Abstract
Carbonic anhydrases (CAs, EC 4.2.1.1) are ubiquitous enzymes that catalyze the hydration of CO2 to bicarbonate and protons. Inhibition of CAs has been clinically exploited for the treatment of various classes of diseases for decades, but investigating new classes of inhibitors continues to be important. We have synthesized a series of 2-amino-3-cyano-4-heteroaryl (5a–l) compounds and characterized the structures by NMR, IR and elemental analyses. We tested the ability of these compounds to inhibit two metalloenzyme human carbonic anhydrase (hCA, EC 4.2.1.1) isozymes, hCA I and hCA II. Compounds 5d and 5b showed the best inhibition activity against hCA I (IC50: 33 and 34 µM, respectively), and compound 5d showed the best activity against hCA II (IC50: 56 µM).
Introduction
Carbonic anhydrase (CA) is a zinc metalloenzyme that catalyzes the reversible reaction of CO2 and water: ![]() . It was discovered in 1932 in the red blood cells following the realization that the uncatalyzed rate of HCO3− dehydration was too low to support CO2 excretion during the time blood spent at the gas exchange surfaceCitation1–4.
. It was discovered in 1932 in the red blood cells following the realization that the uncatalyzed rate of HCO3− dehydration was too low to support CO2 excretion during the time blood spent at the gas exchange surfaceCitation1–4.
As an enzyme, CA is of broad interest because it has one of the fastest known reaction rates. The turnover number (kcat, exceeds 1 × 106 s−1 for some CA isoformsCitation1–4). Furthermore, the reaction catalyzed by CA is fundamental to a wide array of physiological processes, including calcification, photosynthesis, respiration, metabolism, cell growth and ionic, acid-base and fluid balanceCitation1–4. This diversity of functions suggests that CA may have been among the earliest enzymes to appear, and CA seems to be almost ubiquitously expressed in living organisms. Five genetically unrelated families (α, ß, γ, δ and ξ) of CA isoforms existCitation5. The α-CA family is the best studied group, although recent reviews indicate rapid advancement of knowledge about other CA familiesCitation6–11.
The classes of CA inhibitors (CAIs) are known: the metal complexing anions, and the substituted sulfonamides such as acetazol amideCitation1 and their bioesters (sulfamates, sulfamides), which bind to the metal ion of the enzyme either by substituting for the non-protein zinc ligand to generate a tetrahedral adduct or by participating in the metal coordination sphere to generate trigonal-bipyramidal speciesCitation12. PhenolsCitation2 which bind to the zinc-coordinated water molecule/hydroxide ion from active site are also very important inhibitors that display competitive inhibitionCitation13,Citation14. CoumarinesCitation3 and thiocoumarines which have an inhibition mechanism independent of Zn (II) because they don’t have proton-shutting moieties in their molecules are the third class of inhibitors, CAIsCitation15–17. In the recent studies polyamines such as spermine and spermidineCitation4 are reported a new class of CAI inhibitorsCitation18,Citation19. The Zn (II) ion in CAs is critical for the inhibition of these enzymes. Three mechanisms of inhibition have been proposed, one of which involves anchoring of the inhibitor to the Zn (II) bound solvent molecule (a water or hydroxide ion). Phenols and polyamines bind in this wayCitation18–21.
A critical problem in the design of CA inhibitors with pharmacological applications for the treatment and prevention of various diseases is related to the high number of CA isoforms (16 in mammals), the diffuse localization of CAs in many tissues/organs, and the lack of isozyme selectivity of the presently available inhibitorsCitation22,Citation23.
The N-heteroaromatic pyridine is incorporated into the structure of many pharmaceuticals. Among these, cyanopryridines and aminocyanopyridines substituted with different alkyl and aryl groupsCitation5 were found to have antimicrobialCitation24, anti-inflammatory, analgesic and antipyretic propertiesCitation25. 2-Thienyl-4-furyl-pyridine and 2-thienyl-4-furyl-6-aryl pyridine derivatives exhibited strong inhibitory activity against the nuclear enzymes topoisomerase I and II and inhibitory activities and cytotoxicity against several human cancer cell linesCitation26. These compounds are also valuable intermediates in the syntheses of a variety of biologically active heterocyclic compoundsCitation27.
