Abstract
A series of quinoxalin-2-carboxamides were designed as per the pharmacophoric requirements of 5-HT3 receptor antagonists and synthesized by condensing the carboxylic group of quinoxalin-2-carboxylic acid with various amines in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride and 1-hydroxybenzotriazole. The structures of the synthesized compounds were confirmed by physical and spectroscopic data. The carboxamides were evaluated for their 5-HT3 receptor antagonisms in longitudinal muscle-myenteric plexus preparation from guinea pig ileum against 5-HT3 agonist, 2-methy-5-HT. All the synthesized compounds showed 5-HT3 receptor antagonism, (4-benzylpiperazin-1-yl)(quinoxalin-2-yl)methanone was the most potent compound among this series.
Introduction
Nausea and vomiting are the most common adverse effects of the existing chemotherapeutic agents and these side-effects occur in a substantial number of patients whose undergoing surgery or radiation therapyCitation1–4. Although nausea and vomiting seems like harmless side-effects, inadequately controlled emesis can result in serious dehydration, loss of sodium and other electrolytes, nutritional depletion and loss of appetite, weight loss, oesophageal tears, deterioration of patient’s physical, and mental status (anxiety and depression) which prompt them into non-adherence to the life saving anticancer treatmentCitation5–6. The recent incorporation of supportive treatment, that is 5-HT3 antagonists substantially increase the patience compliance of anticancer treatmentCitation3 by decreasing their side-effects (nausea and vomiting), but this 5-HT3 receptor antagonists are ineffective in 10–30% of patientsCitation7–8 and they possess chiral centres, which increases the synthetic cost of these drugsCitation9 and further only a very few selective 5-HT3 antagonist are availableCitation10. With this information one may conclude the need of newer 5-HT3 antagonists. Several new chemical entities for 5-HT3 receptor antagonists have been reported thusCitation10–13 based on the Hibert et al. pharmacophore modelCitation14, which consists of an aromatic ring, a linking carbonyl group and a basic nitrogen centre at specific distances (). In our previous studiesCitation10,Citation13, we evaluated various 3-substituted quinoxalin-2-carboxamides (consists of mannich base as linking unit for piperazine moiety and quinoxaline nucleus) as 5-HT3 receptor antagonists, unfortunately none of the compound showed antagonism greater than or equal to 5-HT3 receptor antagonist, ondansetron (). So in the present study to develop new 5-HT3 receptor antagonists without chiral centre, our attempts were concentrated on developing quinoxlin-2-carboxamides with devoid of mannich base and no substituent at third position of quinoxaline nucleus as potential 5-HT3 receptor antagonists.
Figure 1. Pharmacophore of 5-HT3 receptor antagonists.
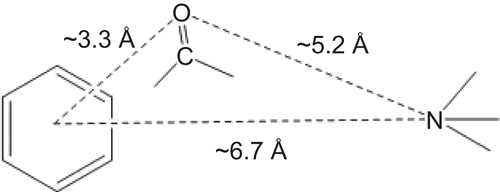
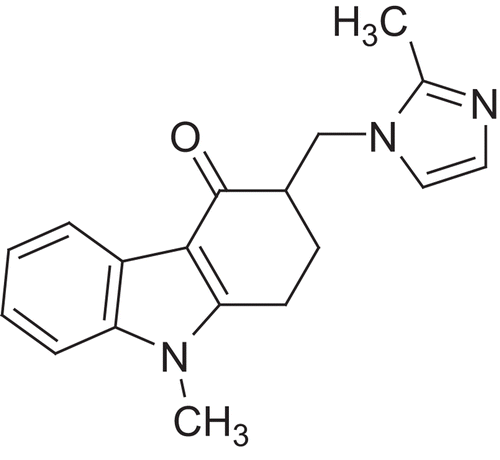
Figure 2. Ondansetron.
