Abstract
The present investigation deals with the synthesis of novel prodrugs of dexibuprofen with amino acids with an aim to achieve potent anti-inflammatory activity and less gastrointestinal toxicity. Structures of synthesized compounds were confirmed by spectral and elemental analyses. In vitro hydrolytic studies in simulated intestinal fluid, 80% plasma and rat faecal matter showed satisfactory release of dexibuprofen due to enzymatic cleavage. The synthesized prodrugs were evaluated for anti-inflammatory activity, analgesia, ulcerogenicity and histopathology. The anti-inflammatory activity of dexibuprofen was 43.3% whereas an improved value of 73.4, 77.3, 72.8 and 64.5% was observed for the synthesized prodrugs. The percentage analgesia of the prodrugs increased, whereas a decrease in the mean ulcer index values than dexibuprofen was observed. The histopathological studies revealed less ulceration in the gastric region when treated with prodrugs. Thus, the prodrugs were proved to be better in action as compared with the parent drug.
Introduction
Dexibuprofen (Dex), a novel non-steroidal anti-inflammatory drug (NSAID), is a single and pharmacologically effective enantiomer of racemic ibuprofen. Racibuprofen and dexibuprofen differ in their physicochemical properties, pharmacological profiles and metabolic activitiesCitation1. The efficacy of dexibuprofen was found to be same as that of other NSAIDs, such as diclofenac, naproxen and celecoxib. Generally, the NSAIDs are associated with various gastrointestinal (GI) side effects like stomach ulceration, bleeding and perforation due to the presence of its acidic group. The GI toxicity is produced either by direct contact mechanism or by generalized systemic action, which occur after absorption. To overcome the GI side effects, prodrugs of NSAIDs can be developed by hiding its acidic group through conjugation with amino acids. Many amino acids possess site specificity, marked anti-inflammatory activity and exhibit profound healing effect on gastric toxicityCitation2. During conjugation, the NSAIDs having free carboxylic group can be derivitized into corresponding amides of amino acid, and it results in altering the physical properties of parent drug with one or more of hydrolase enzymes serving as the in vivo reconversion sites. The new prodrugs were expected to have better lipophilicity, reduced gastric irritancy, improved therapeutic index through the prevention of GI irritation and bleeding, improved anti-inflammatory activity, reduced GI erosive properties and synergistic analgesic effect than the parent drugCitation3,Citation4.
The synthesis of prodrugs of ketoprofen, diclofenac, flubiprofen, naproxen, ibuprofen, etc., with various amino acids via masking the carboxylic acid group to form ethyl ester, methyl ester and glycolamide ester prodrugCitation5–7 were carried out successfully so far. The objectives of this study are (i) to synthesize the amide prodrugs of Dex with methyl esters of l-tryptophan, l-phenylalanine, glycine and l-tyrosine to form Dex-1, Dex-2, Dex-3 and Dex-4, respectively and (ii) to perform a detailed study on their various physicochemical properties, hydrolysis kinetics and pharmacological activities. The compounds synthesized will act as prodrugs of dexibuprofen, which upon administration would release the parent drug as a result of hydrolysis (enzymatic or non-enzymatic) in the body.
Materials and methods
Materials
The amino acids l-tryptophan, l-phenylalanine, glycine and l-tyrosine were obtained from M/s Hi-Media Ltd., India, and drug dexibuprofen was obtained as a gift sample from Alkem Laboratories, India. Other reagents and solvents were of analytical grade. The melting points were recorded using melting point determination apparatus by Sigma Instrument, India, and are uncorrected. The elemental analysis was performed using Carlo-Erba Model 1108 Analyzer (Italy). 1H NMR and 13C NMR spectra were recorded in diemthylsulphoxide (DMSO) on NMR spectrophotometer (Bruker DRX 300, USA). Chemical shifts are expressed as δ (ppm) values. IR spectra were recorded using IR spectrophotometer (Shimadzu FTIR-8201PC (Kyoto, Japan)) in KBr phase and mass spectra were recorded on mass spectrophotometer (Jeol SX-102 (FAB), Japan). The hydrolysis data and drug content determination were performed by ELICO Double Beam UV-VIS Spectrophotometers (Hyderabad, India).
