Abstract
4-Sulfamoyl-N-(3-morpholinopropyl)benzamide (I-1), N-(3-morpholinopropyl)benzene-1,4-disulfonamide (I-2) and N-(4-diethylaminoethoxybenzyl)benzene-1,4-bis(sulfonamide (I-3), were prepared and assayed as inhibitors of four carbonic anhydrase (CA) isoenzymes hCA I, hCA II, hCA IV and hCA XII. These compounds exhibited nanomolar half maximal inhibitory concentration (IC50) ranging from 58 to 740 nmol/L. All three aromatic sulfonamides show different activities for the isoenzymes studied with lowest affinity against isoenzyme hCA XII.
Introduction
Compounds bearing sulfonamide groups have long been known to be potent inhibitors of the carbonic anhydrases (CACitation1–3). Various substituted aromatic and heterocyclic sulfonamides have been synthesized and evaluated for possible therapeutic use as antiglaucoma agentsCitation1,Citation2,Citation4–6. They bind as anions to the Zn2+ ion within the enzyme active siteCitation1–3 (with abnormally high affinities of around 10−6–10−9 M−1 for isozyme CA II, refs 7–9). Clinically used sulfonamide antiglaucoma drugs include orally administered acetazolamide, ophthalmic suspension of brinzolamide and ophthalmic solution of dorzolamideCitation5,Citation6. In the case of these human carbonic acid inhibitors for optimal in vivo activity the balanced hydro- and liposolubility is necessary. It is well establishedCitation10,Citation11 that a water-soluble sulfonamide, also possessing relatively balanced lipid solubility, would be an effective antiglaucoma drug via the topical route. One of the conditions needed for a sulfonamide to act, as an effective intraocular pressure-lowering agent, is to possess modest lipid solubility attributable to its unionized formCitation8,Citation10–12.
In this work, we report the synthesis of a novel drug-like aromatic sulfonamides 4-sulfamoyl-N-(3-morpholinopropyl) benzamide (I-1), N-(3-morpholinopropyl)benzene-1,4-disulfonamide (I-2) and N-(4-diethylaminoethoxybenzyl)benzene-1, 4-bis(sulfonamide (I-3), with favorable structural, physicochemical and some pharmacokinetic properties comparable to those obtained for clinically useful acetazolamide, dorzolamide and brinzolamideCitation13–16. The new compounds reported here were in vitro tested on the enzymatic hCA I, hCA II, hCA IV and hCA XII activities. Affinities in the nanomolar range were found for those compounds against all four isoenzymes studied.
Experimental section
Chemistry
General
1H NMR spectra at 300 MHz were measured on Varian Gemini spectrometer (chemical shifts are expressed as δ values relative to TMS as standard). Melting points were determined on Kofler block and are uncorrected. Elemental analysis: Carlo Erba Model 1106 Instrument Elemental Analyser. The obtained results showed a maximum deviation of 0.3% compared to the theoretical values. All reactions were monitored by thin-layer chromatography (TLC) using silica gel plates (E. Merck) in system acetone–methanol (3:1).
Procedure for the preparation of compounds I-1, I-2 and I-3
4-Sulfamoyl-N-(3-morpholinopropyl)benzamide (I-1) and N-(3-morpholinopropyl)benzene-1,4-disulfonamide (I-2) were prepared according to the procedure described in ref. Citation16 and depicted in and 2. N-(4-diethylaminoethoxybenzyl)benzene-1,4-bis(sulfonamide) (I-3) was prepared as depicted in .
Scheme 1 Synthesis of 4-sulfamoyl-N-(3-morpholinopropyl) benzamide (I-1).
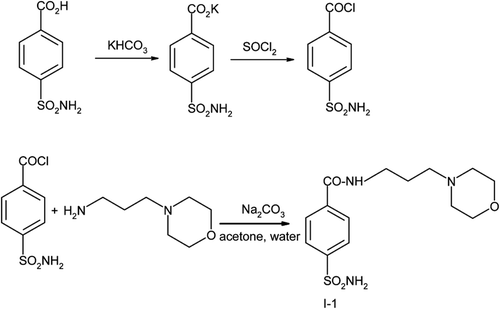
Scheme 2 Synthesis of N-(3-morpholinopropyl)benzene-1,4-disulfonamide (I-2).
