Abstract
Direct interaction between 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid and primary α-amino acids (exemplified by glycine, alanine, and l-valine) in aqueous ethanolic NaHCO3 at 70–80°C for 24–72 h produced the respective N-(4-oxoquinolin-7-yl)-α-amino acids (6a–c). The latter derivatives underwent reductive lactamization upon treatment with Na2S2O4 in aqueous ethanol to afford moderate yields of the corresponding pyrido[2,3-f]quinoxaline-8-carboxylic acids (8a–c). Acetylation of 8a–c using acetyl chloride afforded N4-acetylated hexahydro-2,7-dioxopyrido[2,3-f]quinoxaline-8-carboxylic acids (9a–c). The structures, assigned to these new heterocyclic products, are supported by analytical and spectral data. The synthesized compounds (6a–c/9a–c) showed appreciable antibacterial activity as compared with ciprofloxacin.
Introduction
Synthetic fluoroquinolones (e.g. ciprofloxacin 1a) represent a successful achievement towards the design and development of potent anti-infectious drugs.Citation1–5 Some derivatives such as 7-(4-arylpiperazin-1-yl)-6-fluoro-8-trifluoromethylquinolones, for example 1b, were reported to act as non-nucleoside reverse transcriptase inhibitors (NNRTIs) of HIV-1 reverse transcriptase.Citation6–9
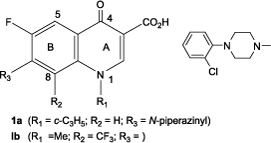
On the other hand, simple or heterocyclic-fused quinoxalinones have become interesting compounds for study as antiviral agentsCitation10–27 exemplified by the 3,4-dihydroquinoxalin-2-one core (2, Y=O), a second generation of NNRTIs, that is opaviraline (2a)Citation22–27 and the related 2-thione talveraline (2b),Citation14–19 are in clinical development.
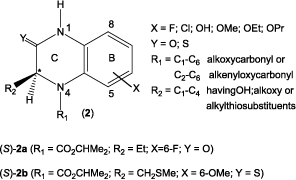
The antibacterial potency of new trihybrid system of both systems (1 and 2), 4-methyl-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido[2,3-f]-quinoxaline-8-carboxylic acid, selected [a]-fused heterocycles, and acyclic precursors (3a and b) has been reported by our group earlier.Citation28
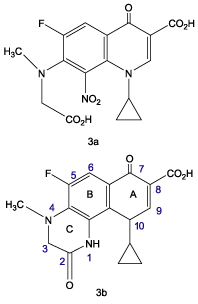
Owing to the potential biological interest in these heterocyclic compounds, the present work aims at the synthesis and characterization of new 3-(substituted)-N4-acetylated-10-cyclopropyl-5-fluoro-2,7-dioxo-hexahydropyrido[2,3-f]quinoxaline-3-carboxylic acids (8a–c/9a–c) (), a tricyclic system encompassing the structural features of both 4-quinolone and 3,4-dihydroquinoxalin-2-one chemotypes. Such hybrid heterocyclic derivatives might have interesting potential bioactive properties such as antimicrobial and/or antiviral activity, which prompted us to prepare an additional set of title compound and to examine the effect of alkylation at C-3 and/or N4-acetylation on antibacterial activity.
Scheme 1. Synthesis of N4-acetylated hexahydro-2,7-dioxopyrido[2,3-f]quinoxaline-8-carboxylic acids (9a-c).
![Scheme 1. Synthesis of N4-acetylated hexahydro-2,7-dioxopyrido[2,3-f]quinoxaline-8-carboxylic acids (9a-c).](/cms/asset/af0eb9b6-29a4-45ec-9b71-51b16b66d4f5/ienz_a_543421_f0001_b.gif)
Materials and methods
Chemistry
The primary α-amino acids, employed in this study, are biochemical grades (Aldrich, UK) and were used as received. 2,4-Dichloro-5-fluoro-3-nitrobenzoic acid was purchased from Acros-Belgium through local agents. Melting points (uncorrected) were determined on a Gallenkamp electrothermal melting temperature apparatus.
1H NMR and 13C NMR spectra were measured on a Bruker DPX-300 instrument with TMS as internal reference. High-resolution MS-ESI (HRMS) data of selected compounds were obtained in positive ion mode using electrospray ion trap technique by collision-induced dissociation on Bruker Apex-IV (7 Tesla) or on Bruker Bio TOF III instruments. IR spectra were recorded as KBr discs on a Nicolet Impact-400 FT-IR spectrophotometer. Microanalyses were preformed at the Microanalytical Laboratory–Medicinal Chemistry Division, Faculty of Pharmacy, University of Jordan, Amman, Jordan.
7-Chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (4)
It was prepared using reported methods with minor modifications.Citation30,Citation31
7-[(Carboxymethyl)amino]-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6a)
A well-stirred mixture of glycine (5a/0.68 g, 9 mmol), 4 (1.0 g, 3 mmol), and sodium hydrogen carbonate (1.5 g, 18 mmol) in aqueous ethanol (140 mL, 1:1 v/v) was heated at 70–80°C. The reaction mixture slowly developed a light yellow colour that changed into bright yellow, then into clear orange solution. The progress of the reaction was monitored by thin-layer chromatography (TLC), and was completed within 40–48 h. The resultant orange solution was first extracted with dichloromethane (50 mL), then the aqueous layer was separated, acidified with 3N HCl to pH 6.5, and re-extracted with dichloromethane (50 mL). The aqueous layer was separated and acidified again with 3N HCl to pH 2–3, whereby the title compound was precipitated as fluorescent yellowish solid, which was filtered, washed with cold water (3 × 20 mL), and recrystallized from ethanol.
