Abstract
The protein kinase field is a very active research area in the pharmaceutical industry and many activities are ongoing to identify inhibitors of these proteins. The design of new chemical entities with improved pharmacological properties requires a deeper understanding of the factors that modulate inhibitor–kinase interactions. In this report, we studied the effect of two of these factors—the magnesium ion cofactor and the protein substrate—on inhibitors of the type I insulin-like growth factor receptor. Our results show that the concentration of magnesium ion influences the potency of adenosine triphosphate (ATP) competitive inhibitors, suggesting an explanation for the observation that such compounds retain their nanomolar potency in cells despite the presence of millimolar levels of ATP. We also showed that the peptidic substrate affects the potency of these inhibitors in a different manner, suggesting that the influence of this substrate on compound potency should be taken into consideration during drug discovery.
Keywords::
Introduction
Protein kinases are very attractive drug targets, and for many years tremendous efforts have been made to identify low-molecular-weight modulators of their catalytic activity. As a consequence, this research area is one of the busiest in the pharmaceutical industry and many protein kinase inhibitors have been identified. Over the last 20–25 years, this field has been through many steps that were all seen at the time as very challenging. It was thought to be impossible to identify selective and potent adenosine triphosphate (ATP) competitive inhibitors in view of both the conservation of the ATP-binding domain and the high ATP concentration present in cells. Today, nanomolar selective inhibitors are availableCitation1,Citation2. The first inhibitors were designed to inhibit protein kinases in their active form. Today, several inhibitors targeting these enzymes in their non-active form have been identifiedCitation3,Citation4. Over many years, inhibitors were designed in such a way that they could compete directly with ATP (and in a few cases with their protein substrate). Today, inhibitors binding to pockets located outside the active site have been discoveredCitation5,Citation6. Three main elements have made these achievements possible: luck (still an important part of drug discovery!), progresses in structural biology/chemistry and a better understanding of the biochemical properties of these enzymes. In order to continue discovering innovative inhibitors, an even deeper understanding of the factors that modulate the inhibitor–protein kinases interactions is required.
In this article, we studied the influence of the metal cofactor, Mg2+, and of the protein substrate on protein kinase–inhibitor interactions. We used as model enzyme the type I insulin-like growth factor receptor (IGF1R) because it is an attractive drug target for oncologyCitation7 and because in enzymatic and biophysical assays we have previously characterized two compounds that inhibit its enzymatic activity: NVP-AEW541 (hereafter AEW541), a selective IGF1R inhibitor, and staurosporine (hereafter STAURO), a non-selective protein kinase inhibitorCitation8.
Materials and methods
Materials
All the chemicals were purchased from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated. The non-phosphorylated GST–IGF1R protein was purified as previously describedCitation9. AEW541 and STAURO stock solutions were made from fresh powder at a concentration of 10 mM in dimethyl sulphoxide (DMSO). The purity of the compounds was checked by liquid chromatography–mass spectrometry and their solubility in assay buffer (see below) was verified by dynamic light scatteringCitation10. ATP was dissolved in water (final pH adjusted to 7.5) at a concentration of 100 mM and dosed spectrophotometrically at 259 nm using a molar absorption coefficient of 15,400 M−1 cm−1. Peptide substrate (hereafter, PEP: Ac-EQEDEPEGDYFEWLE-NH2—American Peptide)Citation11 stock solutions were prepared in DMSO and dosed spectrophotometrically at 280 nm using a molar absorption coefficient of 6970 M−1 cm−1. Lactate dehydrogenase (LDH) and pyruvate kinase (PK) were dosed in assay buffer (see below) at room temperature (RT).
Enzymatic assay
The kinetic activity of GST–IGF1R was measured spectrophotometrically using a coupled enzyme assay. Non-phosphorylated IGF1R (2.5 μM) was autophosphorylated in assay buffer (50 mM EPPS–KOH pH 8.0 (EPPS: 4-(2-hydroxyethyl)-1-piperazinepropanesulphonic acid), 0.01% Brij-35, 1 mM dithiothreitol) for 15 min at RT in the presence of 1 mM ATP and 10 mM MgCl2. The phosphorylated enzyme was immediately used in the assay (final concentration 10 nM) and the reaction was run in a 96-well plate (UV-Star Plate, Flat-Bottom, Half area—Greiner) at RT in assay buffer containing 0.005% bovine serum albumin (BSA) in the presence of 1 mM phosphoenolpyruvate, 14 U/mL PK, 0.25 mM nicotinamide adenine dinucleotide (NADH), 10 U/mL LDH and variable concentrations of ATP, peptide substrate, MgCl2 and/or inhibitors. Absorbance was measured at 340 nM every 9 sec in a SpectraMax 340 spectrophotometer (Molecular Devices, Sunnyvale, CA). The assay was run at pH = 8.0 to ensure that ATP is predominantly present as ATPCitation4–12, and EPPS–KOH was utilized to provide the necessary K+ ions for PK activity.