Synthesis of amide prodrugs of dexibuprofen
Dexibuprofen is 2-[4-(2-methyl propyl) phenyl] propanoic acid and the synthesis of its amide prodrugs was carried out by Schotten Baumann techniqueCitation8 as explained below. The purity of the compounds was checked by thin-layer chromatography on pre-coated silica GF252 plates using iodine vapour as a detecting agent. The structures of all the synthesized prodrugs were confirmed by elemental analysis and spectral analysis such as IR, 1H NMR, 13C NMR and mass spectroscopy.
Step 1: Synthesis of dexibuprofen acid chloride
Dex (1) of 10.31 g (0.05 mol L−1) was dissolved in minimum amount of chloroform and freshly distilled 6 mL (0.05 mol L−1) thionyl chloride was added slowly to it. The mixture was refluxed at 60–70°C with continuous stirring on a magnetic stirrer. The viscous liquid was immediately poured on petridish and was vacuum dried to give yellow coloured crude dexibuprofen acid chloride (1a).
Step 2: Synthesis of methyl ester hydrochlorides of l-tryptophan, l-phenylalanine, glycine and l-tyrosine
Freshly distilled 6 mL (0.05 mol L−1) thionyl chloride was slowly added to methanol (100 mL) with cooling and 20.42 g (0.1 mol L−1) of l-tryptophan (2) was added to it. The mixture was refluxed for 6–8 h at 60–70°C with continuous stirring on magnetic stirrer. Excess thionyl chloride and solvent was removed under reduced pressure giving crude tryptophan methyl ester hydrochloride. It was treated with 20 mL portion of cold ether at 0°C. The resulting solid product was collected and dried under vacuum. It was recrystallized from hot methanol by slow addition of 15–20 mL ether followed by cooling at 0°C. The crystals were collected next day and washed twice with ether: methanol mixture at 5:1 ratio followed by pure ether and dried under vacuum to give pure tryptophan methyl ester hydrochloride (2a). The same procedure was followed to synthesize phenylalanine methyl ester hydrochloride, glycine methyl ester hydrochloride and tyrosine methyl ester hydrochloride.
Step 3: Synthesis of prodrugs of dexibuprofen with methyl esters of l-tryptophan, l-phenylalanine, glycine and l-tyrosine
Ice cold, aqueous sodium hydroxide solution (5 %) was taken in 250 mL beaker and 12.6 g (0.05 mol L−1) of methyl ester of tryptophan hydrochloride was added to it. The reaction mixture was mechanically stirred for 30 min at room temperature, after which the beaker was transferred to an ice bath kept on the mechanical stirrer, maintaining the temperature at 10°C. To this, 22.3 g (0.01 mol L−1) of dex acid chloride was added in small portions with continuous stirring for 7–8 h. The solid that separated out was filtered off. The crude prodrug was recrystallized from methanol. The same procedure was followed for l-phenylalanine, glycine and l-tyrosine. The schematic representation of synthesis of tryptophan-conjugated dexibuprofen (Dex 1), phenylalanine-conjugated dexibuprofen (Dex 2), glycine-conjugated dexibuprofen (Dex 3) and tyrosine-conjugated dexibuprofen (Dex 4) are shown in .
Scheme 1. Synthetic protocol of amide prodrugs of dexibuprofen.
Spectral analysis
The spectral data obtained for Dex 1, Dex 2, Dex 3 and Dex 4 are given as follows.
Dex 1: S(+) Methyl 3-(1H-indol-3-yl)-2-(2-(4-isobutylphenyl)propanamido)propanoate− UV Spectra (λmax) in simulated gastric fluid (SGF) 205 nm, in simulated intestinal fluid (SIF) 225 nm, in 80% human plasma 218 nm; IR (KBr, cm−1) 3290 (NH str. of amide), 3075 (aromatic CH str.), 1677 (C=O str. of ester), 1570 (C=N), 1292 (CO str.of ester); 1H NMR (δ ppm) (DMSO-d6) 1.48 (d, J = 7.1 Hz, 3H, CH3), 1.52 (d, J = 6.8 Hz, 1H, CH), 3.72 (d, J = 7.1 Hz, 2H, CH2), 7.18 (m, 5H, indol ring), 7.20 (m, 4H, aromatic ring), 9.77 (s, 1H, NH), 9.88 (s, 1H, NH); 13C NMR (δ ppm) (DMSO-d6) 17.3, 18.3, 18.3, 36.2, 38.2, 43.5, 47.3, 48.2, 51.9, 125.2, 126.2, 126.2, 126.3, 126.8, 127.2, 127.2, 127.3, 128.2, 129.2, 129.2, 171.3, 178.2; Mass (m/z) 406 (M+).