Yield 0.90 g (82 %), mp 236–238°C (decomposition)
Anal. Calcd for C15H12FN3O7 (365.27): C, 49.32; H, 3.31; N, 11.50; found C, 49.02; H, 3.22; N, 11.41
IR (KBr): ν = 3090, 2915, 1740 (br), 1610, 1455, 1319, 1260, 1210 cm−1
1H NMR (300 MHz, DMSO-d6): δ 0.95 (m, 4H, H2-2′/H2-3′), 3.67 (br m, 1H, H-1′), 4.25 (br, dd, J = 5.4 Hz, 6.2 Hz, 2H, a-CH2–NH), 7.57 (br s, 1H, N-H), 7.95 (d, 3JH-F = 13.9 Hz, 1H, H-5), 8.72 (s, 1H, H-2), 13.43 (br s, 1H, CH2–CO2H), 14.63 (br s, 1H, C(3)–CO2H)
13C NMR (75 MHz, DMSO-d6): δ 10.2 (C-2′/C-3′), 40.6 (C-1′), 47.1 (d, JC-F = 12.5 Hz, a-CH2NH), 109.5 (C-3), 114.4 (d, 2JC-F = 23.0 Hz, C-5), 116.6 (d, 3JC-F = 7.1 Hz, C-4a), 128.0 (d, 3JC-F = 5.6 Hz, C-8), 135.5 (C-8a), 138.8 (d, 2JC-F = 14.3 Hz, C-7), 150.4 (d, 1JC-F = 247 Hz, C-6), 152.0 (C-2), 165.4 (C(3)–CO2H), 171.7 (d, JC-F = 2.5 Hz, CH2–CO2H), 175.5 (d, 4JC-F = 2.6 Hz, C-4).
(S)-7-[(1-Carboxyethyl)amino]-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid [(S)-6b]
A stirred mixture of (S)-α-alanine [(S)-5b/0.8 g, 9 mmol], 4 (1.0 g, 3 mmol), and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50% aqueous ethanol (140 mL) was heated at 75–80°C for 24–30 h under reflux conditions. Workup of the reaction mixture as described for 6a above, produced the title compound as fluorescent yellow solid, which was recrystallized from ethanol.
Yield 0.86 g (76%), mp 219–221°C (decomposition)
Anal. Calcd for C16H14FN3O7 (379.30): C, 50.67; H, 3.72; N, 11.08; found C, 50.44; H, 3.61; N, 10.95
IR (KBr): ν = 3091, 2915, 2880, 1730, 1610, 1450, 1310, 1250, 1215 cm−1
MS (EI): m/z (% rel. int.): 379 (6, M+), 359 (2), 335 (20), 301 (9), 289 (24), 257 (50), 244 (58), 217 (30), 188 (21), 174 (13), 147 (11), 107 (9), 85 (61), 83 (100)
HRMS Calcd for C16H14FN3O7 379.08153, Found 379.08219
1H NMR (300 MHz, DMSO-d6): δ 0.94 (m, 4H, H2-2′/H2-3′), 1.46 (d, J = 6.8 Hz, 3H, CH3), 3.67 (m, 1H, H-1′), 4.62 (dq, J = 6.8 Hz, 7.1 Hz, 1H, α-CHMe), 7.36 (d, J = 7.1 Hz, 1H, N-H), 8.03 (d, 3JH-F = 13.7 Hz, 1H, H-5), 8.75 (s, 1H, H-2), 13.30 (br s, 1H, CH–CO2H), 14.59 (br s, 1H, C(3)–CO2H)
13C NMR (75 MHz, DMSO-d6): δ 10.1 (C-2′/C-3′), 19.8 (d, JC-F = 2.9 Hz, CH3), 40.7 (C-1′), 53.8 (d, JC-F = 12 Hz, α-CHMe), 109.7 (C-3), 114.9 (d, 2JC-F = 22.9 Hz, C-5), 117.3 (d, 3JC-F = 7.1 Hz, C-4a), 128.7 (d, 3JC-F = 5 Hz, C-8), 135.7 (C-8a), 137.9 (d, 2JC-F = 14.7 Hz, C-7), 150.2 (d, 1JC-F = 248 Hz, C-6), 152.1 (C-2), 165.3 (C(3)–CO2H), 174.2 (CH–CO2H), 175.5 (d, 4JC-F = 2.2 Hz, C-4).
(S)-7-(1-Carboxy-2-methylpropylamino)-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid [(S)-6c]
A stirred mixture of (S)-l-valine [1.0 g, 9 mmol], 4 (1.0 g, 3 mmol), and sodium hydrogen carbonate (1.5 g, 18 mmol) in 50% aqueous ethanol (140 mL) was heated at 75–80°C for 100–120 h under reflux conditions. Workup of the reaction mixture as described for 6a above produced the title compound as fluorescent yellow solid, which was recrystallized from ethanol.
Yield 1.1 g (90%), mp 217–222°C (decomposition)
Anal. Calcd for C18H18FN3O7 (407.35): C, 53.07; H, 4.45; N, 10.32; found: C, 52.83; H, 4.69; N, 10.68
IR (KBr): ν = 3308, 3070, 2971, 1733, 1700, 1629, 1549, 1518, 1469, 1321, 1257, 1216, 1144, 1033 cm−1
MS (FAB): m/z (% rel. int.): 408 [100, (M+H)+/Calcd. for C18H18FN3O7 407 (M)]
1H NMR (300 MHz, DMSO-d6): δ 0.89, 0.95 (2d, J = 6.8 Hz, 6.0 H, (CH3)2–CH), 0.92 (m, 4H, H2-2′/H2-3′), 2.21 (m, 1H, CHMe2), 3.68 (m, 1H, H-1′), 4.50 (br d, J = 7.2 Hz, CH–CO2H), 7.21 (d, J = 8.0 Hz, 1H, N-H), 8.07 (d, 3JH-F = 13.5 Hz, 1H, H-5), 8.77 (s, 1H, H-2), 13.39 (br s, 1H, CH–CO2H), 14.55 (br s, 1H, C3–CO2H)
13C NMR (75 MHz, DMSO-d6): δ 10.1 (C-2′/C-3′), 18.0, 18.5 ((CH3)2), 31.6 (CHMe2), 40.9 (C-1′), 63.2 (d, JC-F = 11.3 Hz, CH-NH), 109.8 (C-3), 115.2 (d, 2JC-F = 22.7 Hz, C-5), 117.8 (d, 3JC-F = 7.1 Hz, C-4a), 129.3 (d, 3JC-F = 5.4 Hz, C-8), 135.8 (C-8a), 138.1 (d, 2JC-F = 14.9 Hz, C-7), 150.4 (d, 1JC-F = 248 Hz, C-6), 152.3 (C-2), 165.3 (C3–CO2H), 172.7 (CH–CO2H), 175.6 (d, 4JC-F = 2.5 Hz, C-4).