Experimental
Chemistry (general)
All the chemicals and reagents were obtained from Spectrochem Pvt. Ltd. (Mumbai, Maharashtra, India), S.D. Fine Chem Limited (Mumbai, Maharashtra, India) and Sigma–Aldrich (St. Louis, MO). Solvents used for reactions were dried over molecular sieves (4Å). Reactions were monitored by thin-layer-chromatography (TLC), which was performed with 0.2 mm Merck pre-coated silica gel 60 F254 aluminum sheets. Compounds were detected by ultraviolet, iodine chamber, and by dipping the TLC plates in ethanolic solution of ninhydrin and heating. Melting points (uncorrected) were determined on Buchi 530 melting point apparatus. Infrared (IR) spectra (vmax in cm−1) were recorded on a Jasco IR Report-100 IR spectrophotometer and IR Prestige-21 Fourier transform (FT)-IR spectrophotometer, proton nuclear magnetic resonance (1H NMR) spectra were recorded at 400 MHz on a Bruker Avance-II, FT-NMR spectrometer using tetramethylsilane as internal standard (chemical shifts in δ, ppm). Mass spectra were obtained on a VG 70–250S instrument using electron spray ionization technique (positive and negative mode).
Synthesis of 2-(1,2,3,4-tetrahydroxybutyl)quinoxaline (2)
o-Phenylenediamine (45 g, 0.416 mol) was stirred with 10% aqueous acetic acid for 30 min in 1 L, two-necked round-bottom flask. To the above reaction mass, d-fructose (75 g, 0.416 mol) was added and the mixture heated to 80°C and temperature maintained for 18 h. The reaction mixture was cooled, filtered through Whatmann filter paper using Buchner funnel. The obtained solids were washed with water (2 × 100 mL) and dried under vacuum oven overnight. The target product was obtained in a 45% yield, mp: 180–184°C (Lit 181–184°C); 1H NMR [dimethyl sulfoxide (DMSO)-d6] 9.19 (s, 1H), 8.20 (m, 2H), 8.05–8.11 (m, 2H), 5.63 (d, 2H),5.17 (d, 1H), 4.74 (d, 1H), 4.64 (d, 1H), 4.42 (t, 1H), 3.65–3.72 (m, 3H); IR (KBr) cm−1: 3200 (broad O-H str.) 3040 (aromatic C-H str.), 1570, 1490 (C=C, C=N ring str.), 1100, 1040 (C-O str.).
Synthesis of quinoxalin-2-carboxylic acid (3)
Sodium hydroxide (155.4 g, 3.88 mol) was taken in a 1 L three-necked round-bottom flask equipped with thermometer, reflux condenser, and pressure equalizing dropping funnel and to this water (1575 mL) was added, once the solution temperature reached 40°C, 2-(1,2,3,4-tetrahydroxybutyl)quinoxaline (70.0 g, 280 mmol) was added and stirred for 45 min. To the reaction mixture, 30% hydrogen peroxide (130 mL) was added slowly over the period of 15 min and heated to 60°C for 3 h, the remainder 30% hydrogen peroxide (115 mL) was added in portions for 30 min and stirred for 45 min. The mass was brought to ambient temperature and allowed to stand for 3 h, filtered through Whatmann filter paper using Buchner funnel. The solids were acidified with concentrated hydrochloric acid; the obtained off-white solids were filtered, washed with water and dried in a vacuum oven overnight. The desired acid was obtained in 50% yield, mp: 210–212°C (Lit 209–210°C); 1H NMR (CDCl3) (δ) ppm: 9.54 (s, 1H, quinoxaline), 8.29 (m, 1H, quinoxaline), 8.19 (m, 1H, quinoxaline), 7.95 (m, 2H, quinoxaline); IR (KBr) cm−1: 3440 (broad O-H str.COOH), 3040 (aromatic C-H str.), 1690 (C=O str.) 1570, 1490 (C=C, C=N ring str.), 1300, 1100, (C-O str.).; mass spectra [electrospray ionization (ESI)] of the compound exhibited the molecular ion peak at m/z 174.1 (M+).
