Abstract
A series of flavonoids, such as quercetin, catechin, apigenin, luteolin, morin, were investigated for their inhibitory effects against the metalloenzyme carbonic anhydrase (CA). The compounds were tested against four α-CA isozymes purified from human and bovine (hCA I, hCA II, bCA III, hCA IV) tissues. The four isozymes showed quite diverse inhibition profiles with these compounds. The flavonoids inhibited hCA I with KI-s in the range of 2.2–12.8 μM, hCA II with KI-s in the range of 0.74–6.2 μM, bCA III with KI-s in the range of 2.2–21.3 μM, and hCA IV with inhibition constants in the range of 4.4–15.7, with an esterase assay using 4-nitrophenyl acetate as substrate. Some simple phenols/sulfonamides were also investigated as standard inhibitors. The flavonoids incorporate phenol moieties which inhibit these CAs through a diverse, not yet determined inhibition mechanism, compared to classic inhibitors such as the sulfonamide/sulfamate ones.
Keywords::
Introduction
Flavonoids are polyphenols incorporating diphenylpropanes scaffolds. The four major classes are the 4-oxoflavonoids (flavones, flavonols, etc.), anthocyanins, isoflavones, and the flavan-3-ol derivatives (catechins and tanninsCitation1) About 6500 naturally occurring flavonoids have been identified up to dateCitation2 They are important constituents of the human diet, and the daily intake of flavonoids is approximately 200–1000 mg.Citation3 Major dietary sources of flavonoids include for example: beverages (cola, coffee, tea, fruit juices, red wine), fruits and vegetables, nutsCitation4,Citation5. The proposed health optimizing activities of the flavonoids are due to the following actions: (i) antioxidant properties through the ability to scavenge reactive oxygen species (ROS) or through their influences on the redox status (ii) modulation of signal transduction pathways involved in cell proliferation and angiogenesis (iii) influencing enzymes participating in estrogen biosynthesis (iv) modulation of enzymes necessary for metabolic activation of pro-carcinogens and detoxification of carcinogensCitation5,Citation6.
The hydration of carbon dioxide to bicarbonate and protons, a simple, but essential reaction, is catalyzed by a superfamily of enzymes which have been investigated in detailCitation7. These enzymes, known as carbonic anhydrases (CAs, EC 4.2.1.1) are pH regulatory/metabolic enzymes in all life kingdoms, being found in organisms all over the phylogenetic tree. Sixteen CA isoforms are presently known in mammalsCitation7,Citation8. Mammalian CAs were the first enzymes of this type to be isolated and studied in detail, and many of them are established therapeutic targets with the potential to be inhibited or activated to treat a wide range of disorders.Citation7–11 There are currently known diuretics, antiglaucoma, antiepileptic, antiobesity, and anticancer drugs based on CAIs, and they target various mammalian α-CA isoformsCitation7. CAs are inhibited by four main mechanisms: (i) coordination of the inhibitor to the Zn(II) ion by replacing the zinc-bound water/hydroxide ion and leading to a tetrahedral geometry of Zn(II) (e.g. the sulfonamides, Citation7,Citation12). (ii) Addition of the inhibitor to the metal coordination sphere, when the Zn(II) ion is in a trigonal bipyramidal geometry (e.g. the thiocyanate adduct, Citation7,Citation12). (iii) Anchoring of the inhibitor to the Zn(II)-bound solvent molecule, i.e. a water or hydroxide ion (e.g. phenol, Citation13) (iv) Occlusion of the CA active site entrance, with coumarinsCitation9–11,Citation14 lacosamideCitation9 and fullerenesCitation15 binding in this way, as shown schematically in for a hydrolyzed coumarin derivativeCitation10. All these binding modes have been demonstrated by means of X-ray crystallography of enzyme–inhibitor adductsCitation7–15. It should be noted that passing from sulfonamides and their bioisosteres (sulfamates, sulfamides, etc.) to inhibitors occluding the entrance of the active site, the role of the Zn(II) ion is constantly diminishing in its interaction with the inhibitor moleculeCitation10–15. This phenomenon has important consequences for the drug design of CA inhibitors (CAIs), because the bottom of the active site cavity is very much conserved in the 16 CA isozymes described so far in mammals, whereas the regions with the highest variation in amino acid sequence, therefore the highest degree of structural diversity, are just those at the entrance of the active siteCitation7,Citation8,Citation12. Indeed, phenolsCitation15–20 but also coumarinsCitation9,Citation10 and other types of non-zinc binder inhibitorsCitation11 were recently shown to lead to isozyme-selective CAIs, a goal difficult to achieve with the classical sulfonamide/sulfamate inhibitorsCitation7,Citation22.