10-Cyclopropyl-5-fluoro-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido[2,3-f]quinoxaline-8-carboxylic acid [8a]
To a vigorously stirred solution of 7-[(carboxymethyl)amino]-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6a/0.37 g, 1.0 mmol) and potassium carbonate (0.96 g, 7.0 mmol) in water (20 mL), was added portion wise a solution of sodium dithionite (1.22 g, 7.0 mmol) in water (5 mL). Following each addition, the solution acquired a dark brown colour, which shortly faded away (1–2 min). The reaction mixture was further stirred at room temperature for 25 min. Thereafter, the pH of the resultant solution was adjusted to about 4 by slow addition of 3N HCl, the precipitated product was filtered, washed successively with water and methanol, air-dried, and recrystallized from ethanol.
Yield 0.17 g (54%), mp 290-292°C (decomposition/the crystals darken at 270°C)
Anal. Calcd for C15H12FN3O4 (317.27): C, 56.78; H, 3.81; N, 13.24; found C, 56.54; H, 3.77; N, 13.08
IR (KBr): ν = 3270, 2980, 1730, 1680, 1615, 1583, 1525, 1444, 1380, 1315, 1185, 1082, 1010 cm−1
MS (TOF, ES+): m/z 318 (M + H)+, HRMS Calcd for C15H13FN3O4 318.0890, found 318.0885; m/z 340 (M + Na)+, HRMS Calcd for C15H12FN3O4Na 340.0709, found 340.0704
1H NMR (300 MHz, 3% NaOD in D2O): δ 0.21, 0.69 (2m, 4H, H2-2′/H2-3′), 3.27 (s, 2H, H2-3), 3.98 (m, 1H, H-1′), 7.00 (d, 3JH-F = 10.8 Hz, 1H, H-6), 8.17 (s, 1H, H-9)
13C NMR (75 MHz, 3% NaOD in D2O): δ 9.4 (C-2′/C-3′), 40.1 (C-1′), 42.9 (C-3), 102.3 (d, 2JC-F = 20.2 Hz, C-6), 114.8 (C-8), 119.9 (d, 3JC-F = 7 Hz, C-6a), 128.0 (d, 3JC-F = 3.7 Hz, C-10b), 130.0 (d, 2JC-F = 15.5 Hz, C-4a), 131.9 (C-10a), 148.9 (C-9),149.2 (d, 1JC-F = 240 Hz, C-5), 168.1 (C-2), 173.1 (C(8)–CO2−), 175.6 (d, 4JC-F = 2.9 Hz, C-7).
(S)-10-Cyclopropyl-5-fluoro-3-methyl-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido-[2,3-f]-quinoxaline-8-carboxylic acid [(S)-8b]
To a stirred solution of 6b (0.38 g, 1.0 mmol) and potassium carbonate (0.96 g, 7.0 mmol) in water (20 mL), was added portion wise a solution of sodium dithionite (1.22 g, 7.0 mmol) in water (5 mL). The reaction mixture was further stirred at room temperature for 10 min, and worked up as described for 8a above. The title product was recrystallized from chloroform/methanol (3:1, v/v) to furnish pale yellow fine crystals.
Yield 0.21 g (64%), mp 315–317°C (decomposition/the crystals darken at 290°C)
Anal. Calcd for C16H14FN3O4 (331.30): C, 58.01; H, 4.26; N, 12.68; found C, 57.86; H, 4.18; N, 12.55
IR (KBr): ν = 3270, 2996, 1724, 1675, 1611, 1585, 1520, 1435, 1380, 1311, 1180, 1080, 1017 cm−1
MS (TOF ES+): m/z 354 (M+Na)+, HRMS Calcd for C16H14FN3O4Na 354.0866, found 354.0861
1H NMR (300 MHz, DMSO-d6): δ 0.97, 1.05 (2m, 4H, H2-2′/H2-3′), 1.32 (d, J = 6.6 Hz, 3H, CH3), 3.93 (br q, J = 6.6 Hz, 1H, H-3), 4.48 (m, 1H, H-1′), 7.48 (br s, 1H, N(4)-H), 7.62 (d, 3JH-F = 10.6 Hz, 1H, H-6), 8.65 (s, 1H, H-9), 10.34 (s, 1H, lactam N(1)-H), 15.13 (s, 1H, CO2H)
13C NMR (75 MHz, DMSO-d6): δ 10.2 (C-2′/C-3′), 16.2 (CH3), 39.0 (C-1′), 50.4 (C-3), 105.4 (d, 2JC-F = 19.6 Hz, C-6), 107.2 (C-8), 116.1 (d, 3JC-F = 6.3 Hz, C-10b), 117.2 (d, 3JC-F = 7.1 Hz, C-6a), 129.3 (C-10a), 131.9 (d, 2JC-F = 18.2 Hz, C-4a), 149.4 (d, 1JC-F = 242 Hz, C-5), 151.0 (C-9), 166.2 (CO2H), 166.5 (C-2), 176.6 (d, 4JC-F = 3.1 Hz, C-7).