General procedure for the synthesis of quinoxaline-2-carboxamides(4a–p and 5a–5d)
Quinoxalin-2-carboxylic acid, 0.5 g (2.87 mmol), and 2 equivalent of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (1.10 g, 5.75 mmol) were taken a 100 mL round-bottom flask and stirred with dry tetrahydrofuran in an inert atmosphere (nitrogen) at 0°C for 15 min. To the above reaction mixture, 1-hydroxybenzotriazole (HOBt) (0.88 g, 5.74 mmol) was added and stirred for another 45 min. To the above mixture one equivalent of amine (alkyl/aryl-substituted-piperazines, N,N-dimethylethylenediamine, tryptamine, N,N-diethyl-p-phenylnedaimine and 3-aminopyridine) was added, and stirring continued for overnight. The reaction mixture was concentrated under reduced pressure, the resultant mass, diluted with dichloromethane, washed with aqueous 10% sodium bicarbonate (2 × 50 mL), saturated sodium chloride solution (2 × 50 mL) and dried over anhydrous sodium sulphate. This was then evaporated under reduced pressure to afford the desired compound in 40–80% yield. Results are summarized in the .
Table 1. Physical constants and pharmacological data of quinoxalin-2-carboxamides.
(4-Phenylpiperazin-1-yl)(quinoxalin-2-yl)methanone (4a)
Yield: 70%; mp: 128–130°C; 1H NMR (DMSO-d6) (δ) ppm: 3.26 (t, 2H, NCH2, piperazine), 3.34 (t, 2H, NCH2, piperazine), 3.95 (t, 2H, NCH2, piperazine), 4.04 (t, 2H, NCH2, piperazine), 6.90 (m, 3H, phenyl), 7.26 (m, 2H, phenyl), 7.81 (m, 2H, quinoxaline), 8.10 (m, 1H, quinoxaline), 8.15 (m, 1H, quinoxaline), 9.22 (s, 1H, quinoxaline); IR (KBr, cm−1):3040, 3000, (C-H), 1650 (C=O), 1598, (C=C), 1460 (CH2); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 319.2 (M + 1+).
[4-(2-Methoxyphenyl)piperazin-1-yl](quinoxalin-2-yl)methanone (4b)
Yield: 63%; mp: 148–150°C; 1H NMR (CDCl3) (δ) ppm: 9.23 (s, 1H, quinoxaline), 8.15 (m, 1H, quinoxaline), 8.07 (m, 1H, quinoxaline), 7.92 (m, 2H, quinoxaline), 6.90 (m, 4H, phenyl), 4.06 (t, 2H, NCH2, piperazine), 3.76 (s, 3H, OCH3), 3.56 (t, 2H, NCH2, piperazine), 3.44 (t, 2H, NCH2, piperazine), 3.29 (t, 2H, NCH2, piperazine); IR (KBr, cm−1): 3050, 3000, 2950, 2900 (C-H), 1635 (C=O), 1498 (CH2), 1370 (CH3); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 349.2 (M + 1+).
[4-(4-Methoxyphenyl)piperazin-1-yl](quinoxalin-2-yl)methanone (4c)
Yield: 52%; mp: 94–96°C; 1H NMR (CDCl3) (δ) ppm: 9.25 (s, 1H, quinoxaline), 8.16 (m, 1H, quinoxaline), 8.08 (m, 1H, quinoxaline), 7.97 (m, 2H, quinoxaline), 6.96 (m, 2H, phenyl), 6.90 (m, 2H, phenyl), 4.06 (t, 2H, NCH2, piperazine), 3.73 (s, 3H, OCH3), 3.56 (t, 2H, NCH2, piperazine), 3.44 (t, 2H, NCH2, piperazine), 3.29 (t, 2H, NCH2, piperazine); IR (KBr, cm−1): 3045, 3000, 2950, 2900, 2830 (C-H), 1640 (C=O), 1470 (CH2), 1370 (CH3); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 349.2 (M + 1+).
