Abstract
A novel series of isoindolo[2,1-a]quinoxaline and indolo[1,2-a]quinoxaline derivatives was synthesized and evaluated in vitro against various human cancer cell lines for antiproliferative activity. These new compounds displayed activity against leukemia and breast cancer cell lines in the 3- to 18-µM concentration range.
Introduction
The pyrrolo[1,2-a]quinoxaline heterocyclic framework constitutes the basis of an important class of compounds possessing interesting biological activities. These compounds have been reported to serve as key intermediates for the assembly of several heterocycles including antipsychotic agentCitation1, anti-HIV agentCitation2, adenosine A3 receptor modulatorCitation3, antiparasitic agentsCitation4–7, and antitumor agentsCitation8,Citation9. In this last field, the discovery and development of novel therapeutic agents are one of the most important goals in medicinal chemistry. Recently, we have designed and developed a series of new interesting antiproliferative-substituted pyrrolo[1,2-a]quinoxalines I–IICitation10,11 ().
Figure 1. Structure of compounds I, II and new synthesized substituted isoindolo- or indoloquinoxaline derivatives 1a-h.
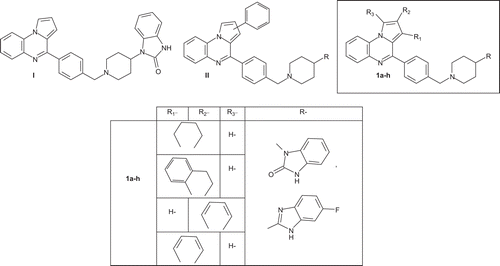
As an extension of our work on the development of anticancer drugs of the pyrroloquinoxaline typeCitation10,Citation11, we synthesized new conformationally restricted analogues of our previously synthesized compounds II such as isoindolo[2,1-a]quinoxaline and indolo[1,2-a]quinoxaline structures 1, which could be defined as new bioisosteres of the pyrrolo[1,2-a]quinoxaline skeleton (). Cyclizing open structures or creating an additional ring system in a given structure represents one of the useful methods in the search for biologically active conformations. The end result is a more constrained molecule, with an imposed conformationCitation12. Moreover, these new isoindolo[2,1-a]quinoxaline and indolo[1,2-a]quinoxaline moieties 1 were substituted by a benzylpiperidinyl benzimidazolone substituent or benzylpiperidinyl fluorobenzimidazole group in analogy to our previously bio-active reference compounds I–IICitation10,Citation11. The antiproliferative profile of the obtained derivatives 1 was then evaluated in vitro against a panel of five leukemic cell lines: U937, K562, Jurkat, U266, and HL60, and against one breast cancer cell line: MCF7. Moreover, to determine their respective cytotoxicity, the new isoindolo[2,1-a]quinoxaline and indolo[1,2-a]quinoxaline derivatives were tested on activated human peripheral blood mononuclear cells.
Materials and methods
Chemistry
Instrumentation
Melting points were determined with an SM-LUX-POL Leitz hot-stage microscope and reported uncorrected. IR spectra were recorded on a BRUKER IFS-25 spectrophotometer. NMR spectra were recorded on a BRUKER AVANCE 300 spectrometer (300 MHz). Chemical shifts refer to tetramethylsilane, which was used as an internal reference. Analytical thin layer chromatography was carried out on 0.25 precoated silica gel plates (POLYGRAM SIL G/UV254) with visualisation by irradiation with a UV lamp. Silica gel 60 (70–230 mesh) was used for column chromatography. Mass spectra were recorded on a Micromass-Waters Q-TOF Ultima spectrometer. Elemental analyses were conducted by CNRS, Vernaison, France. Compound LY-294002 was purchased from Sigma-Aldrich. Microwave experiments were carried out at atmospheric pressure using a microwave reactor (MILESTONE Microwave Laboratory Systems Start S). The instrument consists of a continuous focused microwave power output from 0 to 850 W.
Synthesis of ethyl 2-(2-nitrophenyl)-4,5,6,7-tetrahydroisoindole-1-carboxylate, ethyl 2-(2-nitrophenyl)-4,5-dihydrobenzo[e]isoindole-1-carboxylate and ethyl 1-(2-nitrophenyl)indole-2-carboxylate (3a-c). Method A: To the solution of methyl or ethyl substituted pyrrole-2-carboxylate 2a–c or (3.4 mmol) in 12 mL of dimethylformamide (DMF), cesium carbonate (4.06 mmol) was added. The mixture was stirred at room temperature for 10 min, then 1-fluoro-2-nitrobenzene (5.1 mmol) was added. The reaction mixture was refluxed for 1 h 30 min, then diluted in AcOEt (60 mL) after cooling. The reaction mixture was washed with water (2 × 50 mL), then brine (50 mL) and dried over sodium sulfate. The organic layer was concentrated under vacuum to give products 3a–c. Method B: A suspension of methyl- or ethyl-substituted pyrrole-2-carboxylate 2a–b (3.4 mmol), 1-fluoro-2-nitrobenzene (5.1 mmol), and cesium carbonate (4.06 mmol) in 12 mL of DMF was irradiated for 10 min. The irradiation was programmed to maintain a constant temperature (200°C) with a maximal output power of 850 W. The reaction mixture was then diluted in AcOEt (60 mL), washed with water (2 × 50 mL) and brine (50 mL) and then dried over sodium sulfate. The organic layer was concentrated under vacuum to give products 3a–b as oil.
Ethyl 2-(2-nitrophenyl)-4,5,6,7-tetrahydroisoindole-1-carboxylate (3a). Yield: 95% (method A), 88% (method B), yellow oil; IR vmax (KBr)/cm−1 1705 (CO); 1H NMR (300 MHz, CDCl3) δ 8.04 (dd, 1H, J = 7.95 and 1.55 Hz, H-3′), 7.66 (ddd, 1H, J = 7.95, 7.80 and 1.55 Hz, H-4′), 7.54 (ddd, 1H, J = 7.95, 7.80 and 1.55 Hz, H-5′), 7.38 (dd, 1H, J = 7.95 and 1.55 Hz, H-6′), 6.63 (s, 1H, H-3), 4.10 (q, 2H, J = 7.00 Hz, CH2), 2.91–2.86 (m, 2H, CH2), 2.61–2.58 (m, 2H, CH2), 1.83–1.78 (m, 4H, 2 CH2), 1.16 (t, 3H, J = 7.00 Hz, CH3). Anal. Calcd. for C17H18N2O4: C, 64.96; H, 5.77; N, 8.91. Found: C, 65.12; H, 5.72; N, 8.79.
Ethyl 2-(2-nitrophenyl)-4,5-dihydrobenzo[e]isoindole-1-carboxylate (3b). Yield: 97% (method A), 86% (method B), orange oil; IR νmax (KBr)/cm−1 1710 (CO); 1H NMR (300 MHz, CDCl3) δ 8.19 (dd, 1H, J = 8.00 and 2.00 Hz, H-9), 8.06 (dd, 1H, J = 8.10 and 1.50 Hz, H-3′), 7.69 (ddd, 1H, J = 8.10, 7.50 and 1.50 Hz, H-4′), 7.58 (ddd, 1H, J = 8.10, 7.50 and 1.50 Hz, H-5′), 7.44 (dd, 1H, J = 8.10 and 1.50 Hz, H-6′), 7.31–7.18 (m, 3H, H-6, H-7 and H-8), 4.11 (q, 2H, J = 7.15 Hz, CH2), 2.92–2.88 (m, 2H, CH2), 2.72-2.66 (m, 2H, CH2), 1.05 (t, 3H, J = 7.15 Hz, CH3). Anal. Calcd. for C21H18N2O4: C, 69.60; H, 5.01; N, 7.73. Found: C, 69.85; H, 4.92; N, 7.96.
Ethyl 1-(2-nitrophenyl)indole-2-carboxylate (3c). Yield: 75% (method A), yellow crystals, mp = 88°CCitation13.