(S)-10-Cyclopropyl-5-fluoro-3-isopropyl-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido-[2,3-f]quinoxaline-8-carboxylic acid [(S)-8c]
To a stirred solution of (±)-7-(1-carboxy-2-methylpropylamino)-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (6c, 0.36 g, 1 mmol) and (0.96 g, 7 mmol) of potassium carbonate in 20 mL water, an aqueous solution of sodium dithionite (0.87 g, 5 mmol) in 5 mL water was added drop wise. The reaction mixture was further stirred at room temperature for 15–25 min. Thereafter, the pH of the solution was adjusted to about 4.0. The precipitated product was filtered, washed with water, and air-dried. Then further re-crystallization was carried out from acetone and ethanol (1:1), yielding faint yellow crystals.
Yield 0.33 g (92%), mp 285–291°C (decomposition)
Anal. Calcd for C18H18FN3O4 (359.35): C, 60.16; H, 5.05; N, 11.69; found: C, 60.22; H, 5.04, N, 11.76
IR (KBr): ν = 3282, 2996, 1724, 1683, 1621, 1581, 1530, 1436, 1385, 1322, 1184, 1083, 1027 cm−1
MS (EI): m/z (% rel. int.): 359 (38), 315 (100), 288 (7), 272 (39), 257 (55), 244 (40), 230 (39), 216 (33), 203 (44), 188 (52), 175 (74), 163 (20), 147 (21)¸ 135 (11), 121 (9), 108 (7), 95 (3) HRMS Calcd for C18H18FN3O4 359.12810, found 359.12940
1H NMR (300 MHz, DMSO-d6): δ 0.79, 0.89 (2m, 2H, overlapped with a CH3 doublet), and 1.06, 1.26 (2m, 2H) (H2-2′/H2-3′), 0.90 (d, J = 6.5 Hz, 3H) and 0.94 (d, J = 6.7 Hz, 3H) [CH(CH3)2], 1.88 (m, 1H, CHMe2), 3.53 (dd, J = 8.1, 2.9 Hz, 1H, H-3), 4.50 (br m, 1H, H-1′), 7.60 (d, 3JH-F = 10.5 Hz, 1H, H-6), 7.77 (br s, 1H, N(4)-H), 8.62 (s, 1H, H-9), 10.36 (s, 1H, lactam N(1)-H), 15.11 (s, 1H, CO2H)
13C NMR (75 MHz, DMSO-d6): δ 9.0, 11.3 (C-2′/C-3′), 18.9, 19.2 [CH(CH3)2], 29.5 (CHMe2), 39.0 (C-1′), 61.2 (C-3), 105.4 (d, 2JC-F = 19.4 Hz, C-6), 107.1 (C-8), 115.8 (d, 3JC-F = 6.4 Hz, C-10b), 116.9 (d, 3JC-F = 7.1 Hz, C-6a), 129.3 (C-10a), 130.7 (d, 2JC-F = 18.3 Hz, C-4a), 149.3 (d, 1JC-F = 242 Hz, C-5), 150.9 (C-9), 164.5 (C-2), 166.2 (CO2H), 176.6 (d, 4JC-F = 3.2 Hz, C-7).
4-Acetyl-10-cyclopropyl-5-fluoro-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido[2,3-f]quinoxaline-8-carboxylic acid (9a)
To a stirred solution of 10-cyclopropyl-5-fluoro-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido[2,3-f]quinoxaline-8-carboxylic acid (8a, 0.32 g, 1 mmol) and (0.3 mL) of triethylamine in 20 mL chloroform, acetyl chloride (0.08 mL, 1 mmol) in 5 mL chloroform was added drop wise. The reaction mixture was refluxed for 48 h at 60–65°C. The chloroform was removed under pressure. The product was collected, washed with ice-cold water, and air-dried. Then further re-crystallization was carried out from acetone and ethanol (1:1), yielding white crystals.
Yield 0.19 g (53%), mp 300–306°C (decomposition)
Anal. Calcd for C17H14FN3O5 (359.31): C, 56.83; H, 3.93; N, 11.69; found C, 56.84; H, 3.97; N, 11.58
IR (KBr): ν = 3270, 2996, 1730, 1680, 1620, 1580, 1523, 1440, 1376, 1313, 1183, 1070, 1000 cm−1
HRMS Calcd for C17H13FN3O5Na 381.29039, found 381.29077
1H NMR (300 MHz, 3% NaOD in D2O): δ 0.25, 0.71 (2m, 4H, H2-2′/H2-3′), 1.80 (m, 1H, H-1′), 2.25 (s, 3H,−CO–CH3), 4.07 (s, 2H, H2-3), 7.50 (d, 3JH-F = 10.8 Hz, 1H, H-6), 8.17 (s, 1H, H-9)
13C NMR (75 MHz, 3% NaOD in D2O): δ 9.3 (C-2′/C-3′), 21.2 (–COCH3), 40.2 (C-1′), 42.0 (C-3), 102.6 (d, 2JC-F = 20.2 Hz, C-6), 115.0 (C-8), 120.0 (d, 3JC-F = 7 Hz, C-6a), 128.1 (d, 3JC-F = 3.7 Hz, C-10b), 130.3 (d, 2JC-F = 15.5 Hz, C-4a), 132.1 (C-10a), 148.7 (C-9), 149.1 (d, 1JC-F = 240 Hz, C-5), 168.5 (C-2), 170.2 (–COCH3), 173.2 (C(8)–CO2−), 175.6 (d, 4JC-F = 2.9 Hz, C-7).