(4-(4-Chlorophenyl)piperazin-1-yl)(quinoxalin-2-yl)methanone (4d)
Yield: 71%; mp: 124–126°C; 1H NMR (CDCl3) (δ) ppm: 9.24 (s, 1H, quinoxaline), 8.15 (m, 1H, quinoxaline), 8.08 (m, 1H, quinoxaline), 7.94 (m, 2H, quinoxaline), 7.29 (m, 2H, phenyl), 7.00 (m, 2H, phenyl), 4.03 (t, 2H, NCH2, piperazine), 3.53 (t, 2H, NCH2, piperazine), 3.42 (t, 2H, NCH2, piperazine), 3.29 (t, 2H, NCH2, piperazine); IR (KBr, cm−1): 3038, 3004, 2956, 2905, 2856 (C-H), 1636 (C=O); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 353.0 (M + 1+).
(4-(3-Chlorophenyl)piperazin-1-yl)(quinoxalin-2-yl)methanone (4e)
Yield: 81%; mp: 63–65°C; 1H NMR (CDCl3) (δ) ppm: 9.23 (s, 1H, quinoxaline), 8.10 (m, 1H, quinoxaline), 8.07 (m, 1H, quinoxaline), 7.93 (m, 2H, quinoxaline), 7.36 (t, 1H, phenyl), 7.06 (m, 3H, phenyl), 4.04 (t, 2H, NCH2, piperazine), 3.54 (t, 2H, NCH2, piperazine), 3.41 (t, 2H, NCH2, piperazine), 3.27 (t, 2H, NCH2, piperazine); IR (KBr, cm−1): 3038, 3004, 2956, 2905, 2856 (C-H), 1636 (C=O); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 353.2 (M + 1+).
Quinoxalin-2-yl(4-(3-(trifluoromethyl)phenyl)piperazin-1-yl)methanone (4f)
Yield: 65%; mp: 150–152°C; 1H NMR (CDCl3) (δ) ppm: 9.26 (s, 1H, quinoxaline), 8.14 (m, 1H, quinoxaline), 8.09 (m, 1H, quinoxaline), 7.90 (m, 2H, quinoxaline), 7.40 (t, 1H, phenyl), 7.16 (m, 3H, phenyl), 4.09 (t, 2H, NCH2, piperazine), 3.57 (t, 2H, NCH2, piperazine), 3.43 (t, 2H, NCH2, piperazine), 3.28 (t, 2H, NCH2, piperazine); IR (KBr, cm−1): 3040, 3000, 2950, 2900, 2850 (C-H), 1635 (C=O); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 387.1 (M + 1+).
(4-Benzylpiperazin-1-yl)(quinoxalin-2-yl)methanone hydrochloride (4g)
Yield: 74%; mp: 216–220°C; 1H NMR (DMSO-d6) (δ) ppm:12.33 (s, 1H, salt proton), 9.21 (s, 1H, aromatic proton) 8.13 (m, 2H, aromatic protons), 7.86 (m, 2H, aromatic protons), 7.75 (s, 2H, aromatic protons), 7.44 (s, 3H, aromatic protons), 4.84 (d, 1H, piperazine), 4.55 (d, 1H, piperazine), 4.43 (s, 2H, benzylic CH2), 4.12 (t, 1H, piperazine), 3.77 (t, 1H, piperazine), 3.57 (d, 1H, piperazine), 3.13 (m, 3H, piperazine); IR (KBr, cm−1): 3045, 2930, 2870 (C-H), 1640 (C=O), 1480 (CH2); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 333.2 (M + 1+).
(4-Methylpiperazin-1-yl)(quinoxalin-2-yl)methanone (4h)
Yield: 64%; 1H NMR (CDCl3) δ ppm: 9.18 (s, 1H, quinoxaline) 8.17 (m, 1H, quinoxaline), 8.11 (m, 1H, quinoxaline), 7.88 (m, 2H, quinoxaline), 3.93 (t, 2H, piperazine) 3.84 (t, 2H, piperazine) 2.66 (t, 2H, piperazine), 2.57 (t, 2H, piperazine), 2.40 (s, 3H, methyl); IR (KBr, cm−1): 3045, 2930, 2870 (C-H), 1640 (C=O), 1480 (CH2); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 257.3 (M + 1+).
