisation. The predictive ability of the model was found to be satisfactory and could be used for designing a similar group of compounds.
Introduction
The interleukin-1 receptor associated kinases (IRAKs) are a family of serine/threonine kinases involved in mediating cellular signalling downstream of the IL-1, IL-18 and a number of Toll-like receptors [Citation1]. IRAK-4 is critical for the activation of the intracellular signalling cascades, such as the NFКB and MAPK pathways, which are essential for the production of the inflammatory cytokines [Citation2]. It has been shown that mice lacking IRAK-4 are viable and show complete abrogation of inflammatory cytokine production in response to IL-1, IL-18 or LPS.3 similarly, human patients lacking IRAK-4 are severely immunocompromised and are not responsive to these cytokines [Citation3,Citation4]. The role of IRAK-4 in innate immunity makes it an interesting target for inhibition by small molecules [Citation5]. Recently, a novel series of amides and imidazo[1,2-α] pyridines as inhibitors of IRAK-4 have been reported by Buckley and co-workers. [Citation6–8].
The experimental measurement of the inhibition activity of chemicals is difficult, expensive and time-consuming, thus a great deal of effort has been put into attempting the estimation of activity through statistical modelling. Quantitative structure–activity relationship (QSAR) analysis is an effective method in research into rational drug design and the mechanism of drug actions. In addition, it is useful in areas like the design of virtual compound libraries and the computational-chemical optimisation of compounds. QSAR studies can express the biological activities of compounds as a function of their various structural parameters and also describes how the variation in biological activity depends on changes in the chemical structure [Citation9]. Recently, a QSAR study of biological activity has been published by our group [Citation10–12]. If such a relationship can be derived from the structure-activity data, the model equation allows medicinal chemists to say with some confidence which properties are important in the mechanism of drug action.
The success of a QSAR study depends on choosing robust statistical methods for producing the predictive model and also the relevant structural parameters for expressing the essential features within those chemical structures. Nowadays, genetic algorithms (GA) are well known as interesting and widely used methods for variable selection [Citation11,Citation13–19]. GA are stochastic methods used to solve the optimisation problems defined by the fitness criteria, applying the evolutionary hypothesis of Darwin and also different genetic functions i.e. crossover and mutation. In the present work, we have used a genetic algorithm for the variable selection, and developed an MLR model for the QSAR analysis of the IRAK-4 inhibitors.
In a QSAR study the model must be validated for its predictive value before it can be used to predict the response of additional chemicals. Validating QSAR with external data (i.e. data not used in the model development), although demanding, is the best method for validation [Citation20–21]. However the availability of an independent external validation set of several compounds is rare in QSAR. Thus, the input data set must be adequately split by experimental design or other splitting procedures into representative training and validation/test sets [Citation22–24]. In the present work, the data splitting was performed randomly and was confirmed by the factor spaces of the descriptors, as in our previous work [Citation10,Citation16–18,Citation25,Citation26]. Finally, the accuracy of the proposed model was illustrated using the following: leave one out, bootstrapping and external test set, cross-validations and Y-randomisation techniques.
Methodology
Data set
In this study, the data set of 65 amides and imidazo[1,2-α] pyridines were collected as IRAK-4 inhibitors, as previously reported [Citation6–8]. The inhibitory activity values are expressed as the half maximal inhibitory concentration (IC50). The chemical structures and activity data for the complete set of compounds are presented in . The activity data [IC50 (µM)] was converted to the logarithmic scale pIC50 [-log IC50 (M)] and then used for the subsequent QSAR analyses as the response variables. The data set was randomly divided into two subsets: the training set containing 52 compounds (80%) and the test set containing 13 compounds (20%). The training set was used to build a regression model, and the test set was used to evaluate the predictive ability of the model obtained.
Table 1. Chemical structures and the corresponding observed and predicted pIC50 values by the MLR method.