Dex 2: S(+) Methyl 2-(2-(4-isobutylphenyl)propanamido)-3-phenylpropanoate—UV Spectra (λmax) in SGF 210 nm, in SIF 217 nm, in 80% human plasma 213 nm; IR (KBr, cm−1) 3326 (NH str. of amide), 2930 and 2906 (aromatic CH str.), 1726 (C=O str. of ester), 1630 (C=N), 1272 (CO str.of ester); 1H NMR (δ ppm) (DMSO-d6) 1.42 (d, J = 7.1 Hz, 3H, CH3), 3.68 (q, J = 6.8 Hz, 1H, CH in ring), 3.72 (q, J = 7.2 Hz, 1H, CH), 7.28 (m, 4H, Ar. H), 7.32 (m, 4H, Ar. ring), 9.77 (s,1H, NH); 13C NMR (δ ppm) (DMSO-d6) 17.3, 17.3, 18.3, 35.2, 35.2, 42.3, 47.3, 47.8, 51.9, 119.2, 120.2, 125.2, 125.3, 125.3, 126.2, 126.6, 127.2, 127.3, 127.5, 128.3, 128.4, 171.3, 172.9; Mass (m/z) 367 (M+).
Dex 3: S(+) Methyl-2-(2-(4-isobutyl phenyl)propanamido)acetate—UV Spectra (λmax) in SGF 212 nm, in SIF 219 nm, in 80% human plasma 222 nm; IR (KBr, cm−1) 3280 (NH str. of amide), 3043 (aromatic CH str.), 2932 (C=O str. of ester), 1630 (C=N), 1290 (CO str.of ester); 1H NMR (δ ppm) (DMSO-d6) 1.38 (d, J = 6.7 Hz, 6H, CH3), 2.42 (d, J = 7.3 Hz, 2H, CH2), 3.80 (q, J = 7.1 Hz, 1H, CH), 3.82 (q, J = 7.1 Hz, 1H, CH), 7.48 (d, J = 8 Hz, 4H, Ar.), 9.68 (s, 1H, NH); 13C NMR (δ ppm) (DMSO-d6) 17.3, 17.3, 17.8, 20.2, 35.2, 42.3, 47.8, 51.9, 63.2, 120, 120.8, 128.6, 128.9, 139.6, 172.9; Mass (m/z) 277 (M+).
Dex 4: S(+) Methyl 3-(4-hydroxyphenyl)-2-(2-(4-isobutylphenyl)propanamido) propanoate—UV Spectra (λmax) in SGF 208 nm, in SIF 221 nm, in 80 % human plasma 218 nm; IR (KBr, cm−1) 3362 (NH str. of amide), 3046 (aromatic CH str.), 1745 (C=O str. of ester), 1532 (C=N), 1280 (CO str.of ester); 1H NMR (δ ppm) (DMSO-d6) 1.28 (d, J = 6.7 Hz, 6H, CH3), 3.62 (q, J = 7.1 Hz, 1H, CH), 7.28 (d, J = 8 Hz, 4H, Ar. ring), 7.63 (d, J = 8 Hz, 4H, Ar. ring), 9.68 (s, 1H, NH); 13C NMR (δ ppm) (DMSO-d6) 17.8, 18.2, 18.3, 36.2, 47.3, 48.2, 48.3, 51.9, 120, 120.2, 120.6, 122, 122.2, 125, 125.6, 126, 126.2, 128.2, 128.4, 135.2, 171.2, 172.1; Mass (m/z) 383 (M+).