(4-Ethylpiperazin-1-yl)(quinoxalin-2-yl)methanone hydrochloride (4i)
Yield: 66%; mp: 240–242°C; 1H NMR (CDCl3) (δ) ppm: 9.16 (s, 1H, quinoxaline) 8.15 (m, 1H, quinoxaline), 8.10 (m, 1H, quinoxaline), 7.85 (m, 2H, quinoxaline), 3.91 (t, 2H, piperazine) 3.80 (t, 2H, piperazine) 2.63 (t, 2H, piperazine), 2.54 (t, 2H, piperazine), 2.38 (q, 2H, methylene), 1.38 (t, 3H, methyl). IR (KBr, cm−1): 3050, 3000, 2900, 2860 (C-H), 1635 (C=O), 1470 (CH2), 1360 (CH3); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 271.1 (M + 1+).
(4-Allylpiperazin-1-yl)(quinoxalin-2-yl)methanone (4j)
Yield: 68%; 1H NMR (CDCl3) (δ) ppm: 9.24 (s, 1H, quinoxaline) 8.18 (dd, 1H, quinoxaline), 8.08 (dd, 1H, quinoxaline), 7.90 (m, 2H, quinoxaline), 6.14 (m, 1H, methine), 5.50 (m, 2H, olefinic methylene) 4.23 (s, 4H, piperazine) 3.52 (d, 2H, piperazine) 3.14 (q, 4H, 2H piperazine, 2H methylene). IR (KBr, cm−1): 3050, 3000, 2900, 2860 (C-H), 1635 (C=O), 1470 (CH2), 1360 (CH3); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 283.2 (M + 1+).
N-(2-(dimethylamino)ethyl)quinoxalin-2-carboxamide hydrochloride (5a)
Yield: 54%; mp: 44–46°C; 1H NMR (CDCl3) (δ) ppm: 9.66 (s, 1H, quinoxaline), 8.16 (dd, 1H, quinoxaline), 8.10 (dd, 1H, quinoxaline), 8.08 (s, 1H, NH) 7.86 (m, 2H, quinoxaline), 4.36 (t, 2H, CONHCH2), 2.88 (t, 2H, CH2NMe2), 2.54 (s, 6H, NMe2); IR (KBr, cm−1): 3300 (N-H), 3030, 2980, 2850 (C-H), 1665 (C=O), 1550 (N-H), 1460 (CH2); mass spectra of the compound exhibited the molecular ion peak at m/z 245.1 (M + 1+).
N-(2-(1H-indol-3-yl)ethyl)quinoxalin-2-carboxamide (5b)
Yield: 79%; mp: 146–150°C; 1H NMR (CDCl3) (δ) ppm: 9.61 (s, 1H, quinoxaline) 8.13 (m, 3H, quinoxaline, amide, indole), 8.03 (dd, 1H, quinoxaline), 7.94 (m, 2H, quinoxaline), 7.70 (d, 1H, NH indole), 7.41 (d, 1H, H2 of indole), 7.25 (m, 1H, indole), 7.18 (m, 2H, indole), 3.92 (q, 2H, NCH2) 3.19 (t, 2H, NCH2CH2); IR (KBr, cm−1): 3390 (N-H), 3045, 3020, 2920, 2850 (C-H), 1665 (C=O), 1535 (N-H), 1460 (CH2); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 317.0 (M + 1+).
N-(4-(diethylamino)phenyl)quinoxalin-2-carboxamide (5c)
Yield: 78%, mp 240–242°C; 1H NMR (DMSO-d6) (δ) ppm: 10.66 (s, 1H, CONH), 9.66 (s, 1H, quinoxaline), 8.28 (m, 1H, quinoxaline), 8.20 (m, 1H, quinoxaline), 8.15 (d, 2H, phenyl), 7.92 (m, 2H, quinoxaline) 7.84 (d, 2H, phenyl), 3.63 (q, 4H, (NCH2CH3)2), 1.18 (t, 6H, (NCH2CH3)2); IR (KBr, cm−1): 3450 (N-H), 3020, 2995, 2850 (C-H), 1680 (C=O), 1600 (C=C), 1540 (N-H), 1470 (CH2), 1330 (CH3); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 321.1 (M + 1+).