Calculation of the free Mg2+ concentration
The concentration of free Mg2+ present in the assay was calculated with:
where [Mg2+]f is the calculated concentration of free Mg2+, [Mg2+]0 the total concentration of Mg2+ and X1 is obtained from:
where [ATP]0 is the total concentration of ATP. ATP, phosphoenolpyruvate and NADH bind to Mg2+, with a dissociation constant (Kd) equal to 0.0143, 5 and 19.5 mMCitation13, respectively. Since Kd of phosphoenolpyruvate or NADH are high, most of the bound Mg2+ should be complexed with ATP. A first [Mg2+]f was calculated assuming that only ATP is present (lowest Kd value). This value was then used as new [Mg2+]0 to calculate a second [Mg2+]f assuming that only phosphoenolpyruvate (intermediate Kd) is present. Finally, the same calculation was repeated for NADH (highest Kd) to obtain the final [Mg2+]f present in the assay.
Data analysis
The kinetic parameters of the phosphorylated GST–IGF1R protein were determined by global fit using the rate equation for a Bi–Bi rapid equilibrium random sequential mechanismCitation8:
where v is the measured initial rate, Vmax the maximum rate, [ATP] and [PEP] the concentrations of ATP and peptide substrate, KATP and KPEP the dissociation constant for ATP and the peptide substrate, α defines the degree of interaction between both substrates.
The potency, IC50, of the different inhibitors was determined with:
where s is the measured signal, Smax the maximum signal, [I] the inhibitor concentration and h the Hill factor.
Since AEW541 and STAURO are ATP competitive inhibitors for IGF1RCitation8, the inhibition experiments were analyzed by global fit with:
where v is the measured initial rate, ![]() . the maximum rate, [ATP] the ATP concentrations,
. the maximum rate, [ATP] the ATP concentrations, ![]() and
and ![]() the dissociation constant for ATP and the inhibitor constant. The superscript app indicates apparent kinetic parameters.
the dissociation constant for ATP and the inhibitor constant. The superscript app indicates apparent kinetic parameters.
The two parameters KI and β were calculated with:
where ![]() is the apparent inhibitor constant, [PEP] the peptide substrate concentration, KPEP the dissociation constant for the peptide substrate, β is the degree of interaction between the peptide substrate and the inhibitorCitation14. β and KI were calculated from measurements of
is the apparent inhibitor constant, [PEP] the peptide substrate concentration, KPEP the dissociation constant for the peptide substrate, β is the degree of interaction between the peptide substrate and the inhibitorCitation14. β and KI were calculated from measurements of ![]() realized at two independent peptide concentrations.
realized at two independent peptide concentrations.
Theoretical IC50s were calculated with:
where KI is the inhibitor constant, [ATP] and [PEP] the concentrations of the two substrates, KATP and KPEP the dissociation constant for ATP and the peptide substrate, α defines the degree of interaction between both substrates, β is the degree of interaction between the peptide substrate and the inhibitor. The derivation of Equation 5 is given in Supplementary Information.
The kinetic data were analyzed with Grafit v 5.04 (Erithacus Software Limited, Horley, UK).