However, aminocyanopyridine derivatives bearing cycloalkanes at C5–C6 along with substituted five membered hetereocycles at C4 have not yet been tested as for inhibitory activity against CAs. We sought to fill in this gap in the literature by examining these derivatives.
Materials and methods
Chemicals
Sepharose 6B, protein assay reagents and 4-nitrophenylacetate were obtained from Sigma–Aldrich Co. (Sigma–Aldrich Chemie GmbH, Taufkirchen, Germany). All other chemicals were obtained from Merck (Merck KGaA, Darmstadt, Germany). All the chemicals investigated in the study were reagent grade and were further purified only as necessary. All organic solvents used in this study were purified according to standard methods. Elemental analyses were carried out with a LECO-CHNS-932 instrument. 1H and 13C-NMR spectra were recorded on a Varian 200 MHz spectrometer using TMS (Tetramethylsilane) as an internal standard. IR spectra were recorded on a Mattson-5000 FT-IR instrument in KBr pellets. Melting points were determined with a Gallenkamp melting point apparatus.
General method for preparation of 3a-b
Intermediates 3a–b were prepared according to the methods in the literatureCitation28.
General method for preparation of 5a–l
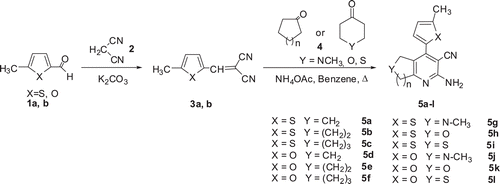
Compound 3a, b (0.005 mol) was suspended in benzene (10 ml), and ammonium acetate (0.0075 mol) and cycloalkanone 4 (0.005 mol) were added. The flask was fitted with a reflux condenser and a water separator, and the mixture was refluxed for 6 h. Then, the solvent was evaporated, and the mixture was re-dissolved in chloroform (150 ml) and washed with water (2 × 50 ml). The organic phase was dried with MgSO4, filtered off and recrystallized from ethyl acetateCitation28. All aminocyanopyridines (5a–l) were prepared by this procedure ().
Scheme 1. Synthesis of 2-amino-3-cyanopyridines (5a–l).
2-Amino-6,8-dihydro-4-(5-methylfuran-2-yl)-5H-thiopyrano[3,4-b]pyridine-3-carbonitrile (5l); m.p: 222–225 °C, 48% yield
IR cm−1 3412–3321 (NH2), 3175 (CH Aryl), 2923 (CH Alkyl), 2210 (CN).
1H-NMR (DMSO-d6, δ, ppm) 2.39 (s, 3H, CH3), 2.92–2.99 (m, 4H, 2CH2), 3.33 (s, 2H, CH2), 6.35–6.37 (dd, J: 3.3, 0.98 Hz, 1H Ar-H), 6.76 (bs, 2H, NH2), 6.84–6.96 (d, J: 3.3 Hz, 1H, Ar-H).
13C-NMR (DMSO-d6, ppm) 15.0, 26.7, 27.7, 35.8, 87.1, 109.9, 117.1, 118.5, 118.8, 141.6, 146.8, 155.9, 160.6, 163.0.
El. Anal. Predicted: C, 61.97; H, 4.83; N, 15.49; S, 11.82. Observed: C, 61.64; H, 4.81; N, 15.44; S, 11.64.
Purification of CA isozymes from human erythrocytes by affinity chromatography
The two CA isozymes were purified via a simple single-step method using Sepharose-4B-l-tyrosine-sulfanilamide affinity gel chromatography.
Erythrocytes were purified from fresh human blood obtained from the blood centre of the research hospital at Ataturk University. The blood samples were centrifuged at 1500 rpm for 15 min, and the plasma and buffy coat were removed. The red cells were isolated and washed twice with 0.9% NaCl and hemolyzed with 1.5 volumes of ice-cold water. The ghost and intact cells were removed by centrifugation at 20.000 rpm for 30 min at 4°C. The pH of the hemolysate was adjusted to 8.7 with solid Tris.