(S)-4-Acetyl-10-cyclopropyl-5-fluoro-3-methyl-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido[2,3-f]quinoxaline-8-carboxylic acid [(S)-9b]
Yield 0.24 g (65%), mp 302–305°C (decomposition)
Anal. Calcd for C18H16FN3O5 (373.34): C, 57.91; H, 4.32; N, 11.26; found C, 57.86; H, 4.35; N, 11.21
IR (KBr): ν = 3270, 2996, 1724, 1675, 1611, 1585, 1520, 1435, 1380, 1311, 1180, 1080, 1017 cm−1
MS (TOF, ES+): m/z 374 (M + H)+, HRMS Calcd for C18H16FN3O5 373.10740, found 373.1108
1H NMR (300 MHz, DMSO-d6): δ 0.90, 1.11 (2m, 4H, H2-2′/H2-3′), 1.35 (d, J = 6.6 Hz, 3H, CH3), 1.89 (m, 1H, H-1′), 2.30 (s, 3H,−CO–CH3), 3.95 (br q, J = 6.6 Hz, 1H, H-3), 7.62 (d, 3JH-F = 10.6 Hz, 1H, H-6), 8.60 (s, 1H, H-9), 9.90 (s, 1H, lactam N(1)-H), 15.13 (s, 1H, CO2H)
13C NMR (75 MHz, DMSO-d6): δ 10.1 (C-2′/C-3′), 16.5 (CH3), 20.9 (–COCH3), 39.0 (C-1′), 51.0 (C-3), 105.0 (d, 2JC-F = 19.6 Hz, C-6), 107.8 (C-8), 116.4 (d, 3JC-F = 6.3 Hz, C-10b), 118.1 (d, 3JC-F = 7.1 Hz, C-6a), 130.1 (C-10a), 132.1 (d, 2JC-F = 18.2 Hz, C-4a), 148.4 (d, 1JC-F = 242 Hz, C-5), 151.2 (C-9), 166.3 (CO2H), 166.8 (C-2), 171.1 (–COCH3), 176.6 (d, 4JC-F = 3.1 Hz, C-7).
(S)-4-Acetyl-10-cyclopropyl-5-fluoro-1,2,3,4,7,10-hexahydro-3-isopropyl-2,7-dioxopyrido[2,3-f]quinoxaline-8-carboxylic acid [(S)-9c]
Yield 0.32 g (80%), mp 305–309°C (decomposition)
Anal. Calcd for C20H20FN3O5 (401.39): C, 59.85; H, 5.02; N, 10.47; found: C, 59.91; H, 5.03, N, 10.50
IR (KBr): ν = 3182, 2996, 1724, 1683, 1621, 1581, 1530, 1436, 1385, 1322, 1184, 1083, 1027 cm−1
MS (TOF, ES+): m/z 402 (M + H)+, HRMS Calcd for C20H20FN3O5 401.13870, found 301.14010
1H NMR (300 MHz, DMSO-d6): δ 0.78, 0.82 (2m, 2H, overlapped with a CH3 doublet), and 0.90, 0.92 (2m, 2H) (H2-2′/H2-3′), 0.96 (d, J = 6.5 Hz, 3H) and 1.14 (d, J = 6.7 Hz, 3H) [CH(CH3)2], 1.52 (m, 1H, CHMe2), 2.05 (br m, 1H, H-1′), 2.65 (s, 3H,−CO–CH3); 3.51 (dd, J = 8.1, 2.9 Hz, 1H, H-3), 5.25 (s, 1H, lactam N(1)-H), 7.55 (d, 3JH-F = 10.2 Hz, 1H, H-6), 8.85 (s, 1H, H-9), 14.01 (s, 1H, CO2H)
13C NMR (75 MHz, DMSO-d6): δ 6.2, 9.5 (C-2′/C-3′), 18.0, 19.1 (CH(CH3)2), 21.1 (–COCH3), 28.5 (CHMe2), 37.0 (C-1′), 64.1 (C-3), 108.1 (d, 2JC-F = 17.4 Hz, C-6), 107.5 (C-8), 115.1 (d, 3JC-F = 6.3 Hz, C-10b), 117.8 (d, 3JC-F = 7.1 Hz, C-6a), 127.7 (d, 2JC-F = 18.3 Hz, C-4a), 130.3 (C-10a), 150.1 (d, 1JC-F = 241 Hz, C-5), 153.1 (C-9), 166.2 (CO2H), 170.5 (C-2), 171.1 (–CO–CH3), 176.5 (d, 4JC-F = 3.2 Hz, C-7).
Antibacterial activity
The minimum inhibitory concentrations (MICs) were determined by the conventional broth dilution method using two serial dilution techniques. The standardization of bacterial test suspension was carried out according to Mcfarland standard method as described by the National Committee for Clinical Laboratories Standard (NCCLS) (1993). Stock solutions of the test compounds were prepared using dimethyl sulphoxide (DMSO) to obtain concentration of 200 μg/cm3. Serial dilutions were prepared to obtain test concentrations ranging from 100 to 0.098 μg/cm3. Each tube was then inoculated with 0.1 cm3 of the cultured bacteria (containing approximately 1 to 2 × 108 CFU/cm3), mixed, and incubated at 37°C for 24 h. Growth inhibitions with concentrations at 10 μg/cm3 or lower were carried out in duplicates.
All test tubes showing positive/negative growth were confirmed by the agar plate method. The results were recorded according to presence and absence of growth. The MICs were calculated as the average concentration of the test agent in the broth tubes showing consecutive positive and negative growth. In vitro antibacterial activity of the model compounds are given in .
Table 1. In vitro antibacterial activity (minimum inhibitory concentration (MIC) values, μg/mL) of model compounds of the formulae 6a–c/9a–c as compared with the reference ciprofloxacin.
Control tests for each experiment were performed. Positive growth control was performed by inoculating 0.1 cm3 of the cultured bacteria to a test tube of the culture medium without the test compound. Negative growth control was also performed using uninoculated tube of medium without the test compound. Both were incubated for 24 h at 37°C for both types of bacteria.