Molecular descriptor generation
All of the molecules were drawn into the HyperChem (Version 7.0 Hypercube, Alberta, Canada) software and pre-optimised using the MM+ molecular mechanics force field. Then a more precise optimisation was performed with the semi-empirical AM1 method in MOPAC [Citation27]. The molecular structures were optimised using the Polak–Ribiere algorithm until the root mean square gradient reached 0.01. The CODESSA [Citation28] and Dragon packages [Citation29] were used for calculating the molecular descriptors. The MOPAC output files were introduced to the CODESSA program to calculate two classes of the descriptors: electrostatic (minimum and maximum partial charges, polarity parameter, charged partial surface area descriptors etc.), and quantum chemical (reactivity indices, dipole moment, HOMO and LUMO energies etc.). The molecular structures were saved by the HIN extension and entered on the DRAGON software for the calculation of the 18 different types of theoretical descriptors for each molecule. They included (a) 0D-constitutional (atom and group counts); (b) 1D-functional groups, 1D-atom centered fragments; (c) 2D-topological, 2DBCUTs, 2D-walk and path counts, 2D-autocorrelations, 2D-connectivity indices, 2D-information indices, 2D-topological charge indices, and 2D-eigenvalue-based indices; and (d) 3D-Randic molecular profiles from the geometry matrix, 3D-geometrical, 3D-WHIM, and 3D-GETAWAY descriptors. These descriptors could represent a variety of aspects of the compounds, and have been successfully used in various QSAR and quantitative structure-property relationship (QSPR) research. Any descriptors with a constant or almost constant value for all the molecules were eliminated. Also, any pairs of variables with a correlation coefficient greater than 0.90 were classified as inter-correlated, and only one of them was considered in developing the model. A total of 557 descriptors were considered for further investigations after discarding the descriptors with constant values and the ones that were inter-correlated.
Genetic algorithm
Genetic algorithms (GAs) are governed by biological evolution rules [Citation30]. These are stochastic optimisation methods that have been inspired by evolutionary principles. The distinctive aspect of a GA is that it investigates many possible solutions simultaneously, each of which explores a different region in the parameter of space [Citation31]. To select the most relevant descriptors, the evolution of the population was simulated [Citation32–34]. The first generation population was randomly selected; each individual member in the population was defined by a chromosome of binary values and represented a subset of descriptors. The number of the genes at each chromosome was equal to the number of the descriptors. A gene was given the value of 1, if its corresponding descriptor was included in the subset; otherwise, it was given the value of zero. The number of the genes with the value of 1 was kept relatively low to have a small subset of descriptors [Citation35]. As a result, the probability of generating 0 for a gene was set greater (at least 60 %) than the value of 1. The operators used here were the crossover and mutation operators. The application probability of these operators was varied linearly with a generation renewal (0–0.1 % for mutation and 60–90 % for crossover). The population size was varied between 50 and 250 for the different GA runs. For a typical run, the evolution of the generation was stopped when 90% of the generations took the same fitness. The fitness function used here was the leave-one-out cross-validated correlation coefficient, Q2LOO. The GA program was written in Matlab 6.5 [Citation36].
Results and discussion
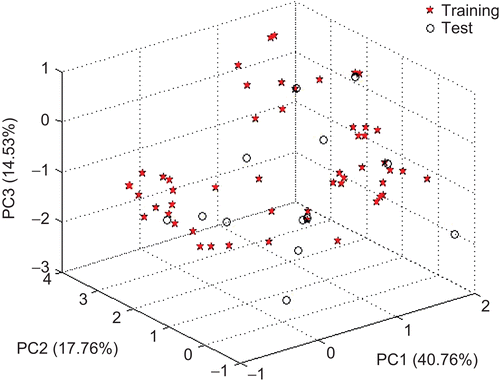
The diversity of the training set and the test set was analysed using the principal component analysis (PCA) method. The PCA was performed with the calculated structure descriptors for the whole data set to detect the homogeneities in the data set, and also to show the spatial location of the samples to assist the separation of the data into the training and test sets. The PCA results showed that three principal components (PC1, PC2, and PC3) described 73.05% of the overall variables, as follows: PC1 = 40.76%, PC2 = 17.76% and PC3 = 14.53%. Since almost all the variables can be accounted for by the first three PCs, their score plot is a reliable representation of the spatial distribution of the points for the data set. The plot of PC1, PC2, and PC3 displayed the distribution of compounds over the first three principal components space (). This figure shows that the samples in both the training and the test sets seemed to be evenly scattered in the 3D space, and therefore it was feasible to split the data set. Moreover, the compounds in the training set were representative of the whole data.