Synthesis of 7,8,9,10-tetrahydro-5H-isoindolo[2,1-a]quinoxalin-6-one, 11,12-dihydro-5H-benzo[e]isoindolo[2,1-a]quinoxalin-6-one and 5H-indolo[1,2-a]quinoxalin-6-one (4a–c). Method A: A suspension of 3a–c (2.5 mmol) and iron powder (10 mmol) in 12 mL of acetic acid was heated under reflux for 2 h. The reaction mixture was cooled, suspended in 35 mL of a 1-M aqueous solution of HCl, agitated, then filtered off, washed with 1 M (25 mL) HCl, followed by water, AcOEt, Et2O and then dried to give 4a–c as a white solid. Method B: A mixture of pyrrole-2-carboxylate ester 2a (0.25 mmol), o-iodotrifluoroacetanilide (0.375 mmol), CuI (0.025 mmol), l-proline (0.05 mmol), and K2CO3 (0.75 mmol) in 5 mL of DMSO was stirred at 80°C for 24 h. To the cooled solution, 1.5 mL of water was added. The mixture was then heated at 60°C for 15 h. The mixture was cooled to room temperature and diluted with 150 mL of ethyl acetate, washed with water, dried over Na2SO4, and concentrated in vacuum. The residue was purified by column chromatography on silica gel (eluting with 1:1 petroleum ether/ethyl acetate) to provide the desired product 4a.
7,8,9,10-Tetrahydro-5H-isoindolo[2,1-a]quinoxalin-6-one (4a). Yield: 81% (method A), 46% (method B), beige crystals, mp = 273°C; IR νmax (KBr)/cm−1 1645 (CO); 1H NMR (300 MHz, DMSO-d6) δ 10.96 (s, 1H, NH), 7.87 (dd, 1H, J = 7.90 and 1.45 Hz, H-1), 7.83 (s, 1H, H-11), 7.21–7.10 (m, 3H, H-2, H-3, and H-4), 2.95–2.91 (m, 2H, CH2), 2.66–2.63 (m, 2H, CH2), 1.74–1.71 (m, 4H, 2 CH2). Anal. Calcd. for C15H14N2O: C, 75.61; H, 5.92; N, 11.76. Found: C, 75.47; H, 6.04; N, 11.71.
11,12-Dihydro-5H-benzo[e]isoindolo[2,1-a]quinoxalin-6-one (4b). Yield: 79% (method A), beige crystals, mp = 297°C; IR νmax (KBr)/cm−1 1650 (CO); 1H NMR (300 MHz, DMSO-d6) δ 11.26 (s, 1H, NH), 8.85 (dd, 1H, J = 8.10 and 1.90 Hz, H-7), 8.12 (s, 1H, H-13), 7.98 (dd, 1H, J = 7.95 and 1.50 Hz, H-1), 7.27–7.17 (m, 6H, H-2, H-3, H-4, H-8, H-9 and H-10), 2.85–2.82 (m, 2H, CH2), 2.76–2.73 (m, 2H, CH2). Anal. Calcd. for C19H14N2O: C, 79.70; H, 4.93; N, 9.78. Found: C, 79.93; H, 4.99; N, 9.67.
5H-Indolo[1,2-a]quinoxalin-6-one (4c). Yield: 94% (method A), beige crystals, mp > 300°CCitation13.
Synthesis of 6-chloro-isoindolo[2,1-a]quinoxalines and 6-chloro-indolo[1,2-a]quinoxaline (5a–d). A solution of 4a–d (30 mmol) in POCl3 (60 mL) was refluxed for 4 h. After removing excess of reactive under vacuum, the residue was carefully dissolved in water at 0°C and the resulting solution was made basic with 32% aqueous ammonium hydroxide solution. The precipitate was filtered, washed with water, dried and extracted with dichloromethane. The organic layer was washed with water, dried over magnesium sulfate, and evaporated to dryness to give 5.
6-Chloro-7,8,9,10-tetrahydroisoindolo[2,1-a]quinoxaline (5a). Yield: 84%, beige crystals, mp = 127°C; 1H NMR (300 MHz, CDCl3) δ 7.79 (dd, 1H, J = 8.0 and 1.40 Hz, H-1), 7.66 (dd, 1H, J = 8.0 and 1.40 Hz, H-4), 7.63 (s, 1H, H-11), 7.45 (ddd, 1H, J = 8.0, 7.75 and 1.40 Hz, H-2), 7.36 (ddd, 1H, J = 8.0, 7.75 and 1.40 Hz, H-3), 3.17 (t, 2H, J = 5.80 Hz, CH2), 2.78 (t, 2H, J = 5.80 Hz, CH2), 1.90–1.80 (m, 4H, 2 CH2). Anal. Calcd. for C15H13ClN2: C, 70.18; H, 5.10; N, 10.91. Found: C, 69.96; H, 5.38; N, 11.05.
6-Chloro-11,12-dihydro-benzo[e]isoindolo[2,1-a]quinoxaline (5b). Yield: 83%, beige crystals, mp = 173°C; 1H NMR (300 MHz, CDCl3) δ 8.07 (dd, 1H, J = 8.20 and 1.40 Hz, H-7), 7.88 (dd, 1H, J = 7.80 and 1.20 Hz, H-1), 7.86 (s, 1H, H-13), 7.77 (dd, 1H, J = 7.80 and 1.20 Hz, H-4), 7.52 (ddd, 1H, J = 7.80, 7.20 and 1.20 Hz, H-2), 7.42 (ddd, 1H, J = 7.80, 7.20 and 1.20 Hz, H-3), 7.40–7.32 (m, 2H, H-8 and H-10), 7.25 (ddd, 1H, J = 8.20, 7.55 and 1.40 Hz, H-9), 2.94–2.89 (m, 2H, CH2), 2.86–2.81 (m, 2H, CH2). Anal. Calcd. for C19H13ClN2: C, 74.88; H, 4.30; N, 9.19. Found: C, 74.74; H, 4.19; N, 9.30.
6-Chloro-indolo[1,2-a]quinoxaline (5c). Yield: 84%, pale-yellow crystals, mp = 129°C; 1H NMR (300 MHz, CDCl3) δ 8.46 (dd, 1H, J = 8.20 and 1.30 Hz, H-11), 8.41 (dd, 1H, J = 8.40 and 1.10 Hz, H-1), 7.98 (dd, 1H, J = 8.40 and 1.10 Hz, H-4), 7.95 (dd, 1H, J = 8.20 and 1.30 Hz, H-8), 7.68–7.57 (m, 2H, H-2 and H-3), 7.51–7.43 (m, 2H, H-9 and H-10), 7.37 (s, 1H, H-7). Anal. Calcd. for C15H9ClN2: C, 71.30; H, 3.59; N, 11.09. Found: C, 71.25; H, 3.82; N, 10.87.
6-Chloro-isoindolo[2,1-a]quinoxaline (5d). Yield: 47%, pale-yellow crystals, mp = 183°C; (Found M+: 253.0522, C15H9ClN2 requires 253.0533); 1H NMR (300 MHz, CDCl3) δ 8.65 (m, 1H, H-1), 8.54 (s, 1H, H-11), 8.12 (m, 1H, H-10), 8.03 (m, 1H, H-7), 7.92 (m, 1H, H-4), 7.65–7.61 (m, 2H, H-2 and H-3), 7.51–7.46 (m, 2H, H-8 and H-9). Anal. Calcd. for C15H9ClN2: C, 71.30; H, 3.59; N, 11.09. Found: C, 71.56; H, 3.54; N, 10.93.