Positive and negative controls were performed with DMSO at the same dilution as in experiment to assure that it is incapable of inhibiting the growth of bacteria. Test tubes were incubated at 37°C 24 h and found to have no influence on microbial growth at tested concentration.Citation31
Results and discussion
Chemistry
The desired 10-cyclopropyl-5-fluoro-2,7-dioxo-hexahydropyrido[2,3-f]quinoxaline-3-carboxylic acids (e.g. 8a–c) and N4-acetylated compounds (9a–c), incorporating masked α-amino acid residues are hitherto undescribed and for which a synthetic route is outlined herein (). This preparative approach entails the construction of 3,4-dihydropyrazinone moiety onto an 1-cyclopropyl-8-nitro-4-quinolone skeleton bearing α-amino acid at C-7 (compounds 6a–c; ), and is analogous to the methodology reported for the preparation of 3,4-dihydroquinoxalinones starting from N-(2-nitrophenyl)- or N-(2,4-dinitrophenyl)amino acids.Citation29 The new compounds 6a–c, required in this study, are readily prepared by direct interaction of 7-chloro-1-cyclopropyl-6-fluoro-8-nitro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid (4) with the corresponding α-amino acid (5a–c) in aqueous ethanolic sodium hydrogen carbonate solution at 60–90°C for 1–5 days as detailed in the “Materials and methods” section. The required synthon (4), substituted 4(1H)-oxoquinoline-3-carboxylic acid, is prepared from 2,4-dichloro-5-fluoro-3-nitrobenzoic acid and ethyl 3-(dimethylamino)-acrylate according to a reported procedure.Citation30,Citation31
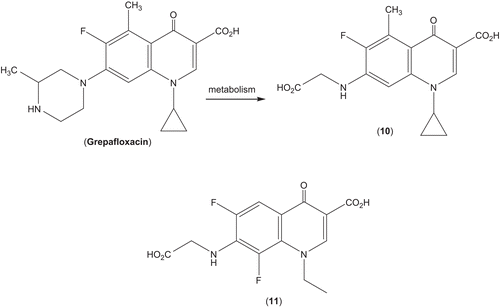
Over the last two decades, a plethora of secondary aminesCitation1–5 and amino alcoholsCitation32 were introduced at C-7 of 6-fluoro-4-oxoquinoline-3-carboxylic acid for enhancement of the antibacterial activity, attainment of good safety profile and optimal blend of favourable bioproperties. However, incorporation of α-amino acids thereat is limited to two reports on the glycine derivatives (10 and 11; ). The former compound (10) was detected as a metabolite (beside others) of grepafloxacin following its oral administration in mammalians (), and was authenticated by a preparative sample obtainable from the reaction of glycine with 7-chloro-1-cyclopropyl-6-fluoro-5-methyl-4-oxo-1,4-dihydroquinoline-3-carboxylic acid.Citation33 The other related glycine derivative (11) has likewise been prepared via the reaction of glycine with 1-ethyl-6,7,8-trifluoro-4-oxo-1,4-dihydroquinoline-3-carboxylic acid.Citation34
Scheme 2. Fluoroquinolones (10, 11) with glycine substituent.
Reduction of 6a–c with sodium dithionite in aqueous potassium carbonate medium converts the nitro group to an amino group, and is followed by spontaneous lactamization of the resultant intermediates (7a–c) to afford good yields of the corresponding target product 8a–c in fairly pure form. Experimentally, this reductive cyclization procedure has added advantages of being quite simple and fast (5–30 min).Citation28,Citation31 Acetylation of 8a–c with acetyl chloride in presence of triethylamine produces N4-acetylated compounds (9a–c).
The constitutions of the new hexahydropyrido[2,3-f]quinoxalines (8a–c/9a–c) and their fluoroquinolone precursors (6a–c) were based on IR, MS, 1H NMR and 13C NMR spectral data, which were fully consistent with the proposed structures. Signal assignments to the various protons and carbons followed from DEPT and 2D (COSY, HMQC, and HMBC) experiments. For 8a–c/9a–c, long-range correlations are observed between H-9 and each of C-10a, C-7, C-1′, and C(3)–CO2H, as well as between H-6 and each of C-10a, C-7, and C-4a. Corresponding long-range correlations are also observed in 6a–c between H-2, H-5, and their neighbour carbons. Those skeletal carbons (4a,5,6,7,8,8a) of the fused benzenoid ring of compounds 6a–c are recognizable by their signal splitting (doublet) arising from coupling with the fluorine atom, while through space 13C…19F coupling is noticeable for the stereogenic α-C*H carbon (d, JC-F δ 12 Hz) in 6a–c.
It turned out that the H-5 in 6a–c, which resonates at ca. 8 ppm (d, 3JH-F δ 13.5 Hz), shows sizable upfield shift in the corresponding annulated products 8a–c and 9a–c [H-6: d δ 7.6 ppm (d, 3JH-F δ10.3 Hz)]. Similarly, the α-C*H proton in 8a–c is shielded as compared with its resonance position in the 1H NMR spectra of the respective uncyclized precursors (6a–c). Yet, the cyclopropyl methine proton (H-1′), which resonates at ca. 3.6 ppm in 6a–c, experiences downfield shift (to about 4.4 ppm) in the annulated compounds (8a–c/9a–c), probably due to the deshielding effect caused by the proximal electron pair of the pyrazinoid ring nitrogen (N1).