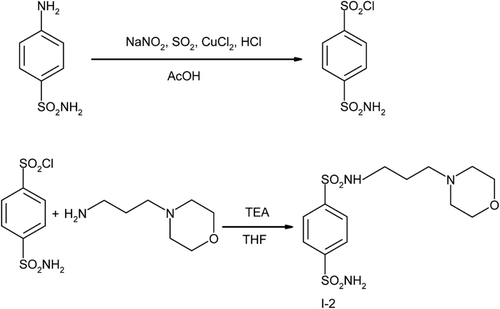
Scheme 3 Synthesis of N-(4-diethylaminoethoxybenzyl)benzene-1,4-bis(sulfonamide (I-3).
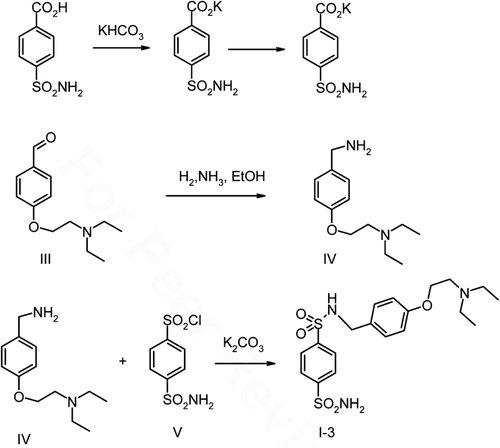
Synthesis of 4-diethylaminoethoxybenzaldehyde III
To the solution of 4-hydroxybenzaldehyde 18.30 g (0.15 mol) in anhydrous acetone (100 ml) N, N-diethyl-2-chloroethylamine II, 21.70 g (0.16 mol) and anhydrous potassium carbonate 22.10 g (0.16 mol) were added. The stirred mixture was refluxed for 12 h, than cooled to 0°C and the precipitated potassium chloride was filtered off and washed with acetone (20 ml). From filtrate the acetone was distilled off and the residual oil was fractionated in vacuum. Colorless liquid, yield 20.02 g (57%), b.p. 150–151°C/2 torr, n20D = 1.5360. Elemental analysis for C13H19N2 (M.r. 221.30), calculated (found, %): C 70.56 (70.80), H 8.65 (8.38), N 6.33 (6.22). 1H NMR (CDCl3) 1.07 (t, 6H, CH3), 2.64 (q, 4H, CH2-N), 4.12 (t, 2H, CH2-O), 7.01 (d, 2H, Har.), 7.83 (d, 2H, Har.) 9.88 (s, 1H, CHO).
Synthesis of 4-diehylaminoethoxy benzylamine IV
Raney nickel (4.00 g) was added to a solution of 4-diethylaminoethoxybenzaldehyde 17.93 g (0.080 mol) in 10% solution of ammonia in ethanol (80 ml). The mixture was hydrogenated at 80°C and pressure of 1.000 psi for 6 h in argon atmosphere. The catalyst was filtered off, washed with ethanol (10 ml). Ethanol was distilled off and liquid residual oil was fractionated in vacuum. Yield 13.10 g (72.8%), yellow liquid b.p. 145–146°C/2 torr, n20D = 1.5471. Elemental analysis for for C13H22N2O (M.r. 222.33), calculated/found, %): C 70.23 (70.39), H 9.98 (9.72), N 12.66 (12.81). 1H NMR (CDCl3) 1.07 (t, 6H, CH3), 1.84 (s, 2H, NH2), 2.64 (q, 4h, CH2-N), 2.87 (t, 2H, CH2-N), 3.79 (s, 2H, CH2-Phenyl), 4.04 (t, 2H, CH2-O), 6.87 (d, 2H, Har.), 7.21 (d, 2H, Har.).
Synthesis of N-[4-(diethylaminoethoxybenzyl)]benzene-1,4-disulfonamide I-3
To the could solution 4-diethylaminoethoxy benzylamine IV in acetone (12 ml) solution of sodium carbonate 2.34 g (0.022 mol) in water (10 ml) in a small portion during 5 min was added. To this stirred mixture 4-sulfamoylbenzenesulfonylchloride 5.12 g (0.02 mol) during 30 min. at 10°C was added. After then the reaction mixture was stirred 12 h at room temperature. The solid inorganic salt was filtered, washed with acetone (5 ml). The solvent from filtrate was evaporated using a vacuum rotatory evaporator. The residue was mixed three times with cold water (3 × 10 ml). The crude solid was filtered and purified by crystallization from 2-propanol. Colorless solid, yield 6.10 g (69.3%), m.p. 72−74°C.