Figure 1. The principal component analysis of the training and test sets.
After analysing splitting the data set into the training set and test set, the next step was to select the main factors which were the most important for the IRAK-4 inhibition activity of amides and imidazo[1,2-α] pyridines. As we do not yet know which descriptors, and which particular combinations, are related to the studied response and can be used in the predictive models, we applied genetic algorithms as the variable selection procedure to select only the best combinations (most relevant) for obtaining the models with the highest predictive power by using the training set. The seven most significant descriptors according to the GA-MLR algorithm are: the maximum atomic orbital electronic population (MAOEP), path/walk 5 - Randic shape index (PW5), the molecular walk count of the order 09 (MWC09), Mor12m, G2p, R4e and nNHR.
The multi-collinearity between the above seven descriptors were detected by calculating their variation inflation factors (VIF), which can be calculated as follows:
where r is the correlation coefficient of the multiple regression between the variables in the model. If VIF equals to 1, then no inter-correlation exists for each variable; if VIF falls into the range of 1–5, the related model is acceptable; and if VIF is larger than 10, the related model is unstable and a recheck is necessary [Citation37]. The corresponding VIF values of the seven descriptors are shown in . As can be seen from this table, most of the variables had VIF values of less than 5, indicating that the obtained model has statistic significance.
Table 2. The linear model based on the seven parameters selected by the GA-MLR method.
To examine the relative importance as well as the contribution of each descriptor in the model, the value of the mean effect (MF) was calculated for each descriptor. This calculation was performed with the equation below:
Where MFj represents the mean effect for the considered descriptor j, βj is the coefficient of the descriptor j, dij stands for the value of the target descriptors for each molecule and, eventually, m is the descriptors number for the model. The MF value indicates the relative importance of a descriptor, compared with the other descriptors in the model. Its sign indicates the variation direction in the values of the activities as a result of the increase (or reduction) of the descriptor values. The mean effect values are shown in .
In a QSAR study, generally, the quality of a model is expressed by its fitting ability and prediction ability, and of these the prediction ability is the more important. In order to build and test the model, a data set of 65 compounds was separated into a training set of 52 compounds, which were used to build the model and a test set of 13 compounds, which were applied to test the built model. With the selected descriptors, we have built a linear model using the training set data, and the following equation was obtained:
Ntrain= 52, R2train= 0.852, RMSEtrain= 0.418, F = 36.083, Q2LOO = 0.804, Q2BOOT = 0.785, Q2ext = 0.747, Ntest = 13, R2test = 0.759, RMSEtest = 0.506
In this equation, N is the number of compounds, R2 is the squared correlation coefficient, Q2LOO, Q2BOOT and Q2ext are the squared cross-validation coefficients for leave one out, bootstrapping and external test set respectively, RMSE is the root mean square error and F is the Fisher F statistic. The figures in parentheses are the standard deviations.