Physicochemical characterization of the synthesized prodrugs
Solubility
Approximately 5 mg of prodrug was dissolved in 5 mL of each solvent at 37 ± 1°C in glass test tubes. The solvents used were 0.1 N NaOH, 0.1 N HCl, ethanol, methanol, ether, ethyl acetate, chloroform, acetone, DMF and water. Test tubes were gently shaken and solubility was observed. In case of any observed insoluble fractions, the known amount of solvent was further added to ascertain the solubility of the compound.
Protein binding studies
A solution of synthesized prodrug (10 mg mL−1) was made in phosphate-buffered saline (PBS, pH 7.4). A 100 mL of this solution was taken in a beaker. The cellophane membrane (molecular weight cutoff in the range of 10,000–12,000 Da obtained from Hi-Media, India) was first washed with distilled water and then with buffer solution (pH 7.4). It was tied at the opening end of dialysis tube; the dialysis tube containing (6%) egg albumin was dipped into the drug solution and covered. The whole assembly was placed on a magnetic stirrer and switched at low revolutions per minute. The temperature was maintained at 37 ± 0.5°C. After every 1 h, 1 mL of the PBS containing drug solution was replaced with fresh 1 mL of PBS. Withdrawn sample was diluted further with 1 mL phosphate buffer and the concentration of prodrug was estimated using spectrophotometer at 223 nm. indicates the physicochemical properties of the synthesized prodrugs.
Table 1. Physicochemical properties of prodrugs.
Hydrolysis rate determination in SGF and SIFCitation9
In vitro hydrolysis studies of synthesized prodrugs were carried out in SGF at pH 1.2 (USP 1970) and SIF at pH 7.4 (USP 1970). A solution of 10 mg of prodrug was prepared in 90 mL of SIF (pH 7.4) or SGF (pH 1.2). An aliquot of 15 mL of this solution was withdrawn repeatedly and kept in test tubes maintained at 37 ± 0.5°C. At a definite interval of time (0.5 h, 1–8 h), an aliquot was withdrawn from different test tubes and was transferred to micro-centrifuge tubes followed by addition of methanol to make up the volume. The tubes were placed in freezing mixture in order to arrest further hydrolysis, followed by vortexing at high speed for 5 min. After vortexing, the tubes were centrifuged at high speed (3000g) for 5 min. A 5 mL of clear supernatant obtained from each tube was measured on UV spectrophotometer for the amount of free Dex released after the hydrolysis of prodrugs in SGF and SIF at 210 nm. The kinetics of hydrolysis was monitored by increase in free drug concentration with time, and order of reaction and half life (t1/2) were also calculated. The rate of hydrolysis was calculated using equation:
r = (2.303/t) log [a/(a − x)]
where r represents hydrolysis constant, t is the time in min, a is the initial concentration of prodrug, x is the amount of prodrug hydrolyzed and (a − x) is the amount of prodrug remaining.
Hydrolysis rate determination in 80% human plasmaCitation10
A solution of 10 mg of prodrug was prepared in methanol (2 mL) and was added to 88 mL of 80% human plasma (pH 7.4 prepared by mixing 80 portion of plasma and 20 portion of phosphate buffer at pH 7.4). An aliquot of 15 mL of this solution was withdrawn and kept in test tubes maintained at 37 ± 0.5°C. At definite interval of time (0–8 h), an aliquot of 22 mL was withdrawn and mixed with 0.5 % ZnSO4 solution. Samples were centrifuged at 6000g for 10 min and a clear supernatant solution was analyzed spectrophotometrically at 223 nm.
Hydrolysis rate determination in rat faecal matterCitation11
The prodrug was dissolved in phosphate buffer so that final concentration of the solution was 250 µg/mL. Fresh faecal material of rats were weighed (about 1 g) and placed in different sets of test tubes. To each test tube, 2 mL of the prodrug solution was added and diluted to 5 mL with phosphate buffer. The sets of test tubes were incubated at 37 ± 0.5°C for different intervals of time (0–8 h). For analysis, the free drug was extracted with 5 mL of methanol and estimated on UV spectrophotometer at 210 nm.