Figure 1. Schematic representation for the three main CA inhibition mechanisms: (A) Sulfonamides (and their isosteres, sulfamate, and sulfamide) substitute the fourth zinc ligand and bind in tetrahedral geometry of the metal ionCitation12; (B) Inorganic anion inhibitors (thiocyanate as an example) add to the metal ion coordination sphere leading to trigonal bipyramidal adductsCitation12; (C) Phenols anchor to the Zn(II) coordinated water molecule/hydroxide ionCitation13; (D) Coumarins (hydrolyzed in situ to 2-hydroxycinnamic acids) occlude the entrance of the active site cavity, interacting both with hydrophilic and hydrophobic amino acid residues. The inhibitor does not interact at all with the catalytically crucial Zn(II) ion which is coordinated by three His residues and a water moleculeCitation10,Citation11.
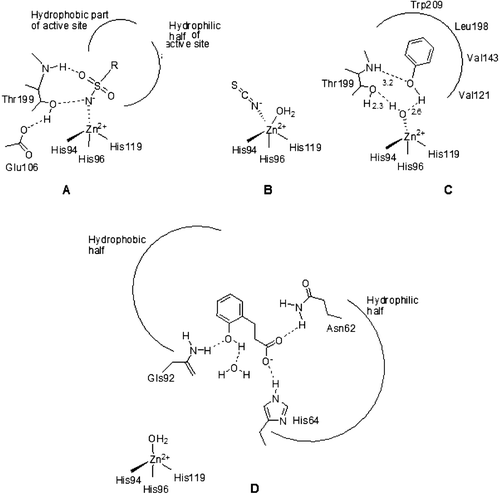
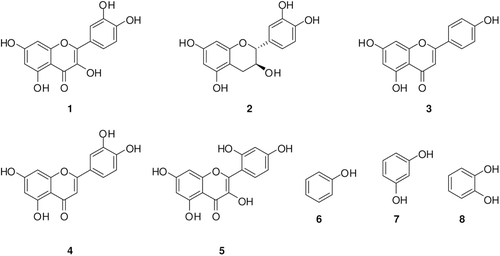
Our groups recently investigated the interaction of hCA I, II, VI isozymes with several types of natural and synthetic compounds, such as the simple phenol, catechol, resorcinol, pyrogallol, gallic acid, tannic acid and several of its substituted derivatives, e.g. salicylates and some of their derivativesCitation19,Citation20. Here we extend these earlier investigations to series of compounds 1–8, some of which are widely used as antioxidant food additives. Among the various natural or synthetic phenolic compounds with antioxidant properties, these compounds are very active in quenching ROSCitation19. They are reported to possess anticancer, anti-carcinogenic, antimutagenic, antibacterial, antiviral or anti-inflammatory activitiesCitation4–6. Quercetin, catechin, apigenin, luteolin and morin, members of the flavonoids family, are ubiquitously present in food, including vegetables, fruits, tea, and red wine. Several biologic properties of these series have been reported to be beneficial to human health, including protection against various diseases, such as osteoporosis, certain forms of cancer, pulmonary and cardiovascular diseases, and agingCitation6 Phenol, resorcinol, and catechol are widely used prodrugs or disinfectantsCitation3,Citation6.
In the present study, we have purified hCA I, hCA II, bCA III, and hCA IV and examined the in vitro inhibition effects of the compounds 1–8, using the esterase activity of these isoenzymes, with 4-nitrophenyl acetate as substrate.