Antibacterial activity
In vitro antibacterial screening of the compounds 9a–c along with 3a and b indicates that acetylation of the−N4– of the quinoxaline ring (9) decreases the over all antibacterial activity, whereas methylation of−N4– produces more active compound (3b), in particular against Gram-negative bacteria.Citation28 The quinoxaline derivative 8b is most active derivative of the non-acetylated series (8a–c) with MIC 5.8 μg/mL against Staphylococcus aureus ATCC 6538a and 11.5 μg/mL against Escherichia coli ATCC 8739. Methylation of the C-3 of quinoxaline increases the activity against S. aureus ATCC 6538a as well as E. coli ATCC 8739 (compound 8b vs. 8a), although introduction of isopropyl group reduces the antibacterial activity (compound 8c vs. 8b). The compound 6a is more active against Gram-positive bacteria (S. aureus ATCC 6538a) than Gram-negative (E. coli ATCC 8739). The fluoroquinolone precursors 6a–c have excellent antibacterial activity than corresponding quinoxaline 8a–c derivatives. It might be due to change in the lipophilicity of the compound. Finally, it has been observed that the preference of quinolone antibacterial for Gram-positive or Gram-negative bacteria is related to the lipophilicity of the side chain at C-7 of the quinolone antibacterials (6a–c). It is generally assumed that the more lipophilic quinolone can penetrate better the lipophilic cell membrane of Gram-positive bacteria, whereas less lipophilic compounds are more liable to penetrate the cell wall of Gram-negative bacteria. However, the overall activity of the compound depends on the hydrophilic/lipophilic balance of the compound.
Acknowledgment
Deanship of Academic Research and Dean, Faculty of Pharmacy, University of Jordan, Amman, Jordan for providing Scifinder and e-library facilities.
Declaration of interest
The authors wish to thank Hamdi Mango Center for Scientific Research-University of Jordan, Amman, for financial support.
References
- Wise R, Andrews JM, Edwards LJ. In vitro activity of Bay 09867, a new quinoline derivative, compared with those of other antimicrobial agents. Antimicrob Agents Chemother 1983, 23, 559–564.
- Grohe K. The chemistry of the quinolones: methods of synthesizing the quinolone ring system. In: Handbook of Experimental Pharmacology. Springer-Verlag, Berlin, Heidelberg, 1998, pp. 13–62.
- Li Q, Mitscher LA, Shen LL. The 2-pyridone antibacterial agents: bacterial topoisomerase inhibitors. Med Res Rev 2000, 20, 231–293.
- Zhanel GG, Ennis K, Vercaigne L, Walkty A, Gin AS, Embil J, Smith H, Hoban DJ. The new fluoroquinolones: focus on respiratory infections. Drugs 2002, 62, 13–59.
- Da Silva AD, De Almeida MV, De Souza MV, Couri MR. Biological activity and synthetic methodologies for the preparation of fluoroquinolones, a class of potent antibacterial agents. Curr Med Chem 2003, 10, 21–39.
- Hagihara M, Kashiwase H, Katsube T, Kimura T, Komai T, Momota K, Ohmine T, Nishigaki T, Kimura S, Shimada K. Synthesis and anti-HIV activity of arylpiperazinyl fluoroquinolones: a new class of anti-HIV agents. Bioorg Med Chem Lett 1999, 9, 3063–3068.
- Kimura F, Katsube T, Nishigaki T, Preparation of trifluoromethyl (piperazinyl)-quinolinecarboxylic acids as anti-HIV agents. Jpn Kokai Tokkyo Koho 1997; JP 09249568. Chem Abstr 1997, 127, 262703.
- Ohmine T, Katsube T, Tsuzaki Y, Kazui M, Kobayashi N, Komai T, Hagihara M, Nishigaki T, Iwamoto A, Kimura T, Kashiwase H, Yamashita M. Anti-HIV-1 activities and pharmacokinetics of new arylpiperazinyl fluoroquinolones. Bioorg Med Chem Lett 2002, 12, 739–742.
- Filipponi E, Cruciani G, Tabarrini O, Cecchetti V, Fravolini A. QSAR study and VolSurf characterization of anti-HIV quinolone library. J Comput Aided Mol Des 2001, 15, 203–217.
- Mikiciuk-Olasik E, Trzebinski P, Nowak R, Kotelko B. Synthesis of new derivatives of 4-acylquinoxalin-2-ones with potential pharmacological activity. Acta Pol Pharm 1994, 51, 231–233.
- Roesner M, Billhardt-Troughton UM, Kirsch R, Kleim JP, Meichsner C, Riess G, Winkler I. Preparation of quinoxalinones as antiviral agents. Eur Pat Appl 1996; EP 708093: A1-19960424, 30 p.
- Meichsner C, Riess G, Kleim JP, Roesner M, Paessens A, Blunck M, Combination of quinoxalines and nucleosides for treating viral infection and preparation of the quinoxalines. Eur Pat Appl 1995; EP 657166. Chem Abstr 1995, 123, 228218.
- Billhardt UM, RoesnerM, Riess G, Winkler I, Bender R. 3,4-Dihydro-2-quinoxalinones, 3,4-dihydro-2-quinoxalinethiones and analogs, methods for their preparation and their use as virucides. Eur Pat Appl 1992; EP 509398. Chem Abstr 1993, 118, 234088.
- Kleim JP, Bender R, Kirsch R, Meichsner C, Paessens A, Rösner M, Rübsamen-Waigmann H, Kaiser R, Wichers M, Schneweis KE. Preclinical evaluation of HBY 097, a new nonnucleoside reverse transcriptase inhibitor of human immunodeficiency virus type 1 replication. Antimicrob Agents Chemother 1995, 39, 2253–2257.
- Rübsamen-Waigmann H, Huguenel E, Paessens A, Kleim JP, Wainberg MA, Shah A. Second-generation non-nucleosidic reverse transcriptase inhibitor HBY097 and HIV-1 viral load. Lancet 1997, 349, 1517.
- Kleim JP, Winkler I, Rösner M, Kirsch R, Rübsamen-Waigmann H, Paessens A, Riess G. In vitro selection for different mutational patterns in the HIV-1 reverse transcriptase using high and low selective pressure of the nonnucleoside reverse transcriptase inhibitor HBY 097. Virology 1997, 231, 112–118.