Pharmacological evaluations
Dex and the synthesized prodrugs were evaluated for anti-inflammatory activity, analgesic activity, ulcerogenicity and histopathology, and a comparative study was performed. Test compounds and standard drugs were administered in the form of a suspension by oral route of administration for anti-inflammatory and analgesic studies (1% carboxymethylcellulose as a vehicle), as well as for ulcerogenicity studies (2% acacia as vehicle). Wistar albino rats of six groups, including a control and a standard group, each with six animals were selected. The selected animals were housed in acrylic cages at standard environmental conditions at 25 ± 2°C, relative humidity of 45–55%, in a well-ventilated room maintained at 12:12 h light:dark cycle, fed with standard rodent diet and water ad libitum. All the animals were acclimatized for a week before experiment. All animal experiments were carried out according to the guidelines of the Committee for the Purpose of Control of Experiments on Animals and approval of the Institutional Animal Ethics Committee, Sree Vidyanikethan College of Pharmacy, Tirupati, India, was obtained.
Anti-inflammatory activity
The anti-inflammatory activity was determined by hind paw oedema methodCitation12 using carrageenan as phlogistic agent. Wistar rats (100–200 g) were divided into six groups, each comprising of six rats, including a control and a standard group. The initial volume of right hind paw of rat was measured by plethysmometer without administration of the prodrug. A 1% sodium carboxymethylcellulose suspension containing a dose equivalent to the 2.4 mg or 12 mg kg−1 body weight of Dex was administered orally. An equivalent quantity of prodrugs 4 mg of Dex-1, 4.36 mg of Dex-2, 3.52 mg of Dex-3 and 4.61 mg of Dex-4 were administered to the test groups. After 30 min of administration of the drugs, carrageenan (0.1 mL, 1% m/v) solution in normal saline was injected into the planter surface of right hind paw of each animal. The volume of right hind paw of albino rat was measured after 2, 4 and 6 h. The mean difference in the volume of the right hind paw of rats was compared with control and standard. The percent inhibition of paw oedema was calculated as
Percent inhibition = (1 − Vt/Vc) × 100
where Vc is the mean relative change in paw edema volume in control group and Vt is the mean relative change in paw edema volume in test group.
Analgesic activity
The analgesic activity was determined by thermal stimulus using tail flick methodCitation13. Analgesiometer was used for the determination of pain threshold of albino mice. Cold water was circulated through the water jackets of the instrument to avoid heating of the area around the hot wire. The rat (100–200 g) was placed in a holder through which the tail of the rat was protruded out. The reaction time was recorded at 1, 2, 3 and 4 h after the treatment and cutoff time was 9 s. The normal reaction time, i.e., the time taken to flick the tail was noted. Animals showing delayed response were rejected. The drug (dose equivalent to 2.4 mg or 12 mg kg−1 body mass) was administered orally in 1% suspension of sodium carboxymethylcellulose. An equivalent quantity of prodrugs 4 mg of Dex-1, 4.36 mg of Dex-2, 3.52 mg of Dex-3 and 4.61 mg of Dex-4 was administered to the test groups. The percent analgesic activity was calculated by the formula given as
% Analgesic activity = [(T2 − T1)/(Tc − T1)] × 100
where T1 is the reaction time (s) before administration of prodrug and T2 the reaction time (s) after administration of prodrug and Tc cutoff time (s).
Ulcerogenicity
GI toxicity of the synthesized prodrugs was measured and compared with the parent drug by measuring mean ulcer index (UI)Citation14. The control group was administered orally in 2% acacia suspension. Test compounds and standard were administered orally (at 10 times higher dose) as a suspension in 2% acacia daily for 5 days. The rats (130–150 g) were fasted after the administration of last dose, thereafter they were killed by decapitation and the stomach was removed, opened and washed with distilled water. The lesions on the gastric mucosa were counted by visual examination using a binocular magnifier. Ulcers greater than 0.5 mm were recorded. The mean UI was calculated by severity of gastric mucosal lesions that are graded as (i) grade 1: >1 mm erosions (ii) grade 2: 1–2 mm erosions and (iii) grade 3: <2 mm erosions.
The UI was calculated as:
UI = [1 × (number of lesions of grade 1) + 2 × (number of lesions of grade 2) + 3 × (number of lesions of grade 3)]/10.