Synthesis of 4-(isoindolo[2,1-a]quinoxalin-6-yl)benzaldehydes and 4-(indolo[1,2-a]quinoxalin-6-yl)benzaldehyde (6a–d). To suspension of compound 5a–d (4.64 mmol) and Pd(PPh3)4 (0.232 mmol) in a mixture of toluene/EtOH (75/4.1 mL) under nitrogen were added K2CO3 (5.1 mmol) and 4-formylphenylboronic acid (5.1 mmol). The reaction mixture was refluxed for 24 h, and the cooled suspension was extracted with CH2Cl2 (3 × 80 mL). The organic layer was washed with a saturated solution of NaCl (95 mL), and the combined organic extracts were dried over sodium sulfate, filtered, and evaporated under reduced pressure. The crude residue was triturated in ethanol. The resulting precipitate when was filtered, washed with ethanol, and purified by column chromatography on silica gel using dichloromethane as eluent gave the pure product 6a–d.
4-(7,8,9,10-Tetrahydroisoindolo[2,1-a]quinoxalin-6-yl)benzaldehyde (6a). Yield: 89%, yellow crystals, mp = 172°C; IR νmax (KBr)/cm−1 1705 (CO); 1H NMR (300 MHz, CDCl3) δ 10.14 (s, 1H, CHO), 8.05 (d, 2H, J = 8.10 Hz, H-2 and H-6), 8.03 (dd, 1H, J = 8.05 and 1.20 Hz, H1′), 7.87 (s, 1H, H-11′), 7.82 (dd, 1H, J = 8.05 and 1.20 Hz, H-4′), 7.79 (d, 2H, J = 8.10, H-3 and H-5), 7.54 (ddd, 1H, J = 8.05, 7.70 and 1.20 Hz, H-2′), 7.44 (ddd, 1H, J = 8.05, 7.70 and 1.20 Hz, H-3′), 2.84 (t, 2H, J = 6.20 Hz, CH2), 2.18 (t, 2H, J = 6.20 Hz, CH2), 1.85–1.75 (m, 2H, CH2), 1.69–1.60 (m, 2H, CH2). Anal. Calcd. for C22H18N2O: C, 80.96; H, 5.56; N, 8.58. Found: C, 81.05; H, 5.70; N, 8.55.
4-(11,12-Dihydrobenzo[e]isoindolo[2,1-a]quinoxalin-6-yl)benzaldehyde (6b). Yield: 86%, yellow crystals, mp = 174°C; IR νmax (KBr)/cm−1 1710 (CO); 1H NMR (300 MHz, CDCl3) δ 10.07 (s, 1H, CHO), 8.09 (dd, 1H, J = 8.10 and 1.35 Hz, H-7′), 7.98 (s, 1H, H-13′), 7.88 (dd, 1H, J = 8.10 and 1.20 Hz, H-1′), 7.86 (d, 2H, J = 8.10 Hz, H-2 and H-6), 7.83 (d, 2H, J = 8.10 Hz, H-3 and H-5), 7.58 (ddd, 1H, J = 8.00, 7.65 and 1.20 Hz, H-2′), 7.55 (ddd, 1H, J = 8.00, 7.65 and 1.20 Hz, H-3′), 7.26 (dd, 1H, J = 8.00 and 1.20 Hz, H-4′), 6.98 (ddd, 1H, J = 8.10, 7.60, and 1.35 Hz, H-8′), 6.58 (ddd, 1H, J = 8.10, 7.60, and 1.35 Hz, H-9′), 6.15 (dd, 1H, J = 8.10 and 1.35 Hz, H-10′), 3.00–2.95 (m, 2H, CH2), 2.92–2.87 (m, 2H, CH2). Anal. Calcd. for C26H18N2O: C, 83.40; H, 4.85; N, 7.48. Found: C, 83.58; H, 4.97; N, 7.62.
4-(Indolo[1,2-a]quinoxalin-6-yl)benzaldehyde (6c). Yield: 83%, yellow crystals, mp = 196°C; IR νmax (KBr)/cm−1 1690 (CO); 1H NMR (300 MHz, CDCl3) δ 10.18 (s, 1H, CHO), 8.57 (d, 1H, J = 8.10 Hz, H-11′), 8.53 (d, 1H, J = 8.30 Hz, H-1′), 8.24 (d, 2H, J = 8.10 Hz, H-2 and H-6), 8.14–8.11 (m, 3H, H-3, H-5 and H-4′), 7.97 (d, 1H, J = 8.10, H-8′), 7.69 (t, 1H, J = 8.30 Hz, H-2′), 7.62 (t, 1H, J = 8.30 Hz, H-3′), 7.53–7.47 (m, 2H, H-9′ and H-10′), 7.29 (s, 1H, H-7′). Anal. Calcd. for C22H14N2O: C, 81.97; H, 4.38; N, 8.69. Found: C, 82.17; H, 4.42; N, 8.89.
4-(Isoindolo[2,1-a]quinoxalin-6-yl)benzaldehyde (6d). Yield: 79%, yellow crystals, mp = 213°C; IR νmax (KBr)/cm−1 1700 (CO); 1H NMR (300 MHz, CDCl3) δ 10.23 (s, 1H, CHO), 8.62 (s, 1H, H-11′), 8.24–8.20 (m, 1H, H-1′), 8.19–8.16 (m, 1H, H-10′), 8.15 (d, 2H, J = 8.40 Hz, H-2 and H-6), 8.01 (d, 2H, J = 8.40 Hz, H-3 and H-5), 7.93 (d, 1H, J = 8.70, H-4′), 7.69–7.65 (m, 2H, H-2′ and H-3′), 7.41–7.35 (m, 1H, H-9′), 7.31–7.28 (m, 1H, H-7′), 7.20–7.14 (m, 1H, H-8′). Anal. Calcd. for C22H14N2O: C, 81.97; H, 4.38; N, 8.69. Found: C, 82.09; H, 4.34; N, 8.77.
Synthesis of 1,3-dihydro-1-{1-[4-(isoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-ones and 1,3-dihydro-1-{1-[4-(indolo[1,2-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-one (1a–d). The pH of a solution of the aldehyde 6a–d (2.5 mmol) and 4-(2-ketobenzimidazol-1-yl)piperidine (3.0 mmol) in 40 mL methanol was adjusted to 6 by the dropwise addition of acetic acid. Powered sodium cyanoborohydride (6.9 mmol) was then added, and the resultant mixture was refluxed for 5 h. The hot precipitate formed was filtered, washed with methanol then with diethyl ether and dried under reduced pressure to give the compounds 1a–d.
1,3-Dihydro-1-{1-[4-(7,8,9,10-tetrahydroisoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-one (1a). Yield: 43%, pale-yellow crystals, mp = 289°C; IR νmax (KBr)/cm−1 3350 (NH), 1685 (C=O); 1H NMR (300 MHz, CDCl3) δ 10.86 (s, 1H, NH), 8.29 (s, 1H, H-11”), 8.18 (dd, 1H, J = 8.00 and 1.25 Hz, H-1”), 7.80 (dd, 1H, J = 8.00 and 1.25 Hz, H-4”), 7.56–7.38 (m, 6H, H-2′, H-3′, H-5′, H-6′, H-2” and H-3”), 7.26–7.23 (m, 1H, H benzimid.), 6.98–6.96 (m, 3H, H benzimid.), 4.18–4.17 (m, 1H, CH pip.), 3.65 (s, 2H, CH2N), 3.02–2.99 (m, 2H, CH2 pip.), 2.75 (t, 2H, J = 5.90 Hz, CH2), 2.43–2.38 (m, 2H, CH2 pip.), 2.18–2.09 (m, 4H, CH2 pip. and CH2), 1.70–1.65 (m, 4H, CH2 and CH2 pip.), 1.57–1.54 (m, 2H, CH2). Anal. Calcd. for C34H33N5O: C, 77.39; H, 6.30; N, 13.27. Found: C, 77.33; H, 6.21; N, 13.45.