Elemental analysis for C19H27N3O5S2 (M.r. 441.57), calculated (found): C 51.68 (51.86) H 6.16 (6.02), N (9.52) (9.38), S 14.52 (14.23). 1H NMR (DMSO) 1.07 (t, 6H, CH3), 2.64 (q, 4H, CH2-N), 2.87 (t, 2H, CH2-N), 4.04 (t, 2H, CH2-O), 6.89 (d, 2H, Har.), 7.12 (d, 2H, Har.-O), 7.63 (s, 2H, SO2-NH2), 8.00 (dd, 4H, Har.-SO2), 8.39 (t, 1H, NH-SO2).
4-Sulfamoylbenzenesulfamoylchloride (V) was prepared by multistep synthesis from 4-aminobenzenesulfonamide as starting compound, treated with sodium nitrite, sulfur dioxide, copper dioxide and hydrogen chloride in acetic acid as solventCitation17. The crude product was crystallized from 1,2-dichloroethane as colorless compound, yield 58%, m.p. 154–156°C. The measured physicochemical characteristic of this product corresponds to the similar data found in ref. 17.
Carbonic anhydrase I, II, IV and XII assay-IC50 determination
An Applied Photophysics stopped-flow instrument has been used for assaying the CA catalysed CO2 hydration activityCitation18. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 10–20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (10 mM) were prepared in distilled–deionised water and dilutions up to 0.01 nM were done thereafter with distilled–deionised water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complex. The inhibition constants were obtained by non-linear least-squares methods using PRISM 3, whereas the kinetic parameters for the uninhibited enzymes from Lineweaver–Burk plots, as reported earlierCitation19–21, and represent the mean from at least three different determinations.
Results and discussion
In despite of fact that several thousand different aromatic sulfonamide CA inhibitors were prepared and tested during the last 50 years in the search of diverse antiglaucoma agentsCitation22 the clinically useful aromatic sulfonamide is not yet available. Orally active heterocyclic acetazolamide can cause several side effects, (including paresthesia of the fingertips and toes, fatigue, depression, kidney stones, nausea and diarrhea, metabolic acidosis, agranulocytosis, aplastic anemia, and Stevens–Johnsons syndrome) and its use rapidly decreased after approval of two heterocyclic sulfonamides (brinzolamide and dorzolamide) given topically. Both topical drugs can cause a bitter taste. Dorzolamide can cause ocular burning. Systemic reactions are very rare but have been documentedCitation2,Citation23.
In our work we applied methods of manual design within the so-called “tail” approachCitation2,Citation24 for identification of a single lead compound. Within this approach a series of new aromatic sulfonamides was modeled. In the early stages of the design it was becoming more important to determine the pKa, water solubility, Lipinsky parameters and other physicochemical properties associated with a sulfonamides, before synthetic work is undertaken, with the aim of avoiding the synthesis of compounds that are predicted to have poor biopharmaceutical characteristicsCitation14,Citation15. This approach led us to the discovery of two new aromatic sulfonamides containing morpholinopropyl “tail” (I-1 and I-2), which exhibit drug-like properties comparable with dorzolamide and/or brinzolamideCitation15. The morpholine oxygen can form a new productive hydrogen bond interaction with complementary CA active site domain. For development of third inhibitor (I-3) previously designed scaffold of the benzene-1,4-disulfonamide type was applied. Extended tail of this derivative contains diethylaminoethoxybenzyl moiety and exploit the strategy of enhanced hydrophobic interactions between hydrophobic moieties of both active site of enzyme and inhibitor. This strategy was successfully applied by Whiteshides’ groupCitation2,Citation12. This strategy led, however, to the increase of lipophilicity for I-3 (1.65) by comparison with I-1 (−0.30) and I-2 (−0.86), XLOGP2 methodCitation15. Tail extension strategy produced also very effective hCA I and hCA II inhibitor I-3 without any appreciable increase of its IC50 values compared to those data for I-2 (). However, it is probable that diethylaminoethoxybenzyl moiety of I-3 is not sufficiently long to interact effectively with hydrophobic regions at the entrance of the enzyme active site and contribute appreciably to the specificity and strength of interaction with the active site of the hCA isozymes studied.