Histopathological studiesCitation15
The histopathological studies of stomach of rats were carried out using haemotoxylin and eosin stain at Pathology Department, Sri Venkateswara Veterinary University, Tirupati, India. The stomach tissues were removed from the rats and fixed in 10% normal saline for at least 48 h. These were then processed routinely and the tissues were embedded in paraffin wax. Histological sections were cut at 5–6 μm and stained with routine haematoxylin and eosin. These were then examined by a consultant histopathologist. The lesions observed were assessed for the following mucosal atrophy, the presence of inflammatory cells in the wall, oesinophils, lymphocytes and plasma cells. Photomicrographs of representative lesions at various magnifications were taken on Zeiss optical microscope (Germany), Stemi 2000-C, with a resolution of 10 × 45X, attached with trinocular camera.
Statistical analysis
Statistical analysis of the pharmacological activity of the synthesized prodrugs on animals was evaluated using a one-way analysis of variance (ANOVA). Student’s t-test was applied for expressing the significance, and the experimental data are expressed as mean ± SD.
Results and discussion
Synthesis and characterization
This study describes the synthesis of amide prodrugs of Dex as Dex-1 {S(+) methyl-3-(1H-indol-3-yl)-2-(2-(4-isobutylphenyl)propanamido)propanoate}, Dex-2 {S(+) methyl 2-(2-(4-isobutylphenyl)propanamido)-3-phenylpropanoate}, Dex-3 {S(+) methyl-2-(2-(4-isobutyl phenyl)propanamido)acetate} and Dex-4 {S(+) methyl 3-(4-hydroxy phenyl)-2-(2-(4-isobutyl phenyl)propanamido)propanoate}as portrayed in . The prodrugs were subjected to solubility, physicochemical characterization, protein binding and hydrolytic studies. All the prodrugs were observed to be highly soluble in organic solvents, sparingly soluble in alkaline solution and insoluble in acidic solutions. The high solubility of the prodrugs in organic solvents indicates their lipophillic behaviour. The physicochemical properties of synthesized prodrugs were determined, as well as the IR, 1H NMR, 13C NMR and mass spectral analyses were carried out. The results of elemental analysis of synthesized prodrugs were in all case within ±0.4% of theoretical value and were in confirmation of desired structure.
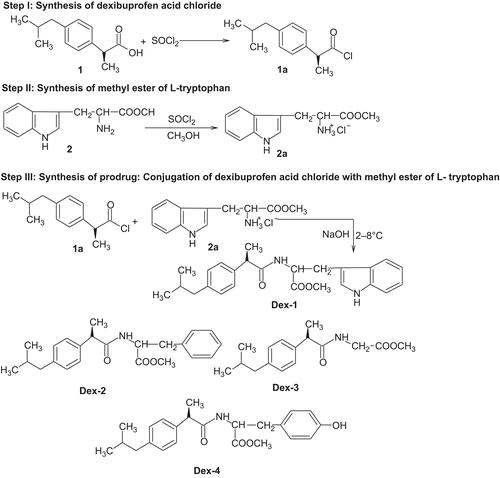
In vitro hydrolysis of Dex-1, Dex-2, Dex-3 and Dex-4 were carried out in SGF (pH 1.2), SIF (pH 7.4), 80% human plasma (pH 7.4) and rat faecal matter (pH 7.4) and their respective half life (t1/2) values in SIF were observed as 4.5, 4.8, 4.5 and 4.6 h, respectively. The comparative hydrolysis patterns of the synthesized prodrugs are depicted in . None of the prodrugs showed significant hydrolysis in SGF. The amount of Dex regenerated by hydrolysis of Dex-1, Dex-2, Dex-3 and Dex-4 in SIF was found as 71, 67, 76 and 73%, respectively, whereas 85, 82, 93 and 89%, respectively, in 80% human plasma and 86, 82, 93 and 89%, respectively, in rat faecal matter. The hydrolysis study revealed satisfactory hydrolysis of prodrugs in SIF and very encouraging hydrolysis in 80% human plasma and rat faecal matter. All the prodrugs followed first-order kinetics. The percentage protein binding of Dex-1, Dex-2, Dex-3 and Dex-4 was found as 74.08, 68.45, 56.78 and 58.78%, respectively. The prodrugs showed low protein binding in comparison with Dex whose value is found to be 85%. This increases the availability of prodrugs for hydrolysis in plasma and the required dose will be less.