1,3-Dihydro-1-{1-[4-(11,12-dihydrobenzo[e]isoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-one. 2 HCl (1b). Yield: 76%, orange crystals, mp = 298°C; IR νmax (KBr)/cm−1 3400 (NH), 2650–2350 (NH+), 1695 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 11.93 (s, 1H, NH+), 11.04 (s, 1H, NH), 9.03 (s, 1H, H-13”), 8.49 (d, 1H, J = 7.50 Hz, H-7”), 8.18 (d, 1H, J = 7.30 Hz, H-1”), 7.86–7.75 (m, 6H, H-2′, H-3′, H-5′, H-6′, H-2” and H benzimid.), 7.62 (t, 1H, J = 7.30 Hz, H-3”), 7.27 (d, 1H, J = 7.30 Hz, H-4”), 7.03–6.98 (m, 4H, H-8” and 3H benzimid.), 6.69 (t, 1H, J = 7.50 Hz, H-9”), 6.11 (d, 1H, J = 7.50 Hz, H-10”), 4.61–4.59 (m, 1H, CH pip.), 4.45 (s, 2H, CH2N), 4.40 (bs, 1H, NH+), 3.47–3.42 (m, 2H, CH2 pip.), 3.21–3.13 (m, 2H, CH2 pip.), 3.08–2.88 (m, 6H, CH2 pip. and 2 CH2), 1.95–1.91 (m, 2H, CH2 pip.). Anal. Calcd. for C38H35Cl2N5O: C, 70.37; H, 5.44; N, 10.80. Found: C, 70.46; H, 5.57; N, 11.04.
1,3-Dihydro-1-{1-[4-(indolo[1,2-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-one (1c). Yield: 69%, yellow crystals, mp = 236°C; IR νmax (KBr)/cm−1 3350 (NH), 1685 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 10.87 (s, 1H, NH), 8.77–8.73 (m, 2H, H-11” and H-1”), 8.05–8.00 (m, 4H, H-3′, H-5′, H-4” and H-8”), 7.71(t, 1H, J = 8.10 Hz, H-2”), 7.61 (t, 1H, J = 8.10 Hz, H-3”), 7.59 (d, 2H, J = 8.10 Hz, H-2′ and H-6′), 7.54-7.48 (m, 2H, H-9” and H-10”), 7.39 (s, 1H, H-7”), 7.28–7.25 (m, 1H, H benzimid.), 7.01–6.96 (m, 3H, H benzimid.), 4.23–4.14 (m, 1H, CH pip.), 3.66 (s, 2H, CH2N), 3.05–3.01 (m, 2H, CH2 pip.), 2.45–2.40 (m, 2H, CH2 pip.), 2.21–2.13 (m, 2H, CH2 pip.), 1.70–1.64 (m, 2H, CH2 pip.). Anal. Calcd. for C34H29N5O: C, 77.99; H, 5.58; N, 13.37. Found: C, 78.20; H, 5.62; N, 13.45.
1,3-Dihydro-1-{1-[4-(isoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-one (1d). Yield: 72%, yellow crystals, mp = 255°C; IR νmax (KBr)/cm−1 3345 (NH), 1690 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 10.83 (s, 1H, NH), 9.24 (s, 1H, H-11”), 8.64 (dd, 1H, J = 8.50 and 1.80 Hz, H-10”), 8.02–7.99 (m, 1H, H-1”), 7.95 (d, 1H, J = 8.50 Hz, H-4”), 7.75 (d, 2H, J = 7.95 Hz, H-3′ and H-5′), 7.73–7.67 (m, 2H, H-2” and H-3”), 7.62 (d, 2H, J = 7.95 Hz, H-2′ and H-6′), 7.36–7.27 (m, 1H, H-9”), 7.26–7.22 (m, 2H, H-7” and H benzimid.), 7.17–7.11 (m, 1H, H-8”), 7.01–6.96 (m, 3H, H benzimid.), 4.26–4.16 (m, 1H, CH pip.), 3.73 (s, 2H, CH2N), 3.09–3.05 (m, 2H, CH2 pip.), 2.50–2.37 (m, 2H, CH2 pip.), 2.25–2.17 (m, 2H, CH2 pip.), 1.73–1.68 (m, 2H, CH2 pip.). Anal. Calcd. for C34H29N5O: C, 77.99; H, 5.58; N, 13.37. Found: C, 78.17; H, 5.43; N, 13.30.
Synthesis of 5-fluoro-2-{1-[4-(isoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-1H-benzimidazoles and 5-fluoro-2-{1-[4-(indolo[1,2-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-1H-benzimidazole (1e–h). The pH of a solution of the aldehyde 6a–d (2.5 mmol) and 4-(5-fluorobenzimidazol-2-yl)piperidine (3.0 mmol) in 40 mL methanol was adjusted to 6 by the dropwise addition of acetic acid. Powered sodium cyanoborohydride (6.9 mmol) was then added, and the resultant mixture was refluxed for 5 h. After removal of the methanol by rotary evaporation, the residue was triturated in water and extracted with dichloromethane. The organic layer was washed with water, dried over magnesium sulfate and evaporated to dryness. Column chromatography of the residue on silica gel using ethyl acetate–cyclohexane (1/1) then methanol–chloroform (1/9) as eluents gave the crude product. This solid was then triturated with diethyl ether, filtered, washed with diethyl ether and dried under reduced pressure to give the compounds 1e–h.
5-Fluoro-2-{1-[4-(7,8,9,10-tetrahydroisoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-1H-benzimidazole (1e). Yield: 63%, pale-yellow crystals, mp = 152°C; IR νmax (KBr)/cm−1 3340 (NH), 1685 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 12.41 (s, 1H, NH), 8.30 (s, 1H, H-11”), 8.19 (d, 1H, J = 8.10 Hz, H-1”), 7.80 (d, 1H, J = 8.10 Hz, H-4”), 7.57–7.31 (m, 7H, H-2′, H-3′, H-5′, H-6′, H-2”, H-3” and H benzimid.), 7.23–7.20 (m, 1H, H benzimid.), 6.99–6.96 (m, 1H, H benzimid.), 3.63 (s, 2H, CH2N), 2.98–2.93 (m, 2H, CH2 pip.), 2.87–2.85 (m, 1H, CH pip.), 2.76 (t, 2H, J = 5.85 Hz, CH2), 2.17–2.12 (m, 4H, CH2 and CH2 pip.), 2.00–1.98 (m, 2H, CH2 pip.), 1.90–1.84 (m, 2H, CH2 pip.), 1.71–1.68 (m, 2H, CH2), 1.56–1.54 (m, 2H, CH2). Anal. Calcd. for C34H32FN5: C, 77.10; H, 6.09; N, 13.22. Found: C, 77.24; H, 6.32; N, 13.15.
5-Fluoro-2-{1-[4-(11,12-dihydrobenzo[e]isoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-1H-benzimidazole (1f). Yield: 40%, yellow crystals, mp = 179°C; IR νmax (KBr)/cm−1 3340 (NH), 1690 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 12.33 (s, 1H, NH), 8.54 (s, 1H, H-13”), 8.29 (d, 1H, J = 7.80 Hz, H-7”), 7.91 (dd, 1H, J = 8.10 and 1.20 Hz, H-1”), 7.61 (ddd, 1H, J = 8.10, 7.90 and 1.20 Hz, H-2”), 7.51 (d, 2H, J = 8.10 Hz, H-3′ and H-5′), 7.50–7.48 (m, 2H, H-3” and H benzimid.), 7.23 (d, 2H, J = 8.10 Hz, H-2′ and H-6′),7.22–7.20 (m, 2H, H-4” and H benzimid.), 7.02–6.91 (m, 2H, H-8” and H benzimid.), 6.52 (t, 1H, J = 7.80 Hz, H-9”), 6.11 (d, 1H, J = 7.80 Hz, H-10”), 3.57 (s, 2H, CH2N), 2.97–2.81 (m, 7H, CH2 pip., CH pip. and 2 CH2), 2.15–2.04 (m, 4H, 2 CH2 pip.), 1.91–1.83 (m, 2H, CH2 pip.). Calcd. for C38H32FN5: C, 79.01; H, 5.58; N, 12.12. Found: C, 78.95; H, 5.57; N, 12.04.