N-(pyridin-3-yl)quinoxalin-2-carboxamide (5d)
Yield: 49%, mp 186–188°C; 1H NMR (CDCl3) (δ) ppm: 9.29 (s, 1H, quinoxaline), 8.56 (d, 1H, pyridine), 8.46 (m, 1H, pyridine), 8.40 (m, 1H, pyridine) 8.13 (m, 2H, quinoxaline and amide), 8.09 (m, 1H, quinoxaline), 7.90 (m, 2H, quinoxaline), 7.44 (m, 1H, pyridine); mass spectra (ESI) of the compound exhibited the molecular ion peak at m/z 251.2 (M + 1+).
Pharmacology (general)
All the animals were obtained from Hissar Agricultural University, Hissar, Haryana, India, and were maintained in colony cages at 23 ± 2°C, relative humidity of 45–55% under a 12-h light/dark cycle, fed with standard animal feed and water ad libitum. The Institutional Animal Ethics Committee of the Birla Institute of Technology and Science, Pilani, India, approved the experimentation on animals (Protocol No.IAEC/RES/04/01/Rev 01, dated 13.08.08). Compounds were assessed for their serotonin type-3 receptor antagonism in male Dunkin Hartley guinea pigs (350–400 g) and for their anti-depressants potentials in Swiss albino mice (18–25 g).
5-HT3 receptor antagonistic activity
For serotonin type-3 receptor antagonistic activity, guinea pigs were sacrificed by mild ether anaesthesia followed by cervical dislocation. The abdomen was cut open and a length of ileum was excised about 2 cm from the ileo-caecal junction. The longitudinal muscle-myenteric plexus, 3–4 cm in length, was removed and mounted as per methodCitation15. The tissue was equilibrated for 30 min. under a resting tension of 500 mg and constant aeration in a 40 mL organ bath containing Tyrode solution maintained at 37°C.
Non-cumulative concentrations of 2-methyl-5-HT (Tocris, UK) were added with a 15-min dosing cycle (to prevent desensitization) and left in contact with the tissue until the maximal contraction had developed. To study the antagonist effect of the test compounds on the response evoked by 2-methyl-5-HT, the compounds were added to the organ bath and left in contact with the tissue for at least 10 min. prior to the addition of 2-methyl-5-HT. The contractions were recorded using a T-305 Force transducer coupled to a Student’s physiograph (Bio Devices, Ambala, India). Antagonism was expressed in the form of pA2 values, which were graphically determinedCitation16. The pA2 values of the test compounds were compared with the standard antagonist ondansetron (Natco Pharma, Hyderabad, India).
Results and discussion
Drug design
The quinoxalin-2-carboxamides were designed based on the Hibert et al. pharmacophoric model. The minimum energy conformation of the designed molecules were generated by ACDLABS-10.0/3D Viewer (CHARMM parameterization) and the pharmacophoric distances were measured from centroid of quinoxaline ring to oxygen of the carbonyl group, carbonyl oxygen to basic nitrogen atom (N4 of piperazines, heterocyclic nitrogen and nitrogen of tertiary amines) and centroid of quinoxaline residue to basic nitrogen. The distances between the pharmacophoric elements of the designed compounds complied with the aforementioned model.
Further, for the better pharmacokinetic (absorption, distribution, metabolism, and excretion) profile, all the molecules were designed according to Lipinski Rule of Five, i.e. hydrogen bond donor atoms not more than 5, hydrogen bond acceptor atoms not more than 10, molecular weight less than 500, and log P value less than 5Citation17.