Materials and methods
Chemicals
Quercetin, catechin, apigenin, luteolin, morin, phenol, resorcinol, catechol, Sepharose 4B, protein assay reagents, 4-nitrophenylacetate were obtained from Sigma-Aldrich Co. All other chemicals were analytical grade and obtained from Merck.
Purification of carbonic anhydrase isozymes by affinity chromatography
Purification of hCA I and hCA II were previously describedCitation17–21. hCA IV was purified following the protocol described by Zhu and SlyCitation22. Bovine CA III was obtained from flank steak and purified using a modification of the method of Tu et al. (1986Citation23). The active fractions comprising the major protein peak were pooled and concentrated in 1 mM mercaptoethanol.
Esterase activity assay
Carbonic anhydrase activity was assayed by following the change in absorbance at 348 nm of 4-nitrophenylacetate (NPA) to 4-nitrophenylate ion over a period of 3 min at 25°C using a spectrophotometer according to the method described by Verpoorte et alCitation24. The enzymatic reaction, in a total volume of 3.0 mL, contained 1.4 mL of 0.05 M Tris-SO4 buffer (pH 7.4), 1 mL of 3 mM 4-nitrophenylacetate, 0.5 mL of H2O and 0.1 mL of enzyme solution. A reference measurement was obtained by preparing the same cuvette without enzyme solution. Enzyme concentrations in the assay system were 9 nM for hCA I, 7 nM for hCA II, 7 nM for IV and 12 nM for bCA III.
In vitro inhibition study
The inhibitory effects of compound 1–8, EZA, ZNA and AZA were examined. All compounds were tested in triplicate at each concentration used. Different inhibitor concentrations were used, starting from 1 nM to 1 mM. Control cuvette activity in the absence of inhibitor was taken as 100%. For each inhibitor, Activity (%)-[Inhibitor] graphs were drawn. To determine KI values, three different inhibitor concentrations were tested. In these experiments, 4-nitrophenylacetate was used as substrate at five different concentrations (0.15–0.75 mM). The Lineweaver-Burk curves were drawnCitation25.
Protein determination
Protein concentration during the purification steps was determined spectrophotometrically at 595 nm according to the Bradford method, using bovine serum albumin as a standardCitation26.
SDS polyacrylamide gel electrophoresis
SDS polyacrylamide gel electrophoresis was performed after purification of the enzymes. It was carried out in 10% and 3% acrylamide for the running and the stacking gel, respectively, containing 0.1% SDS according to Laemmli procedureCitation27.
Results and discussion
Phenol binds to CA in a different manner from the classical inhibitors of the sulfonamide type, for example etazolamide (EZA), which coordinate to the Zn(II) ion from the enzyme active site by substituting the fourth, non-protein ligand, a water molecule or hydroxide ionCitation13,Citation16. The simple phenol has been shown to be the only competitive inhibitor with CO2 as substrate for the main isoform of CA, i.e. human CA II (hCA IICitation13) which is the reason for investigation of these phenolic compounds. The X-ray crystal structure for the adduct of hCA II with phenolCitation13, showed that this compound binds to CA by anchoring its OH moiety to the zinc-bound water/hydroxide ion of the enzyme active site, through a hydrogen bond as well as through a second hydrogen bond to the NH amide of Thr199, an amino acid conserved in all α-CAs and critically important for the catalytic cycle of these enzymesCitation13–20. Our group has recently investigated the interactions of phenol and some of its substituted derivatives with all mammalian CAsCitation18–20, demonstrating some low micromolar/submicromolar inhibitors as well as the possibility to design isozyme selective CAIs. The inhibition profile of various CA isozymes with this class of agents is very variable, with inhibition constants ranging from the millimolar to the submicromolar range for many simple phenolsCitation13,Citation20,Citation28. Thus, it seemed reasonable to us to extend the previous studiesCitation20,Citation28, including in this investigation phenolic compounds and flavonoids with clinical and antioxidant applications as food additives, such as compounds 1–8Citation20,Citation28,Citation29.