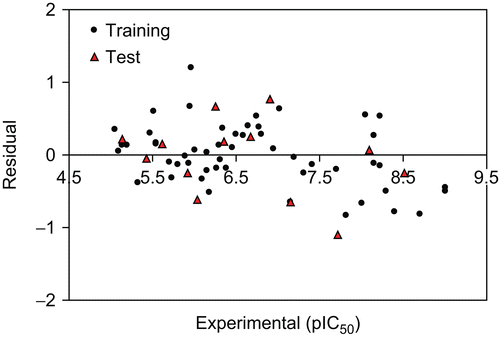
The built model was used to predict the test set data and the prediction results are given in . As can be seen from , the calculated values for the pIC50 are in good agreement with those of the experimental values. The predicted values for pIC50 for the compounds in the training and test sets using equation 1 were plotted against the experimental pIC50 values in . A plot of the residual for the predicted values of pIC50 for both the training and test sets against the experimental pIC50values are shown in . As can be seen the model did not show any proportional and systematic error, because the propagation of the residuals on both sides of zero are random.
Figure 2. The predicted versus the experimental pIC50 by GA-MLR.
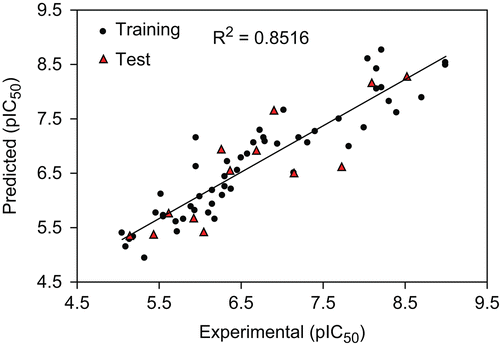
Figure 3. The residual versus the experimental pIC50 by GA-MLR.
The real usefulness of QSAR models is not just their ability to reproduce known data, verified by their fitting power (R2), but is mainly their potential for predictive application. For this reason the model calculations were performed by maximising the explained variance in prediction, verified by the leave-one-out cross-validated correlation coefficient, Q2LOO. To avoid the danger of overfitting and the possibility of overestimating the model predictivity by using Q2LOO, and Q2ext, the internal predictive ability of the models was also verified using the bootstrap Q2BOOT procedure, as is strongly recommended for QSAR modeling. The robustness of the proposed models and their predictive ability was guaranteed by the high Q2BOOT based on the bootstrapping being repeated 5000 times. The Q2LOO, Q2ext and Q2BOOT for the MLR model are shown in Equation 2. This indicates that the obtained regression model has a good internal and external predictive power.
Also, in order to assess the robustness of the model, the Y-randomisation test was applied in this study [Citation20–21]. The dependent variable vector (pIC50) was randomly shuffled and a new QSAR model developed using the original independent variable matrix. The new QSAR models (after several repetitions) would be expected to have low R2 and Q2LOO values (). If the opposite happens then an acceptable QSAR model cannot be obtained for the specific modelling method and data.
Table 3. The R2train and Q2LOO values after several Y-randomisation tests.
Applicability domain
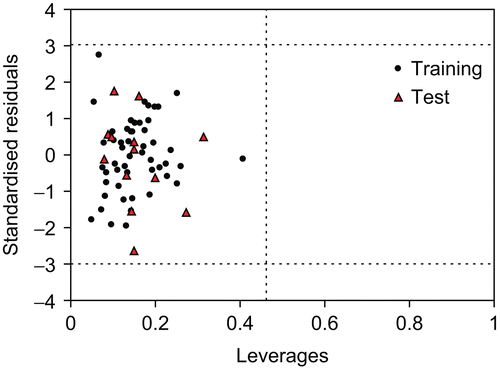
The Williams plot (), the plot of the standardised residuals versus the leverage, was exploited to visualise the applicability domain [Citation38]. The leverage indicates a compound’s distance from the centroid of X. The leverage of a compound in the original variable space is defined as [Citation39]:
Figure 4. The William plot of the GA-MLR model.