Results
Determination of the kinetic parameters of IGF1R at different concentrations of free Mg2+
The kinetic parameters of protein kinases often change when the [Mg2+] present in assays variesCitation15. In the case of IGF1R, [Mg2+] ≥ 10 mM are commonly used in enzymatic assays to ensure maximum enzymatic activity (see, e.g. refs. 11,16–20) However, the concentration of free Mg2+ ([Mg2+]f) found in living cells is in the low millimolar range and close to 0.5 mMCitation21,Citation22. We therefore decided to measure the kinetic parameters of phosphorylated IGF1R at low (0.5 mM) and high (10 mM) [Mg2+]f(). To ensure that the PK/LDH coupled system used in the assay was correctly functioning at both of [Mg2+]f, the catalytic activity of different IGF1R concentrations was measured in the presence of 0.5 and 10 mM [Mg2+]f. In both conditions, the catalytic rates increased linearly in proportion to [IGF1R] suggesting, in agreement with published workCitation13,Citation23–25, that the PK/LDH coupled system is not affected by these changes in [Mg2+]f (data not shown). The measured kinetic parameters of IGF1R are presented in . Varying [Mg2+]f affects both KATP (~16-fold increase from 10 to 0.5 mM [Mg2+]f) and KPEP (~11-fold increase from 10 to 0.5 mM [Mg2+]f) with little effect on Vmax and α. Since the ionic strength of the assay buffer varies between low (0.5 mM) and high (10 mM) [Mg2+]f, the conductivity of these two assay buffers was measured and the ionic strength of the low [Mg2+]f buffer was brought up to the conductivity of the high [Mg2+]f buffer by the addition of NaCl (17 mM). Similar kinetic parameters (<1.5-fold difference) were measured in assay buffer containing [Mg2+]f = 0.5 mM and [Mg2+]f = 0.5 mM + [NaCl] = 17 mM, suggesting that a difference in ionic strength was not at the root of the observed changes in KATP and KPEP (data not shown).
Table 1. Influence of the concentration in free Mg2+ on the kinetic properties of IGF1R.
Figure 1. Determination of the kinetic parameters of IGF1R at two different concentrations of free magnesium. The kinetic parameters of IGF1R were determined in the presence of [Mg2+]f = 0.5 (A) and 10 (B) mM. Experiments were fitted using Equation 1 as described in Material and methods.
![Figure 1. Determination of the kinetic parameters of IGF1R at two different concentrations of free magnesium. The kinetic parameters of IGF1R were determined in the presence of [Mg2+]f = 0.5 (A) and 10 (B) mM. Experiments were fitted using Equation 1 as described in Material and methods.](/cms/asset/0ca7e1b9-11a5-4879-890d-b3a39e663afe/ienz_a_583922_f0001_b.gif)
Inhibitory properties of AEW541 and STAURO and influence of the peptide substrate
Since the cellular concentration of ATP is close to 1 mMCitation26–28, the potency (IC50) of AEW541 and STAURO was measured in the biochemical assay in the presence of 1 mM ATP at 0.5 or 10 mM [Mg2+]f (). The IC50s of AEW541 and STAURO shows an 11- and 8-fold increase, respectively, when [Mg2+]f is increased from 0.5 to 10 mM.
Table 2. Influence of the concentration of free Mg2+ on the inhibitory properties of AEW541 and STAURO.
To estimate the level of interaction between the peptide substrate and AEW541 or STAURO, ![]() was determined at two different [PEP] concentrations () and from these values, β and KI were calculated using Equation 4 (Materials and methods) (). The presence of the bound peptide affects the binding of AEW541 (β0.5 mM = 15; β10 mM = 39) more than that of STAURO (β0.5 mM = 0.9; β10 mM = 5). β values are higher for both compounds when [Mg2+]f increases, suggesting that the binding of a second Mg2+ to IGF1R (at 10 mM) affects the conformation of the protein–peptide complex. In contrast to β, KI is minimally affected (2-fold at most) when [Mg2+]f varies (). This is consistent with the fact that AEW541 and STAURO do not require Mg2+ for binding to IGF1R as previously observed by Surface Plasmon ResonanceCitation8.
was determined at two different [PEP] concentrations () and from these values, β and KI were calculated using Equation 4 (Materials and methods) (). The presence of the bound peptide affects the binding of AEW541 (β0.5 mM = 15; β10 mM = 39) more than that of STAURO (β0.5 mM = 0.9; β10 mM = 5). β values are higher for both compounds when [Mg2+]f increases, suggesting that the binding of a second Mg2+ to IGF1R (at 10 mM) affects the conformation of the protein–peptide complex. In contrast to β, KI is minimally affected (2-fold at most) when [Mg2+]f varies (). This is consistent with the fact that AEW541 and STAURO do not require Mg2+ for binding to IGF1R as previously observed by Surface Plasmon ResonanceCitation8.