Synthesis of quinoxalin-2-carboxamides
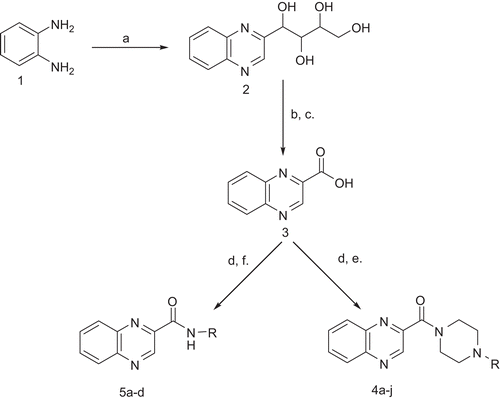
The 2-(1,2,3,4-tetrahydroxybutyl)quinoxaline (2) and quinoxalin-2-carboxylic acid (3) were synthesized as per method reportedCitation18. The oxidative cyclization of o-phenylenediamine with d-fructose afforded the former compound with the yield of 46%, which on oxidation with alkaline hydrogen peroxide gave quinoxalin-2-carboxylic acid with the yield of 50%. The carboxylic acid group of quinoxalin-2-carboxylic acid (3) was coupled with various amines in the presence of 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDC.HCl) and HOBt under nitrogen atmosphere (). All the synthesized compounds were characterized by spectral analysis. Physical constants of the title compounds are represented in .
Scheme 1. Reagents and conditions: (A) D-fructose, 10% aq. AcOH, 80°C, 18 h; (B) NaOH, 30% H2O2, 60°C, 3 h then 90°C, 2 h, (C) Conc. HCl; (D) EDC HCl, HOBt, THF, N2, 0°C–RT, 1 h; (E) piperazine, RT, 5 h; (F) R-NH2, RT, 5 h. THS, tetrahydrofuran
Pharmacology and preliminary structure–activity c of quinoxaline-2-carboxamides
All the synthesized compounds exhibited antagonism towards 5-HT3 receptors, and the pharmacological data of the title compounds are presented in . For the study, first N-phenylpiperazine was coupled with quinoxalin-2-carboxylic acid; the obtained carboxamide 4a showed antagonism (pA2: 7.3) greater than standard drug, ondansetron (pA2: 6.9)Citation19. In order to find out more potent carboxamides, an electron releasing group, a methoxy group was introduced on the phenyl group at second position; the resultant carboxamide 4b drastically lost its potency compared to compound 4a (with no substituent on the phenyl ring) and the observed pA2 value was 4.5. Similar result was observed when methoxy substituent was present on the fourth position of phenyl ring. On the basis of these preliminary results it was decided to change the nature of the substituent present on the phenyl ring i.e. replacing electron releasing substituent with chlorine atom, an electron withdrawing substituent. The obtained carboxamides (4d and 4e) lost their potency, regardless of their substitution pattern. Replacement of chlorine atom with CF3 group which has strong electron withdrawing nature and higher liphophilic nature weakened the antagonism of the obtained compound 4f. With these results, aromatic piperazine was replaced with aliphatic piperazine, methylpiperazine was first coupled with quinoxalin-2-carboxylic acid, the formed carboxamide, 4h, retained its antagonism, but was less potent than phenylpiperazine based carboxamide (4a). Higher homologation, replacement of methylpiperazine with ethylpiperazine increased the lipophilicity of the carboxamides but decreased their potency. Further increase in alkyl chain length by introducing allyl group in place of ethyl group, the resultant amide 4j, showed diminished antagonism. Based on these results methylene group was incorporated between phenyl group and N4 piperazine and the obtained compound 4g exhibited stronger antagonism whose pA2 was greater than standard drug. Various primary amines viz. N,N-dimethylethylenediamine, tryptamine, N,N-diethyl-p-phenylnedaimine and 3-aminopyridine were coupled with quinoxalin-2-carboxylic acid to serve the role of piperazine. The obtained carboxamides (5a–5d) exhibited antagonism towards serotonin type-3 receptor, where compounds 5b (pA2: 7.3), 5c (pA2: 7.0) displayed antagonism greater than the standard drug and compound 5d exert its antagonism (pA2: 6.7) nearly equal to ondansetron.
Conclusion
In summary, 14 compounds were synthesized from the starting material o-phenylenediamine in a sequence of reaction involving oxidative cyclization, oxidation and condensation. The structures of the synthesized compounds were confirmed by physical and spectral data. All the synthesized compounds showed antagonism towards serotonin type-3 receptor and compounds 4a, 4g, 5b, and 5c exhibited antagonism greater than standard drug, ondansetron. Hence, further studies on these molecules are planned to obtain clinically useful antiemetics.