Sepharose 4B was activated with CNBr, then filtered with a Buchner funnel and washed with cold 0.1 M NaHCO3 buffer (pH 10). A saturated solution of l-tyrosine in the same buffer was coupled to the activated Sepharose 4B resin for 90 min, with magnetic stirring. The affinity gel was obtained by coupling diazosulfanilamide to the Sepharose 4B-l-tyrosine. Sulfanilamide (25 mg) was suspended in 10 ml of ice-cold 1 M HCl, and sodium nitrite (75 mg in 5 ml ice-cold water) was added to the suspension. After the mixture had been allowed to react for 10 min, the diazosulfanilamide was mixed with 40 ml of the Sepharose 4B-l-tyrosine suspension. The pH was adjusted to 9.5 with 1 M NaOH and the mixture was stirred gently for 3 h at room temperature. The coupled red Sepharose derivative was then washed with 1 l of water, followed by 200 ml of 0.05 M Tris-sulfate (pH 7.5). After this Sepharose-4B-l tyrosine-sulfanilamide affinity column had been equilibrated with 25 mM Tris–HCl/0.1 M Na2SO4 (pH 8.7), the hemolysate was applied. The affinity gel was washed with 25 mM Tris–HCl/22 mM Na2SO4 (pH 8.7). The human carbonic anhydrase (hCA I and hCA II) isozymes were eluted with 1 M NaCl/25 mM Na2HPO4 (pH 6.3) and 0.1 M CH3COONa/0.5 M NaClO4 (pH 5.6), respectively. All procedures were performed at 4°CCitation29.
Hydratase activity assay
The hydratase activity of hCA I and hCA II was assayed by following the hydration of CO2 according to the method described by Wilbur and AndersonCitation30. The activity of CO2-hydratase in enzyme units (EU) was calculated by using the equation [(to − tc)/tc] where t0 and tc are the times for pH change of the non-enzymatic and the enzymatic reactions, respectively.
Esterase activity assay
CA activity was assayed by following the change in absorbance at 348 nm of 4-nitrophenylacetate (NPA) to 4-nitrophenylate ion over a period of 3 min at 25°C using a spectrophotometer (CHEBIOS UV–Vis) according to the method described by Verpoorte et alCitation31. The enzymatic reaction contained 1.4 mL 0.05 M Tris–SO4 buffer (pH 7.4), 1 mL 3 mM 4-nitrophenylacetate, 0.5 mL H2O and 0.1 mL enzyme solution (total volume, 3.0 mL). A reference measurement was obtained by preparing the mixture without the enzyme solution. All measurements were made in triplicate. The inhibitory effects of 5a, 5b, 5c, 5d, 5e, 5f, 5g, 5h, 5i, 5j, 5k and 5l were examined over a range of concentrations. The activity of hCA I was measured at the following cuvette concentrations of the test compounds: 5a(0.001–1000 mM), 5b (6–49 µM), 5c (6–47 µM), 5d (14–56 µM), 5e (39–88 µM), 5f (38–89 µM), 5g (0.001–1000 mM), 5h (0.001–1000 mM), 5i (155–675 µM), 5j (0.001–1000 mM), 5k (0.001–1000 mM) and 5l (125–756 µM). The activity of hCA II was measured at the following cuvette concentrations of the test compounds: 5a (0.001–1000 mM), 5b (0.037–0.112 µM), 5c (0.035–0.082 µM), 5d (0.042–0.056 µM), 5e (0.001–1000 mM), 5f (0.001–1000 mM), 5g (0.001–1000 mM), 5h (0.001–1000 mM), 5i (0.001–1000 mM), 5j (0.001–1000 mM), 5k (0.001–1000 mM) and 5l (0.001–1000 mM). The activity of the control cuvette (in the absence of inhibitor) was set to 100%. An Activity (%)-[Inhibitor] graph was constructed for each compound. The Ki values were determined from a series of experiments using three different inhibitor concentrations and 4-nitrophenylacetate as the substrate at five different concentrations (0.15–0.75 mM) to construct Lineweaver–Burk curvesCitation32.
Protein determination
The yield of protein during the purification steps was determined spectrophotometrically at 595 nm according to the Bradford method, using bovine serum albumin as the standardCitation33.
SDS polyacrylamide gel electrophoresis
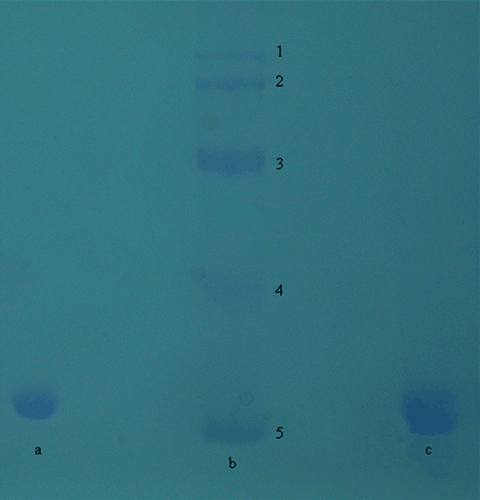
The purity of the enzymes was confirmed using SDS polyacrylamide gel electrophoresis (). It was carried out according to Laemmli procedureCitation34.