Figure 2. Inhibition of IGF1R by AEW541 and STAURO. The kinetic activity of IGF1R was measured in the presence of different concentrations of AEW541 and STAURO at [Mg2+]f = 0.5 (A) and 10 (B) mM. Experiments were globally fitted using Equation 3 (competitive inhibition). A semilogarithmic representation of these experiments is given.
![Figure 2. Inhibition of IGF1R by AEW541 and STAURO. The kinetic activity of IGF1R was measured in the presence of different concentrations of AEW541 and STAURO at [Mg2+]f = 0.5 (A) and 10 (B) mM. Experiments were globally fitted using Equation 3 (competitive inhibition). A semilogarithmic representation of these experiments is given.](/cms/asset/9f9d229b-419e-4305-a27a-175011a76e4b/ienz_a_583922_f0002_b.gif)
The β and KI parameters were used to calculate theoretical IC50s. Recently, the derivation of the relationship between IC50 and KI has been established for various bisubstrate enzyme mechanismsCitation29. Unfortunately, the equation provided for competitive inhibitors of enzymes following rapid equilibrium random mechanisms is only valid on the assumption that inhibitors bind to the free enzyme and the enzyme–peptide complex with roughly equal affinity (β ~ 1). Since this condition does not apply here, a new equation, Equation 5 (Material and methods), was established (see Supplementary Information for details). With the help of this equation and using the calculated values for β and KI and the value of kinetic parameters determined experimentally (), theoretical IC50 values were calculated (). The calculated IC50 values are in very good agreement with the measured IC50 values (less than a 2-fold difference). This supports the validity of the calculated values for β and KI (but also of the other kinetic parameters measured in this work).
Discussion
Biochemists always utilize the lowest possible amount of enzymes in assays because, following the Cheng-Prusoff equationCitation30, it is not possible to measure IC50 values lower than [Enzyme]/2. Accordingly, they identify experimental conditions where enzymes show the highest activity to reduce at best their concentration in assays. In the case of protein kinases, maximum activity is often reached in the presence of high [Mg2+] or Mg2+/Mn2+ mixtures. In such conditions, the affinity of protein kinases for ATP is usually in the micromolar range (for IGF1R: αKATP = 116 μM at 10 mM Mg2+ () or αKATP = 4 μM with an Mg2+/Mn2+ mix8). Balanced assay conditions are also achieved when substrates are present at Km concentrationsCitation31 (for bisubstrate enzymes, see ref. 29). Therefore, kinase assays are frequently run in the presence of micromolar concentrations of ATP, which correspond to Km(ATP) (or αKATP). If nanomolar ATP competitive inhibitors are identified at these low [ATP] concentrations, their potency should decrease in cells because of the presence of millimolar levels of this nucleotide. This is exemplified with AEW541, which shows good potency in the presence of micromolar [ATP] in the presence of a Mg2+/Mn2+ mix (45 nM8) but reduced potency at [ATP] = 1 mM in the presence [Mg2+]f = 10 mM (543 nM; ). However, such a decrease in potency is not observed in cells where AEW541 retains a nanomolar activity (86 nM19). What is at the root of this discrepancy? Our results show that decreasing [Mg2+]f reduces the affinity of IGF1R for ATP. Therefore, in the presence of a physiological concentration of both ATP (1 mM) and free magnesium (0.5 mM), AEW541 (and STAURO) should compete more easily with this nucleotide because it is not present in large excess ([ATP] ~ αKATP). Indeed, the IC50 values obtained in this condition are low () and similar to the IC50 values measured in cells with this compoundCitation19. Therefore, AEW541 is a nanomolar inhibitor in cells despite the presence of millimolar levels of ATP probably because IGF1R has a reduced affinity for this nucleotide at physiological [Mg2+]f. Since other protein kinases show a reduced affinity for ATP in the presence of low [Mg2+]fCitation15, we propose that the observations made here could also apply to ATP competitive inhibitors of these enzymes. However, the affinity of some protein kinases is not affected by [Mg2+]fCitation15, therefore the effect observed here with IGF1R should not be applicable to these enzymes. Furthermore, it should be kept in mind that compounds with poor ADME properties (e.g. low cell permeability, high protein binding, etc.) may show decreased potency once they are tested in cell-based assays.