Where xi is the descriptor vector of the considered compound and X is the descriptor matrix derived from the training set descriptor values. The warning leverage (h*) is defined as [Citation40]:
Where n is the number of training compounds, p is the number of predictor variables. A compound with hi > h* seriously influences the regression performance, but it doesn’t appear to be an outlier because its standardised residual may be small, even though it has been excluded from the applicability domain. Moreover, a value of three for the standardised residual is commonly used as a cut-off value for accepting predictions, because points that lie ± 3 standardised residuals from the mean will cover 99% of normally distributed data [Citation41]. Thus the leverage and the standardised residual were combined for the characterisation of the applicability domain. From , it is obvious that there are no outlier compounds with standard residuals >3δ for both the training and test sets. Also all the chemicals have a leverage lower than the warning h* value of 0.462.
Interpretation of descriptors
As well as demonstrating statistical significance, QSAR models should also provide useful chemical insights into the mechanism of inhibitory activity. For this reason, an acceptable interpretation of the QSAR results is provided below. By interpreting the descriptors contained in the model, it is possible to gain some insights into factors which are related to the IRAK-4 inhibitor activity and brief descriptions of the descriptors are in .
From the seven selected descriptors only the MAOEP descriptor, as calculated by the CODESSA software, appears in the model. The MAOEP which belong to the quantum chemical descriptors is a simplified index to describe the nucleophilicity of molecules. As it is apparent from , the MAOEP mean effect has a negative sign, illustrating a greater mean effect value than that of the other descriptors. Therefore, this descriptor had a significant effect on the IRAK-4 inhibition activity of the studied compounds. The negative sign suggests that the pIC50 value is inversely related to this descriptor. Subsequently, the increase in the nucleophilicity of the molecule results in a decrease in its pIC50.
The second descriptor is the path/walk 5 Randic shape index (PW5), which is one of the topological descriptors. The atomic path/walk indices are defined for each atom as the ratio between the atomic path count and the atomic walk count of the same length. Whereas the number of paths in a molecule is bounded and determined by the molecule’s diameter, the number of walks is unbounded. However, being interested only in quotients, the walk count is terminated when it exceeds the maximum allowed length of the corresponding path [Citation42]. The molecular path/walk indices are defined as the average sum of atomic path/walk indices of equal length. As the path/walk count ratio is independent of molecular size, these descriptors can be considered as shape descriptors. As is apparent from , the PW5 mean effect has a negative sign which indicates that the pIC50 is inversely related to this descriptor; therefore, increasing the PW5 of molecules leads to a decrease in its pIC50 values.
The molecular walk count of the order 09 (MWC09) is the third descriptor, appearing in the model. It is one of the molecular walk counts descriptors. Walk counts are atomic and molecular descriptors obtained from an H-depleted molecular graph. The molecular walk count is related to molecular brunching and size and in general to the molecular complexity of the graph [Citation42]. The MWC09 displays a negative sign, which indicates that the activity of the compounds is inversely related to the complexity of the molecules.
Mor12m is the forth descriptor, appearing in the model. It is one of the 3D-molecule representations of structures based on electron diffraction (3D-MoRSE) descriptors. The 3D-MoRSE descriptors are derived from infrared spectral simulation using a generalised scattering function [Citation42]. This descriptor was proposed as signal 12/weighted by the atomic masses which relates to the atomic masses of the molecule. The Mor12m displays a positive sign, which indicates that the pIC50 is directly related to this descriptor.
The 2st component symmetry directional WHIM index/weighted by atomic polarisabilities (G2p) is the fifth descriptor appearing in the model. It is one of the WHIM descriptors which are based on the statistical indices calculated on the projections of atoms along the principal axes. The algorithm consists of performing a principal components analysis on the centred Cartesian coordinates of a molecule by using a weighted covariance matrix obtained from different weighing schemes for the atoms. Directional WHIM symmetry descriptors are related to the number of central symmetric atoms (along the mth component), the number of unsymmetric atoms and the total number of atoms of the molecule [Citation42]. The atomic polarisabilities are one of the weighting schemes that are used for computing the weighted covariance matrix in this descriptor (G2p). The G2p mean effect has a positive sign which indicates that pIC50 is directly related to this descriptor; therefore, decreasing the G2p of molecules leads to decrease in its pIC50 values.