Figure 1. PAGE of the purified CA isozymes. Lane b Standards: E.coli β-galoksidaz (116 kDa), rabbit muscle phosphorylase b (97 kDa), bovine albumin (66 kDa); ovalbumin (45 kDa), bovine erythrocytes CA (29 kDa), lane a: CA I, lane c: CA II.
Results and discussion
Chemistry
We have previously reported the synthesis and antimicrobial evaluation of various 2-amino-3-cyanopyridine derivativesCitation28. The synthesis of pentasubstituted pyridine derivatives () was based on previously proposed methodsCitation35. First, a Knoevenagel base-catalyzed condensation was used to produce two ylidenemalononitriles (3a, b) in high yield. Then, twelve 2-amino-3-cyanopyridines (5a–I) were synthesized in 30–71% yield by reacting the ylidenemalononitriles (3a, b) with cycloalkanonesCitation4 in the presence of ammonium acetate.
CA purification and assay
CA has been purified from many different organisms and the effects of various chemicals, pesticides, and drugs on its activity have been investigatedCitation36–39. In this study, hCA I and II were purified from human erythrocytes (). The inhibitory effects of a series of 2-amino-3-cyanopyridines were tested in vitro. The inhibitor concentrations that caused 50% inhibition (IC50) were determined from % activity versus [Inhibitor] plots, and the Ki values were calculated from Lineweaver–Burk plots ()Citation33,Citation34.
Table 1. Purification of hCA I and CA II.
Table 2. Inhibitory activity of compounds 5a–l.
CA inhibition
We prepared twelve 2-amino-3-cyanopyridine derivatives with the structures shown in and evaluated their ability to inhibit hCA I and hCA II over a wide range of concentrations (0,001–1000 mM). Seven of the twelve compounds (5b, 5c, 5d, 5e, 5f, 5i, 5l) inhibited hCA I, and three of them (5b, 5c, 5d) inhibited hCA II. When we analyzed the successful inhibitors in detail, we found that five of the six structures that contained a cycloalkyl ring (5a–f) inhibited hCA I, and three of them inhibited hCA II. However, compound 5a did inhibit either hCA I or hCA II. Compounds 5e and 5f inhibited hCA I, but not hCA II. Only two of the six heterocyclic structures (5g–l) inhibited hCA I, and none inhibited hCA II. Although numerous reports have shown that sulfones inhibit CAs, thiopyrano-containing compounds 5i and 5l were only weak inhibitors of hCA I.
Our compounds were weak to moderate inhibitors of hCA I and hCA II. The effects of the six compounds that contain heterocyclic rings were the weakest. Our results are consistent with previous reports that phenols (amines) are weak inhibitors of hCA I and hCA IICitation15–19,Citation40.
Spermine was found bound deep within the hCAII active site, in his inhibition mechanism, nevertheless not directly coordinated to the metal ion. The terminal ammonium group in spermine is anchored to the Zn(II) bond water molecule/hydroxide ion similarly to by means of hydrogen bond phenolCitation2 and this terminal ammonium group forms a second hydrogen bond with the OH of Thr199 at pH 7.4.
pKa values of the polyamines like spermidine and spermine and their derivatives are in the range of 7.9–10.9Citation18–21. Aminopyridines are weak bases with pKa values between 6 and 9. Therefore, they can exist in neutral and protonated for at physiological pH. Aminopyridines pharmacophore and one or more amine groups, being by positive charge on the protonated nitrogen, is suitable for hydrogen bondingCitation41. Possible inhibiton mechanisms of the synthesized compounds can be similar to polyamines like spermine. Ammonium group in synthesized aminocyanopyridines (5b, 5c, 5d, 5e, 5f, 5i, 5l) can be formed both with the Zn(II) bound water/hydroxide ion and second hydrogen bond between OH of Thr199 at physiological pH.