The purification of the CA isozymes was performed with a simple one step method by a Sepharose-4B-aniline-sulfanilamide affinity column chromatoghrapyCitation18. hCA I was purified, 104.7-fold with a specific activity of 917.16 EUmg−1 and overall yield of 54.2 %. hCA II was purified, 732.4-fold with a specific activity of 7045 EUmg−1 and overall yield of 59.36 %. hCA IV was purifed, 89.18-fold with a specific activity of 748.86 EUmg−1 and overall yield of 27.8%. Similarly, bCA III was purifed, 48.8-fold with a specific activity of 456.4 EUmg−1 and overall yield of 46.4%Citation16–20,Citation22,Citation23. Inhibitory effects of compounds 1–8 on enzyme activities were tested under in vitro conditions; KI values were calculated from Lineweaver-Burk graphs and are given in Citation25.
Table 1. KI values (μM) for compound 1–8, EZA, ZNA and AZA of some α-carbonic anhydrase isoforms in human (h) and bovine (b). aRef Citation29.
We investigated the first study on the inhibitory effects of of flavonoids, including quercetin, catechin, apigenin, luteolin, morin, phenol, resorcinol, and catechol on the esterase activity of hCA I, II, IV and bCA III. The sulfonamide CAIs EZA, ZNA and AZA have been used as a negative controls in our experiments, and for comparison reasons. The previous reports by Senturk et alCitation20,Citation29 investigated other antioxidant phenol derivatives (including salicylic acid and gallic acid) by using esterase assay. Data of show the following regarding inhibition of hCA I, II, IV and bCA III with compounds 1–8, EZA, ZNA, and AZA, by an esterase assayCitation24, with 4-nitrophenylacetate (4-NPA) as substrate:
Against hCA I, compound 7 behave as weak inhibitor, with KI value in the range of 828.2 μM. Catechol (8) was also an ineffective hCA I inhibitor (KI of 4014 μM). A second group of derivatives, compounds 1–6, EZA, ZNA and AZA, showed better inhibitory activity with KI values of between 2.2 to 36.2 μM, (). Therefore, the nature of the groups in ortho-, para- and meta- to the phenolic OH moiety strongly influences hCA I inhibitory activity. It is also interesting to note that the pholyphenolic compounds 1–5 were much better hCA I inhibitors as compared to the corresponding other phenolic compounds 7 and 8 from which they were prepared, with KI-s in the range of 2.2–12.8 μM. Acetazolamide AZA is also a medium CA I with this assay and substrate against hCA I (KI of 36.2 μM). Kinetic investigations (Lineweaver-Burk plots, data not shown) indicate that similarly to sulfonamides, metal ions and inorganic anionsCitation28–32, all the investigated compounds act as noncompetitive inhibitors with 4-NPA as substrate, i.e. they bind in different regions of the active site cavity as compared to the substrate. However the binding site of 4-NPA itself is unknown, but it is presumed to be in the same region as that of CO2, the physiological substrate of this enzymeCitation13,Citation29–32.
A better inhibitory activity has been observed with compounds 1 and 3 investigated here for the inhibition of the rapid cytosolic isozyme hCA II (). Five derivatives, i.e. 2, 5–8, showed moderate hCA II inhibitory activity with KI-s in the range of 2.4–11.4 μM, ), whereas the remaining two derivatives 4, EZA, ZNA and AZA were quite effective hCA II inhibitors, with KI-s in the range of 0.32–1.1 μM, (). Structure-activity relationship (SAR) is thus quite sharp for this small series of mono, di, tri-hydroxy compounds and flavonoids ( and ). The best hCA II inhibitor in this series of derivatives was 4, which with a KI of 0.74 μM. It must be stressed that KI-s measured with the esterase method are always in the micromolar range because hCA I and II are weak esterasesCitation28–33.
Compound 7 and AZA were weak inhibitors of bCA III, with KI-s of 219 and 263 μM. However, again compound 5 was medium potency inhibitor (KI of 21.3 μM), and compounds 1, 3, EZA and ZNA, show a higher affinity for this isozyme, with inhibition constants in the range of 8.5–11.6 M, whereas the remaining four derivatives 2, 4, 6, 8 were quite effective bCA III inhibitors, with KI-s in the range of 2.2–6.5 μM, ().