It is often ignored that, for protein kinases following a Bi–Bi rapid equilibrium random sequential mechanism, the binding of the peptide substrate may influence the affinity of ATP competitive inhibitors. Equation 4 (Materials and methods) shows that the peptide substrate affects ![]() in two ways.
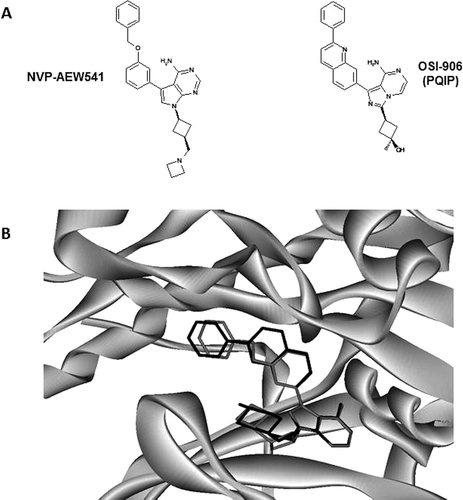
in two ways. ![]() varies with [PEP] and the term β reflects the interaction between the peptide and the inhibitor. When β is <1 the bound peptide substrate enhances the affinity of the inhibitor, and when β is >1 it decreases the affinity. This may have direct implications in drug discovery. For example, if two different pockets/conformations exist for a specific protein kinase, the best choice to obtain the most potent inhibitors will be the pocket/conformation for which the affinity of the compounds is not decreased by the presence of the bound protein substrate (β = 1) or, better, is improved by its presence (β < 1). Therefore, determining β may help medicinal chemists to choose the most appropriate pocket/conformation for the target. Few β values are available in the literature (e.g. refs. 32–34), and some of those concern non-ATP competitive inhibitors. We therefore decided to determine β values for the two ATP competitive inhibitors, AEW541 and STAURO. The bound peptide only moderately affects the binding of STAURO (β ~ 1), whereas it largely reduces (β >> 1) the affinity of AEW541 (). These data show that the peptidic substrate modulates the interaction of these inhibitors with IGF1R in different ways even so they both bind at the ATP-binding site. An interpretation for the high β values measured with AEW541 can be proposed. The structure of AEW541 in complex with IGF1R is not available, but it has been described that OSI906—an inhibitor closely related to AEW541Citation8—adopts a binding mode that is not compatible with an active conformation of IGF1RCitation20. Manual docking of AEW541 into the OSI906–IGF1R structure shows that it can take the same bound conformation as OSI906 (). Therefore, AEW541 may also bind to IGF1R that is not in its active conformation. Since, the peptide substrate interacts with the active conformation of IGF1RCitation16, both molecules compete for two distinct conformations of this protein. This competition explains why the peptide has a negative effect on AEW541 binding and the high β values measured with this inhibitor.
varies with [PEP] and the term β reflects the interaction between the peptide and the inhibitor. When β is <1 the bound peptide substrate enhances the affinity of the inhibitor, and when β is >1 it decreases the affinity. This may have direct implications in drug discovery. For example, if two different pockets/conformations exist for a specific protein kinase, the best choice to obtain the most potent inhibitors will be the pocket/conformation for which the affinity of the compounds is not decreased by the presence of the bound protein substrate (β = 1) or, better, is improved by its presence (β < 1). Therefore, determining β may help medicinal chemists to choose the most appropriate pocket/conformation for the target. Few β values are available in the literature (e.g. refs. 32–34), and some of those concern non-ATP competitive inhibitors. We therefore decided to determine β values for the two ATP competitive inhibitors, AEW541 and STAURO. The bound peptide only moderately affects the binding of STAURO (β ~ 1), whereas it largely reduces (β >> 1) the affinity of AEW541 (). These data show that the peptidic substrate modulates the interaction of these inhibitors with IGF1R in different ways even so they both bind at the ATP-binding site. An interpretation for the high β values measured with AEW541 can be proposed. The structure of AEW541 in complex with IGF1R is not available, but it has been described that OSI906—an inhibitor closely related to AEW541Citation8—adopts a binding mode that is not compatible with an active conformation of IGF1RCitation20. Manual docking of AEW541 into the OSI906–IGF1R structure shows that it can take the same bound conformation as OSI906 (). Therefore, AEW541 may also bind to IGF1R that is not in its active conformation. Since, the peptide substrate interacts with the active conformation of IGF1RCitation16, both molecules compete for two distinct conformations of this protein. This competition explains why the peptide has a negative effect on AEW541 binding and the high β values measured with this inhibitor.