Conclusions
We have synthesized twelve 2-amino-3-cyanopyridine derivatives incorporating biologically active cycloalkyl moieties at the C5–C6 positions and furan and thiophene derivatives at the C4 position of the molecule and evaluated the ability of these compounds to inhibit hCA I and hCA II. Seven of the twelve molecules (5b, 5c, 5d, 5e, 5f, 5i, 5l) inhibited hCA I, and three of them inhibited hCA II (5b, 5c, 5d). The best inhibitors of hCA I were compounds 5d, 5b and 5c, which exhibited IC50 values of 33, 34 and 40 µM, respectively. The best inhibitor of hCA II was compound 5d, with an IC50 value of 56 µM.
The compounds that contained five- and six-membered cycloalkyl rings at C5–C6 were more active than the compounds that contained heterocyclic (O, S, N) rings. The results indicated that the more increase there is in the ring size of cycloalkyl the more decrease there is the inhibition effect of molecules and this may attributed to the steric hindrance.
Of the compounds that contained six-membered heterocyclic rings, only those with sulfur as the heteroatom exhibited an inhibitory effect. Most of the effective inhibitors contained sulfur atoms.
Declaration of interest
The authors thank the Gazi University Scientific Research Fund (Project number: 05/2008-47) for financial support.
References
- Supuran CT. Carbonic anhydrase inhibition/activation: trip of a scientist around the world in the search of novel chemotypes and drug targets. Curr Pharm Des 2010;16:3233–3245.
- Supuran CT. Carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2010;20:3467–3474.
- Supuran CT. Carbonic anhydrase inhibitors and activators for novel therapeutic applications. Future Med Chem 2011;3:1165–1180.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Xu Y, Feng L, Jeffrey PD, Shi Y, Morel FM. Structure and metal exchange in the cadmium carbonic anhydrase of marine diatoms. Nature 2008;452:56–61.
- Burnell JN. (2000). Carbonic anhydrases of higher plants: An overview. In: Chegwidden, WR, Carter, ND, Edwards, YH (Eds.), The Carbonic Anhydrases. New Horizons. Basel: Birkhäuser Verlag, 501–518.
- Forsman, C. (2000). Plant carbonic anhydrases: structure and mechanism. The Carbonic Anhydrases. New Horizons. Basel: Birkhäuser Verlag, 519–533.
- Fukuzawa H, Tsuzuki M, Miyachi S. (2000). The Carbonic Anhydrases. New Horizons. Basel: Birkhäuser Verlag, 535–546.
- Kozliak EI, Guilloton MB, Fuchs JA, Anderson PM. (2000)Bacterial carbonic anhydrases. New Horizons. Bassel: Birkhäuser Verlag, 547–565.
- Zimmerman SA, Ferry JG. The β and g classes of carbonic anhydrase. Curr Pharm Des 2008;14:716–721.
- Rowlett RS. Structure and catalytic mechanism of the ß-carbonic anhydrases. Biochim. Biophys 2010;1804:362–373.
- Supuran CT, Scozzafava A, Conway J. (2004). Carbonic anhydrase− Its inhibitors and activators, Eds. Boca Raton, FL, USA: CRC Press, 1–376.
- Simonsson I, Jonsson BH, Lindskog S. Phenol, a competitive inhibitor of CO2 hydration catalyzed by carbonic anhydrase. Biochem Biophys Res Commun 1982;108:1406–1412.
- Tibell L, Forsman C, Simonsson I, Lindskog S. The inhibition of human carbonic anhydrase II by some organic compounds. Biochim Biophys Acta 1985;829;202–208.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen SA, Scozzafava A et al. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–3062.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarines and thiocoumarines. J Med Chem 2010;53:335–344.
- Temperini C, Innocenti A, Scozzafava A, Parkkila S, Supuran CT. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: The antiepileptic lacosamide as an example and lead molecule for novel classes of carbonic anhydrase inhibitors. J Med Chem 2010;53:850–854
- Carta F, Temperini C, Innocenti A, Scozzafava A, Kaila K, Supuran CT. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–5522.
- Rami M, Winum JY, Innocenti A, Montero JL, Scozzafava A, Supuran CT. Carbonic anhydrese inhibitors: Copper (II) complexes of polyamino-polycarboxyamido aromatic/heterocyclic sulfonamides are very potent inhibitors of tumor-associated isoforms IX and XII. Bioorg Med Chem Lett 2008;18(2):836–841.