The membrane-anchored isoform hCA IV was poorly inhibited by five of the investigated compounds, that is, compounds 1, 3, 5–8 and ZNA which showed KI-s in the range of 9.1–634 μM. These derivatives incorporate either a 3-substituent in meta to the phenol OH moiety (such as resorcinol 7). However, compounds 2 and 4 were more effective hCA IV inhibitors, inhibition constants in the range of 4.4 and 5.6 μM. These compounds are anyhow much weaker inhibitors as compared to the EZA and AZA (KI-s of 0.84 and 0.58 μM against hCA IV, ).
Figure 2. Structures of common used CA inhİbitors.
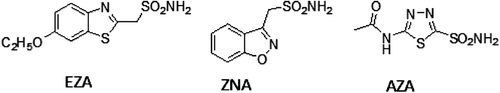
Figure 3. Structures of tested compounds 1–8.
In a recent study it was reported that derivatives of salicylic acid, some natural phenolic compounds, as well as some organic nitratesCitation16–20 acts as a CAIs. These simple compounds are lacking the sulfonamide, sulfamate, or related functional groups that are typically found in classical CAIs. It is critically important to explore further classes of potent CAIs in order to detect compounds with a different inhibition profile as compared to the sulfonamides and their bioisosteres and to find novel applications for the inhibitors of these widespread enzymes.
Conclusions
Phenols 6–8 and similar molecules inhibit the activity of CAs due to the presence of the different functional groups present in their scaffold, first of which the phenolic one which probably is anchored to the non-protein zinc ligandCitation28. Flavonoids 1–5 investigated here were quite effective, micromolar inhibitors of several CA isozymes. Our findings indicate thus another category of CAIs of interest, in addition to the well-known sulfonamides/sulfamates/sulfamides, the phenols/ diphenols bearing bulky ortho or para moieties in their molecules, or the coumarins/polyaminesCitation33. Compound 4 showed the most effective inhibitory activity among derivatives investigated here, with KIs in the low micromolar range, by the esterase method y. These findings point out that substituted phenolic compounds and flavonoids may be used as leads for generating more potent CAIs eventually targeting other isoforms which have not been assayed yet for their interactions with such agents.
Declarations of interest
This study was financed by Turkish Republic Prime Ministry State Planning Organization (DPT), (Project no: 2010K120440) for (MS) and by an FP7 EU grant (Metoxia) to CTS. The authors report no declarations of interest.
References
- Rhodes MJC, Price KR. Analytical problems in the study of flavonoid compounds in onions. Food Chem 1996;57:113–117.
- Teillet F, Boumendjel A, Boutonnat J, Ronot X. Flavonoids as RTK inhibitors and potential anticancer agents. Med Res Rev 2008;28:715–745.
- Kühnau J. The flavonoids. A class of semi-essential food components: Their role in human nutrition. World Rev Nutr Diet 1976;24:117–191.
- Manach C, Williamson G, Morand C, Scalbert A, Rémésy C. Bioavailability and bioefficacy of polyphenols in humans. I. Review of 97 bioavailability studies. Am J Clin Nutr 2005;81:230S–242S.
- Pick A, Müller H, Mayer R, Haenisch B, Pajeva IK, Weigt M et al. Structure-activity relationships of flavonoids as inhibitors of breast cancer resistance protein (BCRP). Bioorg Med Chem 2011;19:2090–2102.
- Havsteen BH. The biochemistry and medical significance of the flavonoids. Pharmacol Ther 2002;96:67–202.
- Supuran CT. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Supuran CT, Scozzafava A. Carbonic anhydrases as targets for medicinal chemistry. Bioorg Med Chem 2007;15:4336–4350.
- Temperini C, Innocenti A, Scozzafava A, Parkkila S, Supuran CT. The coumarin-binding site in carbonic anhydrase accommodates structurally diverse inhibitors: The antiepileptic lacosamide as an example and lead molecule for novel classes of carbonic anhydrase inhibitors. J Med Chem 2010;53:850–854.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen SA, Scozzafava A et al. Non-zinc mediated inhibition of carbonic anhydrases: Coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–3062.
- Maresca A, Temperini C, Pochet L, Masereel B, Scozzafava A, Supuran CT. Deciphering the mechanism of carbonic anhydrase inhibition with coumarins and thiocoumarins. J Med Chem 2010;53:335–344.