5-Fluoro-2-{1-[4-(indolo[1,2-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-1H-benzimidazole (1g). Yield: 86%, yellow crystals, mp = 139°C; IR νmax (KBr)/cm−1 3345 (NH), 1690 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 12.27 (s, 1H, NH), 8.76-8.73 (m, 2H, H-11” and H-1”), 8.04–7.98 (m, 4H, H-3′, H-5′, H-4” and H-8”), 7.70 (ddd, 1H, J = 8.40, 7.20 and 1.20 Hz, H-2”), 7.62–7.54 (m, 3H, H-2′, H-6′ and H-3”), 7.51–7.45 (m, 2H, H-9” and H-10”), 7.39 (s, 1H, H-7”), 7.36-7.31 (m, 1H, H benzimid.), 7.22–7.18 (m, 1H, H benzimid.), 7.02–6.94 (m, 1H, H benzimid.), 3.64 (s, 2H, CH2N), 2.99–2.90 (m, 2H, CH2 pip.), 2.87–2.83 (m, 1H, CH pip.), 2.21–2.13 (m, 2H, CH2 pip.), 2.04–2.00 (m, 2H, CH2 pip.), 1.93–1.82 (m, 2H, CH2 pip.). Calcd. for C34H28FN5: C, 77.69; H, 5.37; N, 13.32. Found: C, 77.56; H, 5.48; N, 13.11.
5-Fluoro-2-{1-[4-(isoindolo[2,1-a]quinoxalin-6-yl)benzyl]piperidin-4-yl}-1H-benzimidazole (1h). Yield: 66%, yellow crystals, mp = 196°C; IR νmax (KBr)/cm−1 3350 (NH), 1690 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 12.48 (bs, 1H, NH), 9.24 (s, 1H, H-11”), 8.64 (dd, 1H, J = 8.10 and 2.10 Hz, H-10”), 8.03–7.99 (m, 1H, H-1”), 7.95 (d, 1H, J = 8.40 Hz, H-4”), 7.72 (d, 2H, J = 7.85 Hz, H-3′ and H-5′), 7.70–7.66 (m, 2H, H-2” and H-3”), 7.58 (d, 2H, J = 7.85 Hz, H-2′ and H-6′), 7.48–7.44 (m, 1H, H benzimid.), 7.35–7.21 (m, 3H, H-7”, H-9” and H benzimid.), 7.16–7.10 (m, 1H, H-8”), 6.99–6.92 (m, 1H, H benzimid.), 3.68 (s, 2H, CH2N), 3.02–2.97 (m, 2H, CH2 pip.), 2.92–2.83 (m, 1H, CH pip.), 2.23-2.15 (m, 2H, CH2 pip.), 2.08–2.01 (m, 2H, CH2 pip.), 1.92–1.78 (m, 2H, CH2 pip.). Anal. Calcd. for C34H28FN5: C, 77.69; H, 5.37; N, 13.32. Found: C, 77.42; H, 5.21; N, 13.57.
2-(2-Aminophenyl)-2H-isoindole-1-carboxylic acid (8). A solution of 1-cyano-2-(2-aminophenyl)isoindole 7 (3 mmol) in acetic acid (10 mL) was refluxed for 30 min. After cooling, the reaction mixture was poured into ice water (150 mL) and stirred for 15 min. The resulting solution was evaporated under reduced pressure. The residue was cooled and triturated in diethyl ether. The precipitate was collected by filtration and dried to give 8. Yield: 98%, orange crystals, mp > 300°C; IR νmax (KBr)/cm−1 3360–2500 (NH2 and OH), 1660 (C=O); 1H NMR (300 MHz, DMSO-d6) δ 12.03 (bs, 1H, COOH), 8.92 (s, 1H, H-3), 8.44 (dd, 1H, J = 8.10 and 0.90 Hz, H-7), 8.37 (dd, 1H, J = 8.10 and 0.90 Hz, H-4), 7.89–7.86 (m, 1H, H-6′), 7.57 (dd, 1H, J = 8.10 and 1.20 Hz, H-3′), 7.46 (ddd, 1H, J = 8.10, 7.50 and 1.20 Hz, H-4′), 7.34–7.28 (m, 3H, H-5, H-6 and H-5′), 6.91 (s, 2H, NH2). Anal. Calcd. for C15H12N2O2: C, 71.42; H, 4.79; N, 11.10. Found: C, 71.37; H, 4.96; N, 11.39.
2-(2-Fluorophenyl)-2H-isoindole-1-carbonitrile (9). To a solution of sodium hydrogen sulfite (0.015 mol) in water (38 mL) was added phthalaldehyde (0.015 mol). The mixture was stirred until the solid was dissolved, and 2-fluoroaniline (0.015 mol) was added. The reaction was heated at 70°C for 30 min, cooled, KCN (0.052 mol) in water (8.0 mL) was added, and the mixture was heated at 70°C for an additional 90 min. The solid formed upon cooling was filtered, washed with water and extracted with dichloromethane. The organic layer was dried over magnesium sulfate and evaporated to dryness. Column chromatography of the residue on silica gel using dichloromethane as eluent gave 9. Yield: 32%, pale-yellow crystals, mp = 120°C; IR νmax (KBr)/cm−1 2200 (C≡N); 1H NMR (300 MHz, CDCl3) δ 7.73 (dd, 1H, J = 8.40 and 0.90 Hz, H-4), 7.69 (dd, 1H, J = 8.40 and 0.90 Hz, H-7), 7.59–7.48 (m, 3H, H-4′, H-5′ and H-6′), 7.38–7.28 (m, 3H, H-3′, H-3 and H-5), 7.17 (ddd, 1H, J = 8.40, 7.80 and 0.90 Hz, H-6). Anal. Calcd. for C15H9FN2: C, 76.26; H, 3.84; N, 11.86. Found: C, 76.36; H, 3.77; N, 11.65.
5H-Isoindolo[2,1-a]quinoxalin-6-one (4d). A suspension of 2-(2-fluorophenyl)-2H-isoindole-1-carbonitrile 9 (3 mmol) and 85% potassium hydroxide (12 mmol) in tert-butanol (21 mL) was heated at 80°C for 4 h, then cooled, poured into crushed ice, and stirred for 10 min. The resulting precipitate was filtered, washed with water then with diethyl ether, and dried under reduced pressure to give 4d. Yield: 80%, white crystals, mp > 260°C; (Found MNa+: 257.0699, C15H10N2OCitation23Na requires 257.0691); IR νmax (KBr)/cm−1 1645 (CO); 1H NMR (300 MHz, DMSO-d6) δ 11.40 (s, 1H, NH), 8.78 (s, 1H, H-11), 8.35 (dd, 1H, J = 8.10 and 1.00 Hz, H-1), 8.31 (dd, 1H, J = 7.90 and 0.90 Hz, H-10), 7.83 (dd, 1H, J = 7.90 and 0.90 Hz, H-7), 7.45–7.39 (m, 2H, H-2 and H-4), 7.33–7.22 (m, 3H, H-3, H-8 and H-9). Anal. Calcd. for C15H10N2O: C, 76.91; H, 4.30; N, 11.96. Found: C, 77.09; H, 4.10; N, 12.07.
Biology
Cell culture. The human leukemic cell lines U937, K562, and HL60, and the breast cancer cell-line MCF7 were grown in RPMI 1640 medium (Life Technology, France) supplemented with 10% fetal calf serum, antibiotics (100 U/mL penicillin, 100 µg/mL streptomycin), and l-glutamin, at 37°C, 5% CO2 in air. The toxicity of various molecules was also evaluated on non-activated, freshly isolated normal human peripheral blood mononuclear cells (PBMNC) and phytohemagglutinin (lymphoproliferative agent) (PHA)-induced cells. PBMNC from blood of healthy volunteers were obtained following centrifugation on Ficoll gradient. Cells were then incubated in medium alone or induced to enter cell cycle by the addition of PHA (5 µg/mL, Murex Biotech Limited, Dartford, UK).