- Hayashi S, Jayesekera D, Jayewardene A, Shah A, Thevanayagam L, Aweeka F. Altered pharmacokinetics of indinavir by a novel nonnucleoside reverse transcriptase inhibitor (HBY-097): a pharmacokinetic evaluation in HIV-positive patients. J Clin Pharmacol 1999, 39, 1085–1093.
- Rübsamen-Waigmann H, Huguenel E, Shah A, Paessens A, Ruoff HJ, von Briesen H, Immelmann A, Dietrich U, Wainberg MA. Resistance mutations selected in vivo under therapy with anti-HIV drug HBY 097 differ from resistance pattern selected in vitro. Antiviral Res 1999, 42, 15–24.
- Rizzo RC, Udier-Blagovic M, Wang DP, Watkins EK, Kroeger Smith MB, Smith RH Jr, Tirado-Rives J, Jorgensen WL. Prediction of activity for nonnucleoside inhibitors with HIV-1 reverse transcriptase based on Monte Carlo simulations. J Med Chem 2002, 45, 2970–2987.
- Balzarini J, Karlsson A, Meichsner C, Paessens A, Riess G, De Clercq E, Kleim JP. Resistance pattern of human immunodeficiency virus type 1 reverse transcriptase to quinoxaline S-2720. J Virol 1994, 68, 7986–7992.
- Kleim JP, Bender R, Billhardt UM, Meichsner C, Riess G, Rösner M, Winkler I, Paessens A. Activity of a novel quinoxaline derivative against human immunodeficiency virus type 1 reverse transcriptase and viral replication. Antimicrob Agents Chemother 1993, 37, 1659–1664.
- Arasteh K, Wood R, Müller M, Prince W, Cass L, Moore KH, Dallow N, Jones A, Klein A, Burt V, Kleim JP. GW420867X administered to HIV-1-infected patients alone and in combination with lamivudine and zidovudine. HIV Clin Trials 2001, 2, 307–316.
- Mutch PJ, Dear GJ, Ismail IM. Formation of a defluorinated metabolite of a quinoxaline antiviral drug catalysed by human cytochrome P450 1A2. J Pharm Pharmacol 2001, 53, 403–408.
- Moore KH, Cass LM, Dallow N, Hardman TC, Jones A, Boyce M, Prince WT. Pharmacokinetics and safety of escalating single and repeat oral doses of GW420867X, a novel non-nucleoside reverse transcriptase inhibitor. Eur J Clin Pharmacol 2001, 56, 805–811.
- Thomas S, Cass L, Prince W, Segal M. Brain and CSF entry of the novel non-nucleoside reverse transcriptase inhibitor, GW420867X. Neuroreport 2000, 11, 3811–3815.
- Balzarini J, De Clercq E, Carbonez A, Burt V, Kleim JP. Long-term exposure of HIV type 1-infected cell cultures to combinations of the novel quinoxaline GW420867X with lamivudine, abacavir, and a variety of nonnucleoside reverse transcriptase inhibitors. AIDS Res Hum Retroviruses 2000, 16, 517–528.
- GraulA, Rabasseda X, Castañer J. GW-420867X. Anti-HIV, Reverse transcriptase inhibitor. Drugs Fut 2000, 25, 24–28.
- Al-Hiari YM, Qaisi AM, AbuShuheil MY, El-Abadelah MM, Voelter W. Synthesis and antibacterial potency of 4-methyl-2,7-dioxo-1,2,3,4,7,10-hexahydropyrido[2,3-f]-quinoxaline-8-carboxylic acid, selected [a]-fused heterocycles and acyclic precursors. Zeitschrift fuer Naturforschung B: Chem Sci 2007, 62B, 1453–1458.
- Chicharro R, De Castro S, Reino RL, Aran JV. Synthesis of tri- and tetracyclic condensed quinoxalin-2-ones fused across the C-3–N-4 bond. Eur J Org Chem 2003, 12, 2314–2326.
- Abu Shuheil MY, Hassuneh MR, Al-Hiari YM, Qaisi AM, El-Abadelah MM, Heterocyclyes [h]-fused onto 4-oxoquinoline-3-carboxylic acid III—facile synthesis and antitumor activity of model heterocycles [a] fused onto pyrido[2,3-f]-quinoloxaline-3-carboxylic acids. Heterocycles 2007, 71, 2155–2172.
- Al-Hiari YM, Al-Mazari IS, Shakya AK, Darwish RM, Abu-Dahab R. Synthesis and antibacterial properties of new 8-nitrofluoroquinolone derivatives. Molecules 2007, 12, 1240–1258.
- Sakae N, Yazaki A, Kuramoto Y, Yoshida J, Niino Y, Oshita Y, Hirao Y, Hayashi N, Amano H. Preparation of quinolinecarboxylic acid derivatives as bactericides. PCT Int. Appl. 1998; WO 98 23, 592. Chem Abstr 1998, 129, 41087j.
- Morita S, Otsubo K, Matsubara J, Ohtani T, Kawano Y, Ohmori K, Ohguro K, Uchida M. Synthesis of possible metabolites of 1-cyclopropyl-1,4-dihydro-6-fluoro-5-methyl-7-(3-methyl-1-piperazinyl)-4-oxo-3-quinolinecarboxylic acid (Grepafloxacin, OPC-17116). Chem Pharm Bull 1995, 43, 2246–2252.
- Sarma MR, Kumar NV, Prasad ASR, Eswaraiah S, Prabhakar C, Reddy GO, Kumar MS, Sadhukhan AK, Venkateswarlu A, Reddy KA. Synthesis and biological activity of novel antibacterial quinolones. Indian J Chem 2001, 40B, 331–335.