The sixth descriptor of the GA-MLR model was the R autocorrelation of lag 4/weighted by the atomic Sanderson electronegativities (R4e). This descriptor is a GETAWAY type and is related to the electronegativity of the atoms in the molecule. This descriptor displays a positive sign, which indicates that the pIC50 is directly related to the electronegativity of molecules.
The final descriptor is nNHR, which is the number of secondary amines. The nNHR mean effect demonstrates a positive sign, revealing that the IRAK-4 inhibition activity is directly related to the number of secondary amines in molecule.
Summarising, it can be concluded that nucleophilicity, molecular size, molecular complexity, atomic mass, atomic polarisability, atomic electronegativity and the number of secondary amines, all play an important role in the IRAK-4 inhibition activity of compounds.
Conclusion
In this article, a QSAR study of 65 IRAK-4 inhibitors was performed based on the theoretical molecular descriptors calculated by the DRAGON and CODESSA software and selected by genetic algorithm. The built model was assessed comprehensively (internal and external validation) and all the validations indicated that the QSAR model built was robust and satisfactory, and that the selected descriptors could account for the structural features responsible for the IRAK-4 inhibition activity of the compounds. By interpreting the molecular descriptors in the regression model, we can conclude that the activity of the studied compounds mainly depends on nucleophilicity, molecular size, molecular complexity, atomic mass, atomic polarisability, atomic electronegativity and the number of secondary amines. The QSAR model developed in this study can provide a useful tool to predict the activity of new compounds and also to design new compounds with high activity.
Acknowledgements
We gratefully acknowledge generous allocations of computing from the Institute of Petroleum Engineering, University of Tehran for Advanced Computing and Supercomputing Facilities.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Janssens S, Beyaert R. Functional diversity and regulation of different interleukin-1 receptor-associated kinase (IRAK) family members. Mol Cell 2003;11:293–302.
- Li S, Strelow A, Fontana EJ, Wesche H. IRAK-4: A novel member of the IRAK family with the properties of an IRAK-kinase. Proc Natl Acad Sci USA 2002;99:5567–5572.
- Medvedev AE, Lentschat A, Kuhns DB, Blanco JC, Salkowski C, Zhang S, Arditi M, Gallin JI, Vogel SN. Distinct mutations in IRAK-4 confer hyporesponsiveness to lipopolysaccharide and interleukin-1 in a patient with recurrent bacterial infections. J Exp Med 2003;198:521–531.
- Picard C, Puel A, Bonnet M, Ku CL, Bustamante J, Yang K, Soudais C, Dupuis S, Feinberg J, Fieschi C, Elbim C, Hitchcock R, Lammas D, Davies G, Al- Ghonaium A, Al-Rayes H, Al-Jumaah S, Al-Hajjar S, Al-Mohsen IZ, Frayha HH, Rucker R, Hawn TR, Aderem A, Tufenkeji H, Haraguchi S, Day NK, Good RA, Gougerot-Pocidalo MA, Cassanova JL. Pyogenic bacterial infections in humans with IRAK-4 deficiency. Science 2003;299:2076–2079.
- Li X. IRAK4 in TLR/IL-1R signaling: possible clinical applications. Eur J Immunol 2008;38:614–618.
- Buckley GM, Ceska TA, Fraser JL, Gowers L, Groom CR, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, Sabin V. IRAK-4 inhibitors. Part II: A structure-based assessment of imidazo[1,2-a]pyridine binding. Bioorg Med Chem Lett 2008;18:3291–3295.