Figure 3. Model for the interaction between AEW541 and IGF1R. (A) OSI906 is structurally related to AEW541Citation35 and it exhibits similar kinetic propertiesCitation8. (B) AEW541 has been docked manually into the structure of the OSI906–IGF1R complex (pdb number 3D94Citation20). The region surrounding the inhibitors is represented in light grey solid ribbons, AEW541 in dark grey and OSI906 in black.
Vanderpool et al.Citation34 have measured β > 1 for CHK1 inhibitors and they have proposed that the binding of the substrate (in their case ATP) affects the volume of the allosteric pocket where these molecules bind. These results together with our data show that measuring β brings valuable additional information on the protein kinase–inhibitor interaction, and we propose that the β parameter should be used by medicinal chemists to study the binding mode of compounds in more detail. It should be borne in mind that to get maximum benefit from β values, the assays should preferentially be carried out with physiological substrates and not synthetic peptidesCitation14. However, many natural substrates possess multiple phosphorylation sites, possibly rendering enzymatic studies more complex.
Declaration of interest
The authors declare that they do not have any conflicts of interest.
Supplementary Material
Download PDF (76.7 KB)References
- Liao JJL. Molecular recognition of protein kinase binding pockets for design of potent and selective kinase inhibitors. J Med Chem 2007;50:410–424.
- Thaimattam R, Banerjee R, Miglani R, Iqbal J. Protein kinase inhibitors: structural insights into selectivity. Curr Pharm Des 2007;13:2751–2765.
- Kufareva I, Abagyan R. Type-II kinase inhibitor docking, screening, and profiling using modified structures of active kinase states. J Med Chem 2008;51:7921–7932.
- Liu Y, Gray NS. Rational design of inhibitors that bind to inactive kinase conformations. Nat Chem Biol 2006;2:358–364.
- Bogoyevitch MA, Fairlie DP. A new paradigm for protein kinase inhibition: blocking phosphorylation without directly targeting ATP binding. Drug Discov Today 2007;12:622–633.
- Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer 2009;9:28–39.
- Li R, Pourpak A, Morris SW. Inhibition of the insulin-like growth factor-1 receptor (IGF1R) tyrosine kinase as a novel cancer therapy approach. J Med Chem 2009;52:4981–5004.
- Chène P, Hau JC, Blechschmidt A, Fontana P, Bohn J, Zimmermann C et al. Study of the selectivity of insulin-like growth factor-1 receptor (IGF1R) inhibitors. Open Enzyme Inh J 2010;3:27–37.
- Erdmann D, Zimmermann C, Fontana P, Hau JC, De Pover A, Chène P. Simultaneous protein expression and modification: an efficient approach for production of unphosphorylated and biotinylated receptor tyrosine kinases by triple infection in the baculovirus expression system. J Biomol Tech 2010;21:9–17.
- Shoichet BK. Screening in a spirit haunted world. Drug Discov Today 2006;11:607–615.
- Bell IM, Stirdivant SM, Ahern J, Culberson JC, Darke PL, Dinsmore CJ et al. Biochemical and structural characterization of a novel class of inhibitors of the type 1 insulin-like growth factor and insulin receptor kinases. Biochemistry 2005;44:9430–9440.
- Hekmat-Nejad M, Cai T, Swinney DC. Steady-state kinetic characterization of kinase activity and requirements for Mg2+ of interleukin-1 receptor-associated kinase-4. Biochemistry 2010;49:1495–1506.
- Saylor P, Wang C, Hirai TJ, Adams JA. A second magnesium ion is critical for ATP binding in the kinase domain of the oncoprotein v-Fps. Biochemistry 1998;37:12624–12630.
- Chène P. The new challenges of biochemistry in identifying the next generation of protein kinase inhibitors. Drug Discov Today 2008;13:522–529.
- Adams JA. Kinetic and catalytic mechanisms of protein kinases. Chem Rev 2001;101:2271–2290.
- Favelyukis S, Till JH, Hubbard SR, Miller WT. Structure and autoregulation of the insulin-like growth factor 1 receptor kinase. Nat Struct Biol 2001;8:1058–1063.