Figure 1. Comparative hydrolysis pattern of prodrugs of dexibuprofen.
Pharmacological studies
Anti-inflammatory activity
shows that after 6 h of administration of Dex, the percentage anti-inflammatory activity was found as 43.3%, whereas an improved value of 73.4, 77.3, 72.8 and 64.5% were observed for prodrugs Dex-1, Dex-2, Dex-3 and Dex-4, respectively. The maximum anti-inflammatory activity of prodrugs was observed at 6 h and remained practically constant upto 8 h. The anti-inflammatory activity of free Dex decreased with time, whereas that of its prodrugs increased with time due to its higher bioavailability compared with the parent drug. Statistical significant testing using one-way ANOVA showed that anti-inflammatory activity of Dex and its prodrugs were effective in comparison with control group. Thus, the amino acid prodrugs proved to be a suitable promoiety for Dex.
Table 2. Comparative anti-inflammatory and analgesic activity of prodrugs.
Analgesic activity
The percentage protection in mice brought about by administration of drug is shown in . After 4 h of the administration of Dex, the percentage analgesia was observed as 46.4%, whereas for Dex-1, Dex-2, Dex-3 and Dex-4, corresponding values of 61.4, 59.7, 63.6 and 52.7% were observed. The study revealed that analgesic activity of Dex decreased with time, while the prodrugs showed an improved analgesic activity.
Ulcerogenicity
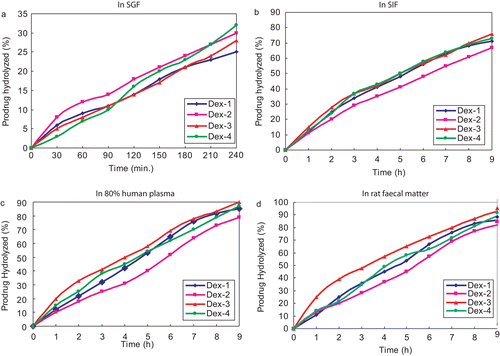
Dex has produced a mean UI of 25.3 while that of its prodrugs resulted in producing very less values and is shown in . The study revealed that the minimized side effect obtained in the prodrugs might be due to the inhibition of direct contact of carboxylic acid group of the drug to the gastric mucosa, which is mainly responsible for the damage.
Figure 2. Comparative ulcer index of dexibuprofen and its prodrugs.
Histopathological studies
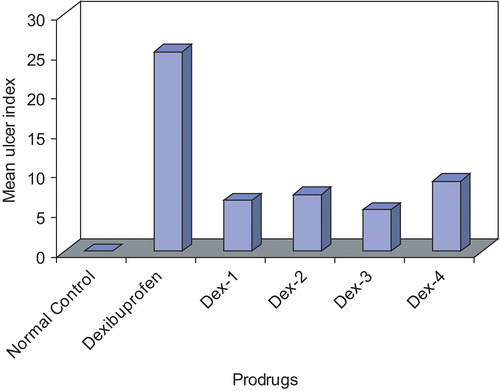
The histopathological studies were attempted and summarized as follows. The gastric tissues were investigated microscopically and the tissue samples of control group rats showed normal histological findings (). Microscopic investigation of Dex group revealed a focal erosive area in gastric mucosa and a zone (clear zone) in the basal regions of the gastric glands stained pale. This zone was parallel to the surface of the stomach lumen. In this zone, the structures of the gland were destroyed. They had disintegrated from the basal lamina and fallen into the lumen. The nuclei of these cells became smaller and dense, and their cytoplasms were stained as dark eosinophilic bodies. Small hemorrhagic areas and patches of inflammatory cell infiltrations were present in the lumen of the glands and lamina propria. Normal histological findings were displayed for Dex-1, Dex-2, Dex-3 and Dex-4 revealing that the prodrugs are not producing any ulceration in the gastric region.
Figure 3. Histopathological studies of dexibuprofen and its prodrugs on rat stomach.