- Buckley GM, Fosbeary R, Fraser JL, Gowers L, Higueruelo AP, James LA, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, Sabin V. IRAK-4 inhibitors. Part III: A series of imidazo[1,2-a]pyridines. Bioorg Med Chem Lett 2008;18:3656–3660.
- Buckley GM, Gowers L, Higueruelo AP, Jenkins K, Mack SR, Morgan T, Parry DM, Pitt WR, Rausch O, Richard MD, Sabin V, Fraser JL. IRAK-4 inhibitors. Part 1: a series of amides. Bioorg Med Chem Lett 2008;18:3211–3214.
- Sammes PG, Taylor JB. Comprehensive Medicinal Chemistry. Oxford: Pergamon Press, 1990:766.
- Riahi S, Pourbasheer E, Dinarvand R, Ganjali MR, Norouzi P. Quantitative structure-activity relationship study on the anti-HIV-1 activity of novel 6-naphthylthio HEPT analogs. Chem Biol Drug Des 2008;74:165–172.
- Riahi S, Pourbasheer E, Ganjali MR, Norouzi P. Investigation of different linear and nonlinear chemometric methods for modeling of retention index of essential oil components: Concerns to support vector machine. J Hazard Mater 2009;166:853–859.
- Riahi S, Pourbasheer E, Ganjali MR, Norouzi P. Support vector machine-based quantitative structure-activity relationship study of cholesteryl ester transfer protein inhibitors. Chem Biol Drug Des 2009;73:558–571.
- Depczynski U, Frost VJ, Molt K. Genetic algorithms applied to the selection of factors in principal component regression. Anal Chim Acta 2000;420:217.
- Alsberg BK, Marchand-Geneste N, King RD. A new 3D molecular structure representation using quantum topology with application to structure-property relationships. Chemometr Intel Lab 2000;54:75–91.
- Jouanrimbaud D, Massart DL, Leardi R, Denoord OE. Genetic algorithms as a tool for wavelength selection in multivariate calibration. Anal Chem 1995;67:4295–4301.
- Riahi S, Ganjali MR, E Pourbasheer Divsar, F, Norouzi P, Chaloosi M. Development and validation of a rapid chemometrics-assisted spectrophotometry and liquid chromatography methods for the simultaneous determination of the phenylalanine, tryptophan and tyrosine in the pharmaceutical products. Curr Pharm Anal 2008;4:231–237.
- Riahi S, Ganjali MR, Pourbasheer E, Norouzi P. QSRR study of GC retention indices of essential-oil compounds by multiple linear regression with a genetic algorithm. Chromatographia 2008;67:917–922.
- Riahi S, Pourbasheer E, Ganjali MR, Norouzi P, Zeraatkar Moghaddam A. QSPR study of the distribution coefficient property for hydantoin and 5-arylidene derivatives. A genetic algorithm application for the variable selection in the MLR and PLS methods. J Chin Chem Soc 2008;55:1086–1093.
- Riahi S, Ganjali MR, Moghaddam AB, Pourbasheer E, Norouzi P. Development of a new combined chemometrics method, applied in the simultaneous voltammetric determination of cinnamic acid and 3, 4-dihydroxy benzoic acid. Curr Anal Chem 2009;5:42–47.
- Tropsha A, Gramatica P, Gombar VK. The importance of being earnest: validation is the absolute essential for successful application and interpretation of QSPR models. QSAR Comb Sci 2003;22:69–77.
- Riahi S, Ganjali MR, Norouzi P, Jafari F. Application of GA-MLR, GA-PLS and the DFT quantum mechanical (QM) calculations for the prediction of the selectivity coefficients of a histamine-selective electrode. Sens. Actuators B 2008;132:13–19.
- Eriksson L, Johansson E, Muller M, Wold S. On the selection of the training set in environmental QSAR analysis when compounds are clustered. J Chemometr 2000;14:599–616.