- Supuran CT. Carbonic anhydrase inhibition with natural products: Novel chemotypes and inhibition mechanisms. Mol Divers 2011;15(2):305–316.
- Davis RA, Innocenti A, Poulsen SA, Supuran CT. Carbonic anhydrase inhibitors. Identification of selective inhibitors of the human mitochondrial isozymes VA and VB over the cytosolic isozymes I and II from a natural product-based phenolic library. Bioorg Med Chem 2010;18:14–18.
- Abbate F, Casini A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with a topically acting antiglaucoma sulfonamide. Bioorg Med Chem Lett 2004;14:2357–2361.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–189.
- Moussa HH, Chabaka LM, Zaki D. Synthesis and evaluation of antifungal properties of a series of the novel 2-amino-5-oxo-4- phenyl-5,6,7,8-tetrahydroquinoline-3-carbonitrile and its analogues. Egypt J Chem 1983;26:469–477.
- Manna F, Chimenti F, Bolasco A, Bizzarri B, Filippelli W, Fiilppelli A, Gagliardi L. Eur. Anti-inflammatory, analgesic and antipyretic 4,6-disubstituted 3-cyano-2-aminopyridines. J Med Chem 1999;34:245–254.
- Thapa P, Karki R, Thapa U, Jahng Y, Jung MJ, Nam JM et al. 2-Thienyl-4-furyl-6-aryl pyridine derivatives: synthesis, topoisomerase I and II inhibitory activity, cytotoxicity, and structure-activity relationship study. Bioorg Med Chem 2010;18:377–386.
- Doe K, Avasthi K, Pratap R, Bakuni DS, Joshi MN. Synthesıs of 2,4-bis(methylthio)-5,7-disubstıtuted-7H-pyrrolo-(2,3-D)pyrimidines and their biological-activity. Indian J Chem Sect B 1991;30:499–506.
- Altundas A, Ayvaz S, Logoglu E. Synthesis and evaluation of a series of aminocyanopyridines as antimicrobial agents. Med Chem Res 2011;1:1–8.
- Abell AD, Nabbs BK, Battersby AR. The reaction of N -magnesium derivatives of pyrroles with N -mesylchloromethylpyrroles: A synthesis of dipyrrylmethanes. J Org Chem 1998;63:8163–8169.
- Wilbur KM, Anderson NG. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 1948;176:147–154.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–4229.
- Lineweaver H, Burk DJ. The determination of enzyme dissociation constants. Am Chem Soc 1934;56:658–666.
- Bradford M. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye-binding. Anal Biochem 1976;72:248.
- Laemmli DK. Clevage of structual proteins during in assembly of the head of bacteriophage T. Nature 1970;227:680–683.
- Elgemeie CEH, Regaila HA, Shehata N. Novel syhthesis of condensed 4-(2-thienyl)-and 4-(2-furyl)3-cyano-pyridi-2-(1H)-1–8 7 ones and their conversions into the correponding pyridin-2-(1H)- thione derivatives.Sulfur Lett 1989;9:253–264.
- D’Ambrosio K, Vitale RM, Dogné JM, Masereel B, Innocenti A, Scozzafava A et al. Carbonic anhydrase inhibitors: bioreductive nitro-containing sulfonamides with selectivity for targeting the tumor associated isoforms IX and XII. J Med Chem 2008;51:3230–3237.
- Barrese AA 3rd, Genis C, Fisher SZ, Orwenyo JN, Kumara MT, Dutta SK et al. Inhibition of carbonic anhydrase II by thioxolone: a mechanistic and structural study. Biochemistry 2008;47:3174–3184.
- Innocenti A, Maresca A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: thioxolone versus sulfonamides for obtaining isozyme-selective inhibitors? Bioorg Med Chem Lett 2008;18:3938–3941.
- Rigaku /MSC, Inc., 9009 new Trails Drive, The Woodlands, TX 77381.
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–1587.
- Caballero NA, Melendez FJ, Muñoz-Caro C, Niño A. Theoretical prediction of relative and absolute pKa values of aminopyridines. Biophys Chem 2006;124:155–160.
- Vomasta D, Innocenti A, König B, Supuran CT. Carbonic anhydrase inhibitors: two-prong versus mono-prong inhibitors of isoforms I, II, IX, and XII exemplified by photochromic cis-1,2-α-dithienylethene derivatives. Bioorg Med Chem Lett 2009;19:1283–1286.