- Li W, Favelyukis S, Yang J, Zeng Y, Yu J, Gangjee A et al. Inhibition of insulin-like growth factor I receptor autophosphorylation by novel 6-5 ring-fused compounds. Biochem Pharmacol 2004;68:145–154.
- Wang Y, Malkowski M, Hailey J, Turek-Etienne T, Tripodi T, Pachter JA. Screening for small molecule inhibitors of insulin-like growth factor receptor (IGF-1R) kinase: comparison of homogeneous time-resolved fluorescence and 33P-ATP plate assay formats. J Exp Ther Oncol 2004;4:111–119.
- García-Echeverría C, Pearson MA, Marti A, Meyer T, Mestan J, Zimmermann J et al. In vivo antitumor activity of NVP-AEW541-A novel, potent, and selective inhibitor of the IGF-IR kinase. Cancer Cell 2004;5:231–239.
- Wu J, Li W, Craddock BP, Foreman KW, Mulvihill MJ, Ji QS et al. Small-molecule inhibition and activation-loop trans-phosphorylation of the IGF1 receptor. Embo J 2008;27:1985–1994.
- McCarthy JT, Kumar R. Divalent Cation Metabolism: Magnesium. Blackwell Publishing, Ames, IA, 1999.
- McDermott M. The intracellular concentration of free magnesium in extensor digitorum longus muscles of the rat. Exp Physiol 1990;75:763–769.
- Adams JA, Taylor SS. Divalent metal ions influence catalysis and active-site accessibility in the cAMP-dependent protein kinase. Protein Sci 1993;2:2177–2186.
- Cook PF, Neville ME Jr, Vrana KE, Hartl FT, Roskoski R Jr. Adenosine cyclic 3′,5′-monophosphate dependent protein kinase: kinetic mechanism for the bovine skeletal muscle catalytic subunit. Biochemistry 1982;21:5794–5799.
- Lin L, Czerwinski R, Kelleher K, Siegel MM, Wu P, Kriz R et al. Activation loop phosphorylation modulates Bruton’s tyrosine kinase (Btk) kinase domain activity. Biochemistry 2009;48:2021–2032.
- Gribble FM, Loussouarn G, Tucker SJ, Zhao C, Nichols CG, Ashcroft FM. A novel method for measurement of submembrane ATP concentration. J Biol Chem 2000;275:30046–30049.
- Kennedy HJ, Pouli AE, Ainscow EK, Jouaville LS, Rizzuto R, Rutter GA. Glucose generates sub-plasma membrane ATP microdomains in single islet beta-cells. Potential role for strategically located mitochondria. J Biol Chem 1999;274:13281–13291.
- Wang RH, Tao L, Trumbore MW, Berger SL. Turnover of the acyl phosphates of human and murine prothymosin alpha in vivo. J Biol Chem 1997;272:26405–26412.
- Yang J, Copeland RA, Lai Z. Defining balanced conditions for inhibitor screening assays that target bisubstrate enzymes. J Biomol Screen 2009;14:111–120.
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099–3108.
- Copeland RA. Evaluation of enzyme inhibitors in drug discovery. Wiley Interscience, Hoboken, NJ, 2005.
- Yagi YI, Abe K, Ikebukuro K, Sode K. Kinetic mechanism and inhibitor characterization of WNK1 kinase. Biochemistry 2009;48:10255–10266.
- Liu M, Choi S, Cuny GD, Ding K, Dobson BC, Glicksman MA et al. Kinetic studies of Cdk5/p25 kinase: phosphorylation of tau and complex inhibition by two prototype inhibitors. Biochemistry 2008;47:8367–8377.
- Vanderpool D, Johnson TO, Ping C, Bergqvist S, Alton G, Phonephaly S et al. Characterization of the CHK1 allosteric inhibitor binding site. Biochemistry 2009;48:9823–9830.
- Ji QS, Mulvihill MJ, Rosenfeld-Franklin M, Cooke A, Feng L, Mak G et al. A novel, potent, and selective insulin-like growth factor-I receptor kinase inhibitor blocks insulin-like growth factor-I receptor signaling in vitro and inhibits insulin-like growth factor-I receptor dependent tumor growth in vivo. Mol Cancer Ther 2007;6:2158–2167.