Acknowledgements
The authors thank the University Grants Commission (UGC), India, Birla Institute of Technology & Science (BITS), Pilani, India and SAIF, Panjab University, Chandigarh, India for providing financial support, laboratory and analytical facilities, respectively.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Tralongo P, Dimari A, Conti G, Aiello R, Mauceri G. Central nervous system side-effects of 5-HT3-receptor antagonists in elderly cancer patients treated with chemotherapy. Ann Oncol 2004;15:987–988.
- Coates A, Abraham S, Kaye SB, Sowerbutts T, Frewin C, Fox RM, Tattersall MH. On the receiving end–patient perception of the side-effects of cancer chemotherapy. Eur J Cancer Clin Oncol 1983;19:203–208.
- Hesketh PJ. Chemotherapy-induced nausea and vomiting. N Engl J Med 2008;358:2482–2494.
- Jordan K, Schmoll HJ, Aapro MS. Comparative activity of antiemetic drugs. Crit Rev Oncol Hematol 2007;61:162–175.
- Craig JB, Powell BL. The management of nausea and vomiting in clinical oncology. Am J Med Sci 1987;293:34–44.
- Cubeddu LX. Mechanisms by which cancer chemotherapeutic drugs induce emesis. Semin Oncol 1992;19:2–13.
- Jordan K, Sippel C, Schmoll HJ. Guidelines for antiemetic treatment of chemotherapy-induced nausea and vomiting: past, present, and future recommendations. Oncologist 2007;12:1143–1150.
- Ohtaka H, Fujita T., In: Jucker E, ed. Progress in Drug Research, Chap. 4, Vol. 41. Switzerland: Birkhauser Verlag Basel, 1993:338.
- Mahesh R, Perumal RV, Pandi PV. Pharmacophore based synthesis of 3-chloroquinoxaline-2-carboxamides as serotonin3 (5-HT3) receptor antagonist. Biol Pharm Bull 2004;27:1403–1405.
- Clark RD, Miller AB, Berger J, Repke DB, Weinhardt KK, Kowalczyk BA, Eglen RM, Bonhaus DW, Lee CH, Michel AD. 2-(Quinuclidin-3-yl)pyrido[4,3-b]indol-1-ones and isoquinolin-1-ones. Potent conformationally restricted 5-HT3 receptor antagonists. J Med Chem 1993;36:2645–2657.
- Ohta M, Suzuki T, Koide T, Matsuhisa A, Furuya T, Miyata K, Yanagisawa I. Novel 5-hydroxytryptamine (5-HT3) receptor antagonists. I. Synthesis and structure-activity relationships of conformationally restricted fused imidazole derivatives. Chem Pharm Bull 1996;44:991–999.
- Venkatesha Perumal R, Mahesh R. Synthesis and biological evaluation of a novel structural type of serotonin 5-HT3 receptor antagonists. Bioorg Med Chem Lett 2006;16:2769–2772.
- Hibert MF, Hoffmann R, Miller RC, Carr AA. Conformation-activity relationship study of 5-HT3 receptor antagonists and a definition of a model for this receptor site. J Med Chem 1990;33:1594–1600.
- Paton WD, Zar MA. The origin of acetylcholine released from guinea-pig intestine and longitudinal muscle strips. J Physiol (Lond) 1968;194:13–33.
- MacKay D. How should values of pA2 and affinity constants for pharmacological competitive antagonists be estimated? J Pharm Pharmacol 1978;30:312–313.
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Del Rev 1997;23:3–25.
- Harms AE. An efficient synthesis of 2-quinoxalinecarboxylic acid. Org Process Res Dev 2004;8:666–669.
- Mahesh R, Perumal RV, Pandi PV. Microwave assisted synthesis of 2-(4-substituted piperazin-1-yl)-1,8-naphthyridine-3-carbonitrile as a new class of serotonin 5-HT3 receptor antagonists. Bioorg Med Chem Lett 2004;14:5179–5181.