- Golbraikh A, Shen M, Xiao Z, Xiao Y-D Lee, K-H, Tropsha A. Rational selection of training and test sets for the development of validated QSAR models. J Comput Aided Mol Des 2003;17:241–253.
- Gramatica P, Pilutti P, Papa E. Validated QSAR prediction of OH tropospheric degradability: splitting into training-test set and consensus modeling. 2004;44:1794–1802.
- Riahi S, Pourbasheer E, Dinarvand R, Ganjali MR, Norouzi P. Exploring QSARs for antiviral activity of 4-alkylamino-6-(2-hydroxyethyl)-2-methylthiopyrimidines by support vector machine. Chem Biol Drug Des 2008;72:205–216.
- Riahi S, Pourbasheer E, Dinarvand R, Ganjali MR, Norouzi P. QSAR Study of 2-(1-Propylpiperidin-4-yl)-1H-Benzimidazole-4-Carboxamide as PARP Inhibitors for Treatment of Cancer. Chem Biol Drug Des 2008;72:575–584.
- Stewart JPP. MOPAC 6.0: Quantum Chemistry Program Exchange QCPE. No. 455. Bloomington, IN:Indiana University, 1989;250–260.
- Katritzky AR. http://www.codessa-pro.com.
- Todeschini R, Consonni V, Pavana M. http://www.disat.unimib.it/chm/.
- Holland H. Adaption in Natural and Artificial Systems. Ann Arbor, MI: The University of Michigan, 1975;342–375.
- Cartwright HM. Applications of Artificial Intelligence in Chemistry. Oxford: Oxford University, 1993;760–765.
- Hunger J, Huttner G. Optimization and analysis of force field parameters by combination of genetic algorithms and neural networks. J Comput Chem 1999;20:455–471.
- Ahmad S, Gromiha MM. Design and training of a neural network for predicting the solvent accessibility of proteins. J Comput Chem 2003;24:1313–1320.
- Waller CL, Bradley MP. Development and validation of a novel variable selection technique with application to multidimensional quantitative structure-activity relationship studies. J Chem Inf Comput Sci 1999;39:345–355.
- Aires-de-Sousa J, Hemmer MC, Casteiger J. Prediction of H-1 NMR chemical shifts using neural networks. Anal Chem 2002;74:80–90.
- The Mathworks. Genetic Algorithm and Direct Search Toolbox Users Guide. Massachusetts: MathWorks, 2002;50–65.
- Agrawal VK, Khadikar PV. QSAR prediction of toxicity of nitrobenzenes. Bioorg Med Chem 2001;9:3035–3040.
- OECD. Guidance Document on the Validation of (Quantitative) Structure–Activity Relationships [(Q)SAR] Models. Paris: Organisation for Economic Co-Operation and Development, 2007;256–278.
- Netzeva TI, Worth AP, Aldenberg T, Benigni R, Cronin MTD, Gramatica P, Jaworska JS, Kahn S, Klopman G, CA Marchant Myatt, G, Nikolova- Jeliazkova N, Patlewicz GY, Perkins R, Roberts DW, Schultz TW, Stanton DT, van de Sandt JJM, Tong W, Veith G, Yang C. Current status of methods for defining the applicability domain of (quantitative) structure-activity relationships. The report and recommendations of ECVAM Workshop 52. ATLA-Altern Lab Anim 2005;33:155–173.
- Eriksson L, Jaworska J, Worth AP, Cronin MTD, McDowell RM, Gramatica P. Methods for reliability and uncertainty assessment and for applicability evaluations of classification- and regression-based QSARs. Environ Health Perspect 2003;111:1361–1375.
- Jaworska JS, Nikolova JN, Aldenberg T. QSAR applicability domain estimation by projection of the training set in descriptor space: A review. ATLA-Altern Lab Anim 2005;33:445–459.
- Todeschini R, Consonni V. Handbook of Molecular Descriptors. Weinheim, Germany: Wiley-VCH, 2000;1–667.