- Stams T, Christianson DW. X-ray Crystallographic Studies of Mammalian Carbonic Anhydrase Isozymes. In The Carbonic Anhydrases—New Horizons; Chegwidden, WR, Edwards, Y, Carter, N, Eds.; Birkha¨user Verlag: Basel, 2000; pp 159–174.
- Nair SK, Ludwig PA, Christianson DW. Two-site binding of phenol in the active site of human carbonic anhydrase II: Structural implications for substrate association. J Am Chem Soc 1994;116:3659–3660.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–777.
- Innocenti A, Durdagi S, Doostdar N, Strom TA, Barron AR, Supuran CT. Nanoscale enzyme inhibitors: Fullerenes inhibit carbonic anhydrase by occluding the active site entrance. Bioorg Med Chem 2010;18:2822–2828.
- Durdagi S, Sentürk M, Ekinci D, Balaydin HT, Göksu S, Küfrevioglu ÖI,et al. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg Med Chem 2011;19:1381–1389.
- Ekinci D, Cavdar H, Talaz O, Sentürk M, Supuran CT. NO-releasing esters show carbonic anhydrase inhibitory action against human isoforms I and II. Bioorg Med Chem 2010;18:3559–3563.
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–9105.
- Sentürk M, Gülçin I, Dastan A, Küfrevioglu OI, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–3211.
- Oztürk Sarikaya SB, Topal F, Sentürk M, Gülçin I, Supuran CT. In vitro inhibition of a-carbonic anhydrase isozymes by some phenolic compounds. Bioorg Med Chem Lett 2011;21:4259–4262.
- Sentürk M, Ekinci D, Göksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzyme Inhib Med Chem 2011. DOI: 10.3109/14756366.2011.591290.
- Zhu XL, Sly WS. Carbonic anhydrase IV from human lung. Purification, characterization, and comparison with membrane carbonic anhydrase from human kidney. J Biol Chem 1990;265:8795–8801.
- Tu CK, Thomas HG, Wynns GC, Silverman DN. Hydrolysis of 4-nitrophenyl acetate catalyzed by carbonic anhydrase III from bovine skeletal muscle. J Biol Chem 1986;261:10100–10103.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–4229.
- Lineweaver H, Burk D. The determination of enzyme dissocation constants. J Am Chem Soc 1934;56:658–666.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
- Laemmli DK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 1970;227:680–685.
- Innocenti A, Vullo D, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: Interactions of phenols with the 12 catalytically active mammalian isoforms (CA I-XIV). Bioorg Med Chem Lett 2008;18:1583–1587.
- Sentürk M, Gülçin I, Beydemir S, Küfrevioglu OI, Supuran CT. In Vitro Inhibition of Human Carbonic Anhydrase I and II Isozymes with Natural Phenolic Compounds. Chem Biol Drug Des 2011;77:494–499.
- Alp C, Ekinci D, Gültekin MS, Sentürk M, Sahin E, Küfrevioglu OI. A novel and one-pot synthesis of new 1-tosyl pyrrol-2-one derivatives and analysis of carbonic anhydrase inhibitory potencies. Bioorg Med Chem 2010;18:4468–4474.
- Ekinci D, Ceyhun SB, Sentürk M, Erdem D, Küfrevioglu OI, Supuran CT. Characterization and anions inhibition studies of an a-carbonic anhydrase from the teleost fish Dicentrarchus labrax. Bioorg Med Chem 2011;19:744–748.
- Ceyhun SB, Sentürk M, Yerlikaya E, Erdogan O, Küfrevioglu OI, Ekinci D. Purification and characterization of carbonic anhydrase from the teleost fish Dicentrarchus labrax (European seabass) liver and toxicological effects of metals on enzyme activity. Environ Toxicol Pharmacol 2011;32:69–74.
- Carta F, Temperini C, Innocenti A, Scozzafava A, Kaila K, Supuran CT. Polyamines inhibit carbonic anhydrases by anchoring to the zinc-coordinated water molecule. J Med Chem 2010;53:5511–5522.