Table 1. Biochemical activity IC50 (nmol/L) and solubility of the CA inhibitors investigated.
Second main goal of design was improvement of solubility of new compounds. Insoluble compounds can plague discovery. Good drug solubility is especially needed for topical eye drop formulation. Solubility of designed compounds was evaluated using AB/logS method implemented in the ACD/Labs softwareCitation25. Our strategy to improve solubility was to introduce polarity on the “tail” part of molecule by polar morpholine substituent. In comparison with the parent brinzolamide and dorzolamide three basic compounds (I-1, I-2, I-3) exhibit improved solubility () and can be easily prepared in the form of corresponding saltsCitation16.
All three new compounds were each assayed for hCA I, hCA II, hCA IV and hCA XII isozymes binding by stopped flow technique and results of which are presented in . According to in vitro assays these compounds can be characterized as isozyme-specific inhibitors. They were a relatively weak inhibitors of hCA XII with IC50 of about 520–740 nmol/L, derivative I-2 containing benzene-1,4-disulfonamide moiety being most active. The activity of benzene-1,4-disulfonamide derivatives I-2 and I-3 towards isoenzyme hCA I is very high and comparable with their analogous activity for hCA II (). However, while the pharmacological function of hCA I is still not clear, for effective lowering of intraocular pressure, 99.99% inhibition of CA II and 98% inhibition of CA IV is requiredCitation26. The activity of compounds I-1, I-2 and I-3 towards hCA II and hCA IV is different, but this difference is not regular. I-1 being 2.8 times, I-2 7.5 times and I-3 6.2 times more active for hCA II. The small preference of aromatic sulfonamides towards CA II in comparison with CA IV was observed for the largest majority of these derivativesCitation2. Compounds I-2 and I-3 containing two sulfonamide moieties interact with pharmacologically important hCA II and hCA IV much stronger ().
Conclusions
Three novel potent inhibitors of hCA I, hCA II, hCA IV and hCA XII has been discovered using operator directed drug design techniques and synthesis of limited number of new aromatic sulfonamides. This strategy led to synthesis of new derivatives with improved solubility while maintaining CA activity. Derivatives containing benzene-1,4-disulfonamide group being more active than structure with the 4-sulfamoylbenzamide moiety. The inhibitors studied have been shown a net preference for hCA II in comparison with hCA IV.
Declaration of interest
The authors report no conflicts of interest.
References
- Maren TH. (2000). Carbonic anhydrase inhibition in ophthalmology: Aqueous humour secretion and development of sulphonamide inhibitors. In Chegwidden WR, Carter ND, Edwards YH. The Carbonic Anhydrases New Horizonts. Basel: Birkhauser Verlag. pp. 425–436.
- Supuran CT, Scozzafava A, Casini A. Carbonic anhydrase inhibitors. Med Res Rev 2003;23:146–189.
- Supuran CT, Scozzafava A, Conway J. (2004). Carbonic anhydrase: Its inhibitors and activators. London: CRC Press.
- Sugrue MF. New approaches to antiglaucoma therapy. J Med Chem 1997;40:2793–2809.
- Sugrue MF. Pharmacological and ocular hypotensive properties of topical carbonic anhydrase inhibitors. Prog Retin Eye Res 2000;19:87–112.
- Siesky B, Harris A, Brizendine E, Marques C, Loh J, Mackey J et al. Literature review and meta-analysis of topical carbonic anhydrase inhibitors and ocular blood flow. Surv Ophthalmol 2009;54:33–46.
- Ponticello GS, Freedman MB, Habecker ChN Lyle, PA, Schwam H, Varga SL et al. Thienothiopyran-2-sulfonamides: A novel class of water-soluble carbonic anhydrase inhibitors. J Med Chem 1987;30:591–597.
- Chen HH, Gross S, Liao J, McLaughlin M, Dean T, Sly WS et al. 2H-Thieno[3,2-e]- and [2,3-e]-1,2-thiazine-6-sulfonamide 1,1-dioxides as ocular hypotensive agents: Synthesis, carbonic anhydrase inhibition and evaluation in the rabbit. Bioorg Med Chem 2000;8:957–975.