Cytotoxicity Test. The 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethonyphenol)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) cell proliferation assay (Promega, France) is a colorimetric assay system, which measures the reduction of a tetrazolium component (MTS) into formazan produced by the mitochondria of viable cells. Cells were washed twice in phosphate buffer saline and plated in quadruplicate into microtiter-plate wells in 100-µL culture media without or with our various compounds at increasing concentrations (0, 1, 5, 10, 20, and 50 µM). After 3 h of incubation at 37°C with 20 µL MTS/well, the plates were read using an ELISA microplate reader (Thermo, Electrocorporation) at 490 nm wavelength. The amount of colour produced was directly proportional to the number of viable cells. The results are expressed as the concentrations inhibiting cell growth by 50% after a 3 days incubation period. The 50% inhibitory concentrations (IC50) were determined by linear regression analysis, expressed in μM ± SD (Microsoft Excel).
Results and discussion
Chemistry
The synthesis of the new isoindolo- or indoloquinoxaline derivatives 1a–h has been accomplished according to the sequence depicted in . Condensation of nitrocyclohexenes with ethyl isocyanoacetate in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) afforded the corresponding fused pyrroles 2a–b using a Barton-Zard reactionCitation14–16. The not commercially available, starting material 2-nitro-3,4-dihydronaphthalene was readily prepared by the nitration of the corresponding alkeneCitation17. The preparation of N-aryl pyrroles 3a-c was then obtained by nucleophilic substitution of the various alkyl pyrrole-2-carboxylates 2a–c with 1-fluoro-2-nitrobenzene using cesium carbonate as the base in refluxing DMF solutionCitation3,Citation10. The preparation of 3a and 3b was also performed under microwave irradiation.
Scheme 1. Reagents and conditions: (i) DBU, THF/t-BuOH, 50°C; (ii) 1-fluoro-2-nitrobenzene, Cs2CO3, DMF, Δ; (iii) Fe, CH3COOH, Δ; (iv) 1) o-iodotrifluoroacetanilide, K2CO3, CuI, L-proline, DMSO, 80°C; 2) H2O, 60°C (v) POCl3, Δ; (vi) OHC-C6H4-B(OH)2, Pd[P(C6H5)3]4, K2CO3, toluene, EtOH, Δ; (vii) 4-(2-ketobenzimidazolin-1-yl)piperidine or 4-(5-fluorobenzimidazolin-2-yl)piperidine, NaBH3CN, MeOH, Δ.
![Scheme 1. Reagents and conditions: (i) DBU, THF/t-BuOH, 50°C; (ii) 1-fluoro-2-nitrobenzene, Cs2CO3, DMF, Δ; (iii) Fe, CH3COOH, Δ; (iv) 1) o-iodotrifluoroacetanilide, K2CO3, CuI, L-proline, DMSO, 80°C; 2) H2O, 60°C (v) POCl3, Δ; (vi) OHC-C6H4-B(OH)2, Pd[P(C6H5)3]4, K2CO3, toluene, EtOH, Δ; (vii) 4-(2-ketobenzimidazolin-1-yl)piperidine or 4-(5-fluorobenzimidazolin-2-yl)piperidine, NaBH3CN, MeOH, Δ.](/cms/asset/cc5a9a61-5be8-40e4-8359-e6791beb5acd/ienz_a_548326_f0002_b.gif)
Reduction of the nitro moiety of 3a–c with iron in hot glacial acetic acid produced the spontaneous ring closure onto the ester to afford the desired isoindolo- or indoloquinoxalinones 4a–c through a one-pot reduction–cyclization stepCitation3,Citation13. CuI/L-proline-catalyzed coupling of 2-iodotrifluoroacetanilide with pyrrole-2-carboxylate ester 2a in DMSO at 80−90°C followed by in situ hydrolysis at 60°C also afforded the isoindolo[2,1-a]quinoxalin-6-one 4aCitation18. The lactams 4a–d were subsequently chlorodehydroxylated with phosphorous oxychloride, leading to the 6-chloroquinoxalines 5a–d. Coupling chloro derivatives 5a–d with 4-formylphenylboronic acid in the presence of Pd(PPh3)4 as a catalyst under Suzuki-Miyaura cross-coupling conditions proceeded to afford the substituted benzaldehydes 6a–dCitation19,Citation10. Reductive amination of benzaldehydes 6a–d with 4-(2-ketobenzimidazolin-1-yl)piperidine or 4-(5-fluorobenzimidazol-2-yl)piperidine using NaBH3CN provided the final products 1a–h. The 3D spatial determinations of 1a and 1c were established by X-ray crystallographyCitation20 and confirmed the structures in the solid state as anticipated on the basis of IR and 1H NMR data ().
Figure 2. The ORTEP drawing of 1,3-dihydro-1-{1-[4-(pyrrolo[1,2-a]quinoxalin-4-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-ones 1a and 1c with thermal ellipsoids at 30% level.
![Figure 2. The ORTEP drawing of 1,3-dihydro-1-{1-[4-(pyrrolo[1,2-a]quinoxalin-4-yl)benzyl]piperidin-4-yl}-2H-benzimidazol-2-ones 1a and 1c with thermal ellipsoids at 30% level.](/cms/asset/78a1dae4-a122-4600-89ea-4f2362461e93/ienz_a_548326_f0003_b.gif)
The synthesis of the isoindoloquinoxalinone derivative 4d was first supported by the work of the Diana et al.Citation21,Citation22 using 2-(2′-aminophenyl)-1-cyano-isoindole 7 as a key intermediate (). The isolation of derivative 7 was possible by a Strecker-type synthesis between substituted 1,2-phenylendiamine and phthalaldehyde in water and in the presence of potassium cyanide and sodium hydrogensulfiteCitation21,Citation22. Refluxing compound 7 in acetic acid followed by an in situ hydrolysis failed in the synthesis of lactam 4d, and only furnished the 2-(2-aminophenyl)-2H-isoindole-1-carboxylic acid 8. The structural identification of compound 8 was unambiguously established on the basis of IR and 1H NMR data. The zwitterionic structure of 8 has been confirmed by IR spectrometry. The IR spectrum indicated the presence of a carboxylate COO− band and a sharp ammonium NH3+ band, respectively, observed at 1660 and 1910 cm−1. Moreover, the 1H NMR spectrum showed a broad singlet at 12.03 ppm (1H), highly characteristic for a carboxylic acid proton, and a singlet at 6.91 ppm (2H), attributed to the amine group.
Scheme 2. Reagents and conditions: (i) NaHSO3, KCN, H2O, 70°C; (ii) CH3COOH, Δ; (iii) KOH, tert-BuOH, 80°C.
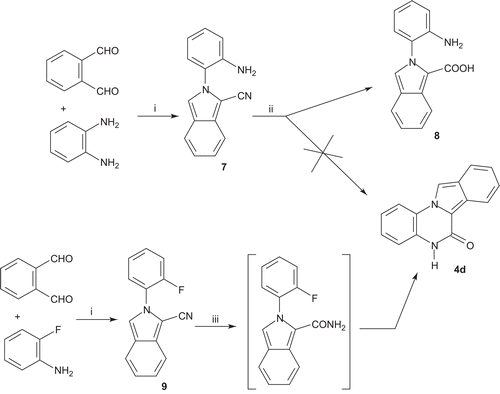
The preparation of 4d was achieved by using the following strategy via the 2-(2-fluorophenyl)-2H-isoindole-1-carbonitrile 9 (). This cyano-substituted isoindole 9 was also preliminary prepared by a Strecker-type synthesis between 2-fluoroaniline and o-phthalodicarboxaldehyde in water and in the presence of potassium cyanide and sodium hydrogensulfiteCitation23. Treatment of 9 with potassium hydroxide in tert-butanol at 80°C led directly to the tetracyclic lactam 4d involving selective hydrolysis to amide, which was able to carry out intramolecular displacement of aromatic fluorine through an anionic ring closureCitation24–27. Structure elucidation of 4d was performed using high-resolution mass spectrometry analysis (experimental mass 257.0699, theoretical mass for C15H10N2OCitation23Na 257.0691). Structural confirmation of compound 4d was also achieved by IR and 1H NMR spectroscopy on the basis of previous results for 5H-pyrrolo[1,2-a]quinoxalin-4-one series published by our groupCitation4–7,Citation10,Citation11. In the 1H NMR spectrum, we observed a D2O exchangeable singlet at 11.40 ppm characteristic of the NH lactam function. Moreover, the data also showed a singlet at 8.78 ppm attributed to the proton in position 11 of the isoindolo[2,1-a]quinoxaline skeleton. In the IR spectrum, lactam 4d showed a strong stretching carbonyl band at 1645 cm−1.