Conclusions
In this study, the amide prodrugs of dexibuprofen were successfully synthesized and the structures were confirmed by elemental and spectral analyses. The hydrolysis studies carried out indicated that the prodrugs possess high yield, high solubility in organic solvents, encouraging hydrolysis rate in 80% human plasma and rat faecal matter as well as low protein binding properties. The prodrugs released dexibuprofen quantitatively in 80% human plasma, rat faecal matter and SIF, but were resistant to hydrolysis at SGF indicating that the prodrugs are resistant to acidic conditions. In vivo studies showed sustained release of Dex upto 8 h as also evident from the longer anti-inflammatory activity observed with prodrugs treated animals compared with Dex-treated animals. The prodrugs were found to be significantly less ulcerogenic and with more analgesia than parent drug. The histopathological study revealed limited ulcer formation in stomach by the prodrugs. These findings suggested that the prodrugs are better in action as compared with dexibuprofen and are advantageous in having less GI side effects without loss of desired anti-inflammatory and analgesic activity of the drug.
Acknowledgements
The authors thank M/s. Alkem Laboratories, Mumbai, India, for providing gift sample of dexibuprofen. The authors are grateful to Padmashree Dr. M. MohanBabu, Chairman, Sree Vidyanikethan Educational Trust, Tirupati, India, for providing the necessary facilities to carry out this work.
Declaration of interest
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Kaehler ST, Phleps W, Hesse E. Dexibuprofen: pharmacology, therapeutic, use and safety. Inflammaopharm 2003;11:371–383.
- Halen PK, Murumkar PR, Giridhar R, Yadav MR. Prodrug designing of NSAIDs. Mini-Rev Med Chem 2009;9:124–139.
- Bajpai AK, Mishra A. Carboxymethyl cellulose (CMC) based semi-IPNs as carriers for controlled release of ciprofloxacine: an in-vitro dynamic study. J Mater Sci Mater Med 2008;19:2121–2130.
- Mishra A, Ravichandran V, Jain PK, Dixit VK, Agrawal RK. Synthesis, characterization and pharmacological evaluation of amide prodrugs of ketorolac. Europ J Med Chem 2008;43:2464–2472.
- Bhosle D, Bharambe S, Gairola N, Dhaneshwar SS. Mutual prodrug concept: fundamentals and applications. Indian J Pharm Sci 2006;68:286–294.
- Rasheed A, Kumar CKA. Novel approaches on prodrug based drug design, Pharm Chem J 2008;42:677–686
- Roy SD, Manoukian E. Permeability of ketorolac acid and its ester analogs (prodrug) through human cadaver skin. J Pharm Sci 1994;83:1548–1553.
- Idle JR, Millburn P, Williams RT. Taurine conjugates as metabolites of arylacetic acids in the ferret. Xenobiotica 1978;8:253–264.
- Nielsenw NW, Bundgaard H. Glycolamide esters as biolabile prodrugs of carboxylic acid agents: synthesis, stability, bioconversion, and physicochemical properties. J Pharm Sci 1988;77:285–298.
- Mohan R, Ramaa CS. Ester prodrugs of Flubiprofen: Synthesis, plasma hydrolysis and gastrointestinal toxicity. Indian J Chem 2007;46B:1164–1168.
- Dev S, Deepali VM, Kadam SS, Dhaneshwar SR. Synthesis and pharmacological evaluation of cyclodextrin conjugate prodrug of Mefenamic acid. Indian J Pharm Sci 2007;69: 69–72.
- Winter CA, Risely EA, Nuss GW. Carregeenan induced oedema in hind paw of the rat as assay for anti-inflammatory drugs. Proc Soc Exp Biol Med 1962;111:544–547.
- Kulkarni SK. Handbook of Experimental Pharmacology, 3rd ed., Vallabh Prakash, New Delhi, 2001;128–130.
- Coili V, Putzolu S, Rossi V, Barcellona PS, Corradino C. The role of direct tissue contact in the production of gastrointestinal ulcers by anti-inflammatory drugs in rats. Toxicol Appl Pharmacol 1979;50:283–289.
- Yagmurca M, Ucar M, Fadillioglu E, Erdogan H, Ozturk F. The effects of nitric oxide on rat stomach injury induced by acetylsalicylic acid. Turk J Med Sci 2009; 39:13–19.