- Mincione F, Starnotti M, Menabuoni L, Scozzafava A, Casini A, Supuran CT. Carbonic anhydrase inhibitors: 4-sulfamoyl-benzenecarboxamides and 4-chloro-3-sulfamoyl-benzenecarboxamides with strong topical antiglaucoma properties. Bioorg Med Chem Lett 2001;11:1787–1791.
- Maren TH. The development of topical carbonic anhydrase inhibitors. J Glaucoma 1995;4:49–62.
- Maren TH, Jankowska L, Sanyal G, Edelhauser HF. The transcorneal permeability of sulfonamide carbonic anhydrase inhibitors and their effect on aqueous humor secretion. Exp Eye Res 1983;36:457–479.
- Jain A, Whitesides GM, Alexander RS, Christianson DW. Identification of two hydrophobic patches in the active-site cavity of human carbonic anhydrase II by solution-phase and solid-state studies and their use in the development of tight-binding inhibitors. J Med Chem 1994;37:2100–2105.
- Remko M. Theoretical study of molecular structure and gas-phase acidity of some. Biologically active sulfonamides. J Phys Chem A 2003;107:720–725.
- Remko M, von der Lieth CW. Theoretical study of gas-phase acidity, pKa, lipophilicity, and solubility of some biologically active sulfonamides. Bioorg Med Chem 2004;12:5395–5403.
- Remko M. Molecular structure, pKa, lipophilicity, solubility and absorption of biologically active aromatic and heterocyclic sulfonamides. Theochem J Mol Struct 2010;944:34–42.
- Remko M, Kožíšek J, Semanová J, Gregáň F. Synthesis, crystal and molecular structure of two biologically active aromatic sulfonamides and their hydrochloride salts. J Mol Struct 2010;973:18–26.
- Cross PE, Gadsby B. Cerebrovasodilatation through selective inhibition of the enzyme carbonic anhydrase. 1. Substituted benzenedisulfonamides. J Med Chem 1978;21:845–850.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–2573.
- Pacchiano F, Carta F, Vullo D, Scozzafava A, Supuran CT. Inhibition of β-carbonic anhydrases with ureido-substituted benzenesulfonamides. Bioorg Med Chem Lett 2011;21:102–105.
- Kolayli S, Karahalil F, Sahin H, Dincer B, Supuran CT. Characterization and inhibition studies of an α-carbonic anhydrase from the endangered sturgeon species Acipenser gueldenstaedti. J Enzyme Inhib Med Chem 2011;26:895–900.
- Innocenti A, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of cytosolic isoforms I, II, III, VII and XIII with less investigated inorganic anions. Bioorg Med Chem Lett 2009;19:1855–1857.
- Scozzafava A, Briganti F, Ilies MA, Supuran CT. Carbonic anhydrase inhibitors: Synthesis of membrane-impermeant low molecular weight sulfonamides possessing in vivo selectivity for the membrane-bound versus cytosolic isozymes. J Med Chem 2000;43:292–300.
- Soltau JB, Zimmerman TJ. Changing paradigms in the medical treatment of glaucoma. Surv Ophthalmol 2002;47:2–5.
- Scozzafava A, Menabuoni L, Mincione F, Briganti F, Mincione G, Supuran CT. Carbonic anhydrase inhibitors. Synthesis of water-soluble, topically effective, intraocular pressure-lowering aromatic/heterocyclic sulfonamides containing cationic or anionic moieties: Is the tail more important than the ring? J Med Chem 1999;42:2641–2650.
- Available at http://www.acdlabs.com/products/phys_chem_lab.
- Brechue WF. Topical carbonic anhydrase inhibitors: Physicochemical properties and aqueous humor dynamics. Rom Chem Quart Rev 1994;2:301–311.
- Abbate F, Coetzee A, Casini A, Ciattini S, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: X-ray crystallographic structure of the adduct of human isozyme II with the antipsychotic drug sulpiride. Bioorg Med Chem Lett 2004;14:337–341.
- Kim ChY Wittington, DA, Chang JS, Liao J, May JA, Christianson DW. Structural aspects of isozyme selectivity in the binding of inhibitors to carbonic anhydrases II and IV. J Med Chem 2002;45:888–893.
- Vernier W, Chong W, Rewolinski D, Greasley S, Pauly T, Shaw M et al. Thioether benzenesulfonamide inhibitors of carbonic anhydrases II and IV: Structure-based drug design, synthesis, and biological evaluation. Bioorg Med Chem 2010;18:3307–3319.