Biology
Antiproliferative Effect. The eight new compounds 1a–h were tested in MTS assay for their in vitro antiproliferative activity against five human leukemic cell lines (U937, K562, Jurkat, U266, and HL60), and one breast cancer cell line (MCF7). Compound LY-294002 (), which showed antiproliferative activity against the HL60, U937 and K562 cell linesCitation28–31, was applied as a referential cytotoxic agent. The results of the antiproliferative activity studies are summarized in .
Table 1. In vitro activity of compounds 1a-h on U937, K562, HL60, Jurkat, U266, and MCF7 cells, and cytotoxicity on human peripheral blood mononuclear cells PBMNC + PHA.
Figure 3. Structure of compound LY-294002.
Among compounds 1a–h, the benzylpiperidinyl fluorobenzimidazole derivatives 1e–g exhibited the best antiproliferative activity on the growth of human myeloid U937 cell line (IC50 from 3.5 to 4 µM) in comparison with their benzylpiperidinyl benzimidazolone analogues 1a–c that were found inactive (IC50 > 50 µM). Surprisingly, the isoindoloquinoxaline 1h, the structural aromatic analogue of the tetrahydroisoindoloquinoxaline 1e, showed moderate antiproliferative activity with an IC50 value of 42 µM. Moreover, in terms of structure–activity relationships discussion, it could be also noticed that the IC50 of compound 1g bearing an indole moiety (IC50 = 4 µM) was found 10 times lower than those of its isoindolo analogue 1h (IC50 = 42 µM). Introduction of a fused benzene nucleus (compound 1f) onto the tetrahydroisoindole ring (compound 1e) did not lead to significant modification in the antiproliferative activity (IC50 = 4 µM for 1f versus 3.5 µM for 1e).
Similar profile of antiproliferative activity was observed against the human myeloma cell line U266. Hence, the three quinoxalines 1e–g, bearing the benzylpiperidinyl fluorobenzimidazole moiety in their 4-position, were always found the most active compounds with an IC50 of 3–4 µM. The fourth fluorobenzimidazole derivative 1h also showed significant antiproliferative activity (IC50 = 14 µM). Concerning the fused substitutions on the quinoxaline ring, we noticed a similar biological profile as those observed against the U937 cell line.
The antiproliferative potencies of these new derivatives 1a–h were also examined toward the human myeloid leukaemia cell lines K562 and HL60. On K562 cell line, 1e–g were also found as the most active compounds (IC50 = 3–4 µM). The replacement of the benzylpiperidinyl fluorobenzimidazole substituent by a benzylpiperidinyl benzimidazolone group in position 4 of the fused heterocyclic skeleton (compounds 1a–c) led to a decrease in the activity (IC50 ~ 46 µM). However, it could be noticed that the nature of the substitution at position 4 in the isoindoloquinoxaline derivatives 1d and 1h was less detrimental for the activity (IC50 of 11 and 13 µM, respectively). The benzylpiperidinyl fluorobenzimidazole compounds possessing a fused tetrahydroisoindole, dihydrobenzo[e]isoindole, or indole nucleus (compounds 1e–g) exhibited better antiproliferative activities than their isoindolo analogue 1h. In a general way, this kind of structure–activity profile was also observed against the HL60 and Jurkat cell lines.
Against the HL60 human acute promyeloid leukemia cell line, most of the tested compounds showed antiproliferative activity with IC50 values from 3 to 18 µM, except 1b and 1c that were found inactive (IC50 > 50 µM). As in a general way, isoindolo- and indoloquinoxalines having a benzylpiperidinyl fluorobenzimidazole moiety at position 4 exhibited better activities than their benzylpiperidinyl benzimidazolone homologues (i.e., IC50 = 4 µM for 1e versus 13.5 µM for 1a, 3 µM for 1f compared >50 µM for 1b, and 4 µM for 1g versus >50 µM for 1c). Surprisingly, this kind of observation could not be applied to compounds 1d and 1h. Hence, the IC50 of 1d (9 µM) was twice lower than those of compound 1h (IC50 = 18 µM).
The antiproliferative activities of compounds 1a–h against the T-lymphocyte Jurkat cell line were similar to those observed against the HL60 cell line. The same observation concerning antiproliferative activity of benzimidazolone 1d versus fluorobenzimidazole 1h on Jurkat cell line could also be noticed (IC50 = 5 µM for 1d versus 12 µM for 1h).
In MCF7 cell line, the pyrrolo[1,2-a]quinoxaline derivatives 1e–g, bearing a benzylpiperidinyl fluorobenzimidazole moiety in position 4 exhibited potent cytotoxicity with IC50 ranging from 3 to 4 µM. However, the isoindolo derivative 1h showed low activity (IC50 = 33 µM) against this breast cancer line. Moreover, two of their benzylpiperidinyl benzimidazolone analogues bearing an hydrogenated isoindole ring (compounds 1a and 1b) have shown significant antiproliferative activity with an IC50 of 11 and 9 µM, respectively. Aromatization of the tetrahydroisoindole nucleus of 1a (compound 1d) led to a decrease in the antiproliferative activity; i.e., IC50 = 11 µM for 1a and >50 µM for 1d.
Against each human cancer cell lines, the antiproliferative activities of compounds 1e–g were always found superior to those of the reference drug LY-294002.
Cytotoxicity. All compounds 1a–h were tested on activated human peripheral blood mononuclear cells (). As expected, most of the pyrrolo[1,2-a]quinoxalines 1a–h showed significant level of cytotoxicity against lymphocytes with IC50 ranging from 4 to >50 µM. These preliminary results were used to determine their respective range of toxic concentration.
In a general way, the less cytotoxic molecules were the less active on the various tumor cell lines except compounds 1d and 1h. Moreover, the benzylpiperidinyl benzimidazolone derivatives 1a–d were found less cytotoxic than their benzylpiperidinyl fluorobenzimidazole analogues 1e–g in the exception of compound 1h that presented an IC50 superior to 50 µM against lymphocytes. Indexes of selectivity were defined as the ratio of the IC50 value on the human mononuclear cells to the IC50 value on the U937, K562, HL60, Jurkat, U266, or MCF7 lines. Compounds that demonstrated high selectivity (high index of selectivity) should offer a potential of safer therapy. This led to identify compounds with index of selectivity >10 for compound 1d on the human leukemic cell lines Jurkat, and >5.5 for compound 1b against the MCF7 breast adenocarcinoma.
Conclusion
In the present report, we described the synthesis of a new series of substituted isoindolo- or indoloquinoxaline derivatives and presented their antiproliferative activities on the human leukemic cell lines U937, K562, Jurkat, U266, and HL60, and the breast cancer cell line MCF7. From the biological activity data, some structure–activity relationships can be inferred. The presence at C-4 position of the benzylpiperidinyl fluorobenzimidazole moiety on the isoindolo- and indoloquinoxaline skeletons resulted in active compounds. Nevertheless, the nature of this substitution at position 4 in the isoindolo[2,1-a]quinoxalines 1d and 1h seems less detrimental for the antiproliferative activity. In addition, the two antiproliferative compounds 1b and 1d showing interesting index of selectivity may constitute suitable candidates for further pharmacological studies. Moreover, it would be also interesting to enlarge the biological evaluation of these two new pyrrolo[1,2-a]quinoxaline derivatives 1b and 1d in order to precise now their mechanism of action.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Campiani G, Butini S, Fattorusso C, Trotta F, Franceschina S, De Angelis M, Nielsen KS. Novel aryl piperazine derivatives with medical utility. 2006, WO2006072608.
- Campiani G, Aiello F, Fabbrini M, Morelli E, Ramunno A, Armaroli S et al. Quinoxalinylethylpyridylthioureas (QXPTs) as potent non-nucleoside HIV-1 reverse transcriptase (RT) inhibitors. Further SAR studies and identification of a novel orally bioavailable hydrazine-based antiviral agent. J Med Chem 2001;44:305–315.
- Schann S, Mayer S, Gardan S. Pyrrolo[1,2-a]quinoxaline derivatives as Adenosine A3 receptor modulators and uses thereof. 2007, EP1798233.
- Guillon J, Grellier P, Labaied M, Sonnet P, Léger JM, Déprez-Poulain R et al. Synthesis, antimalarial activity, and molecular modeling of new pyrrolo[1,2-a]quinoxalines, bispyrrolo[1,2-a]quinoxalines, bispyrido[3,2-e]pyrrolo[1,2-a]pyrazines, and bispyrrolo[1,2-a]thieno[3,2-e]pyrazines. J Med Chem 2004;47:1997–2009.
- Guillon J, Forfar I, Mamani-Matsuda M, Desplat V, Saliège M, Thiolat D et al. Synthesis, analytical behaviour and biological evaluation of new 4-substituted pyrrolo[1,2-a]quinoxalines as antileishmanial agents. Bioorg Med Chem 2007;15:194–210.
- Guillon J, Forfar I, Desplat V, Fabre SB, Thiolat D, Massip S et al. Synthesis of new 4-(E)-alkenylpyrrolo[1,2-a]quinoxalines as antileishmanial agents by Suzuki-Miyaura cross-coupling reactions. J Enzyme Inhib Med Chem 2007;22:541–549.
- Guillon J, Moreau S, Mouray E, Sinou V, Forfar I, Fabre SB et al. New ferrocenic pyrrolo[1,2-a]quinoxaline derivatives: synthesis, and in vitro antimalarial activity. Bioorg Med Chem 2008;16:9133–9144.
- Milne J, Normington KD, Milburn M. Tetrahydroquinoxalinone sirtuin modulators. 2006, WO2006094210.
- Grande F, Aiello F, Grazia OD, Brizzi A, Garofalo A, Neamati N. Synthesis and antitumor activities of a series of novel quinoxalinhydrazides. Bioorg Med Chem 2007;15:288–294.
- Desplat V, Geneste A, Begorre MA, Fabre SB, Brajot S, Massip S et al. Synthesis of new pyrrolo[1,2-a]quinoxaline derivatives as potential inhibitors of Akt kinase. J Enzyme Inhib Med Chem 2008;23:648–658.
- Desplat V, Moreau S, Gay A, Fabre SB, Thiolat D, Massip S et al. Synthesis and evaluation of the antiproliferative activity of novel pyrrolo[1,2-a]quinoxaline derivatives, potential inhibitors of Akt kinase. Part II. J Enzyme Inhib Med Chem 2010;25:204–215.
- Wermuth CG. Ring transformations. In: Wermuth CG, ed. The Practice of Medicinal Chemistry. Amsterdam: Elsevier Netherland; 2003. P 215–231.
- Beach MJ, Hope R, Dieter HK, Rusell RK. Two step synthesis of substituted indolo[1,2-a]-quinoxalin-6-ones. Synth Commun 1995;25:2165–2183.
- May DA, Lash TD. Porphyrins with exocyclic rings. 2. Synthesis of geochemically significant tetrahydrobenzoporphyrins from 4,5,6,7-tetrahydro-2H-isoindoles. J Org Chem 1992;57:4820–4828.
- Finikova OS, Cheprakov AV, Beletskaya IP, Carroll PJ, Vinogradov SA. Novel versatile synthesis of substituted tetrabenzoporphyrins. J Org Chem 2004;69:522–535.
- Ono N, Hironaga H, Ono K, Kaneko S, Murashima T, Ueda T, Tsukamura C, Ogawa T. A new synthesis of pyrroles and porphyrins fused with aromatic rings. J Chem Soc, Perkin Trans 1 1996;5:417–423.
- Jew SS, Kim HD, Cho YS, Cook CH. A practical preparations of conjugated nitroalkenes. Chem Lett 1986;15:1747–1748.
- Yuan Q, Ma D. A one-pot coupling/hydrolysis/condensation process to pyrrolo[1,2-a]quinoxaline. J Org Chem 2008;73:5159–5162.
- Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron compounds. Chem Rev 1995;95:2457–2483.
- Supplementary X-ray crystallographic data of compounds 1a and 1c (CCDC-746052 and CCDC-746053): Cambridge Crystallographic Data Centre, University Chemical Lab, 12 Union Road, Cambridge, CB2 1EZ, U.K.; E-mail: [email protected].
- Diana P, Martorana A, Barraja P, Montalbano A, Dattolo G, Cirrincione G et al. Isoindolo[2,1-a]quinoxaline derivatives, novel potent antitumor agents with dual inhibition of tubulin polymerization and topoisomerase I. J Med Chem 2008;51:2387–2399.
- Diana P, Martorana A, Barraja P, Lauria A, Montalbano A, Almerico AM et al. Isoindolo[2,1-c]benzo[1,2,4]triazines: a new ring system with antiproliferative activity. Bioorg Med Chem 2007;15:343–349.
- Paolini JP, Palopoli FP, Lendvay LJ, Huffman J. Pyrido[2′1′:2,3]imidazo[4,5-c]isoquinoline and the alkylation of pyrido[2′,1′:2,3]imidazo[4,5-c]isoquinolin-5(6H)-one. J Heterocyclic Chem 1987;24:549–553.
- Campiani G, Nacci V, Corelli F, Anzini M. Polycondensed heterocycles. VII. A convenient synthesis of pyrrolo[1,2-a]quinoxaline derivatives by intramolecular aromatic nucleophilic displacement. Synth Commun 1991;21:1567–1576.
- Cailly T, Fabis F, Rault S. A new, direct, and efficient synthesis of benzonaphthyridin-5-ones. Tetrahedron 2006;62:5862–5867.
- Cailly T, Fabis F, Legay R, Oulyadi H, Rault S. The synthesis of three new heterocycles: the pyrido[4,3 or 3,4 or 2,3-c]-1,5-naphthyridines. Tetrahedron 2007;63:71–76.
- Dubost E, Magnelli R, Cailly T, Legay R, Fabis F, Rault S. General method for the synthesis of substituted phenanthridin-6(5H)-ones using a KOH-mediated anionic ring closure as the key step. Tetrahedron 2010;66:5008–5016.
- Liu P, Xu B, Li J, Lu H. LY294002 inhibits leukemia cell invasion and migration through early growth response gene 1 induction independent of phosphatidylinositol 3-kinase-Akt pathway. Biochem Biophys Res Commun 2008;377:187–190.
- Moon DO, Park C, Heo MS, Park YM, Choi YH, Kim GY. PD98059 triggers G1 arrest and apoptosis in human leukemic U937 cells through downregulation of Akt signal pathway. Int Immunopharmacol 2007;7:36–45.
- Jørgensen HG, Allan EK, Graham SM, Godden JL, Richmond L, Elliott MA et al. Lonafarnib reduces the resistance of primitive quiescent CML cells to imatinib mesylate in vitro. Leukemia 2005;19:1184–1191.
- Zhao S, Konopleva M, Cabreira-Hansen M, Xie Z, Hu W, Milella M et al. Inhibition of phosphatidylinositol 3-kinase dephosphorylates BAD and promotes apoptosis in myeloid leukemias. Leukemia 2004;18:267–275.