Abstract
The synthesis, characterization and biological evaluation of novel pyrazole carboxamide derivatives (2–9) are presented. 1H and 13C NMR have been used for the structure description, possible tautomeric structures determination and hydrogen bonding observation. FT-IR results have confirmed the synthesis of the pyrazole derivatives while thermal gravimetric analysis has confirmed thermal stability up to 300°C. The melting temperatures are strongly dependent on their crystal structure as confirmed by differential scanning calorimetry and X-ray diffraction measurements. Impacts of 2–9 as possible antiglaucoma agents were investigated on carbonic anhydrase I and II (CA-I and II) isozymes purified from human erythrocytes in vitro. Compounds 3 and 9 had the highest inhibitory effect while compounds 6 and 8 showed the lowest inhibition.
Introduction
Human carbonic anhydrase (EC, 4.2.1.1., CA) is a metalloenzyme containing the Zn+2 ion associated with glaucoma, a sight-threatening optic neuropathic diseaseCitation1,Citation2. Glaucoma is caused by the elevation of the intraocular pressure which leads to the destruction of optic nerve cells and the consequent deterioration of eyesightCitation3,Citation4. The pressure arises partially from the production of carbondioxides in the eye. The enzyme increasing carbondioxide production is CA that catalyzes the reversible reaction of the hydration of carbon dioxide and dehydration of bicarbonate: CO2 + 2H2O ↔ HCO3− + H3O+5–7. The inhibition of CA decreases sodium and aqueous humor secretion, thus lowering intraocular pressureCitation3. There are 16 distinct CA isoenzymes, and among these enzymes, cytoplasmic CA-I and CA-II are the mostly investigated onesCitation8. These isoenzymes can be obtained easily from red blood cellsCitation9.
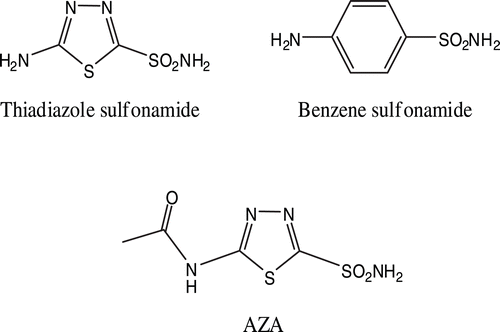
Potent inhibitors of CA-II can be synthesized by having key sulfonamide functionality, R-SO2NH2, on a suitable organic scaffold such as a heterocyclic structureCitation10,Citation11. The sulfonamides gain their ionic structure easily. This feature is extremely important for the inhibition effect on the CA enzyme. Interactions of sulfonamides with the enzyme start with the formation of and ionic bond between the N atom of the R-SO2NH-compound and the Zn+2 in the active site of the CA enzyme. The binding process is completed via hydrophobic interactions between the inhibitor and the enzyme. As a result of these two effects, sulfonamides bind to CA enzyme stronglyCitation12. Through this process, the potent inhibitor displaces the water/hydroxide ligand involved in carbondioxide hydrationCitation10,Citation13. Acetazolamide (AZA) (see ), one the most powerful inhibitors of CA, is used in the treatment of glaucoma. This drug is usually administered orally and there are a lot of metabolic side effects associated to it. In order to reduce the side effects of acetazolamide, alternative sulfonamide derivatives have been investigatedCitation14,Citation15.
Figure 1. Chemical structures of 5-amino-1,3,4-thiadiazole-2-sulfonamide, 4-aminobenzenesulfonamide and AZA.
This study was conducted in order to design, synthesize, characterize and evaluate the biological activity of 4-benzoyl-1-(3-nitrophenyl)-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (2) and its derivatives in order to extend our previous researches in synthesizing novel pyrazole derivativesCitation16,Citation17. In this current attempt, our main aim has been to check whether the new derivatives studied during the course of the current work could show even stronger inhibition effect on CA enzymes, with respect to the compounds previously investigated by our groupCitation16,Citation17. Hence, we want to contribute to the family of pyrazole derivatives by depicting new synthetic routes having excellent yield percentages. The synthesized molecules have been characterized both structurally and thermally by various techniques including NMR, FT-IR, X-ray diffraction (XRD), differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA). Since CA inhibitors have potential for therapeutical and pharmacological applications, the sulfonamide derivatives in the current study have also been investigated in terms of the activities of human erythrocyte CA-I and CA-II isozymes (Schemes 1 and 2).
Experimental
Chemicals
Deuterated dimethyl sulfoxide (DMSO-d6) with 99.98% purity, ethyl benzoyl acetate, benzoyl acetone, CNBr-activated Sepharose-4B, protein assay reagents and chemicals for electrophoresis were purchased from Aldrich. Acetyl acetone, 2-naphtol, sodium azide, sodium nitrite, sodium acetate, para-aminobenzene sulfonamide and l-tyrosine, and solvents such as propanol, ethanol and tetrahydrofuran (THF) were purchased from Merck, Germany. THF was freshly distilled before use. Dibenzoyl methane and 4-aminobenzene sulphonamide were obtained from Fluka, Germany. All reactions were monitored by analytical thin-layer chromatography (TLC) on 0.25-mm pre-coated Kieselgel 60F 254 plates (Merck); compounds were visualized by Camag TLC devices UV (254 and 366 nm).
Characterization
All the compounds were dissolved in DMSO-d6 for NMR measurements. Both proton (1H) and carbon-13 (13C) NMR experiments were performed on a JEOL 500 MHz Lambda instrument with a multi-nuclei 5-mm solution probe (50TH5/FG2). The proton spectra were internally referenced to the signal of the solvent at 2.50 ppm. The 13C NMR spectra were acquired by cross-polarization pulse sequence including proton decoupling. The 13C NMR spectra were again internally referenced to the solvent signal at 39.5 ppm. The FT-IR transmission spectra of each compound were recorded on a Bruker Optics, Vertex 70 FT-IR spectrometer. For each sample, 100 scans were taken at a resolution of 4 cm−1 with N2 exposure. The thermal decomposition temperatures were measured with a Thermogravimetric analyzer. TGA measurements were performed on a TGA 851 (Mettler-Toledo, Greifensee, Switzerland) at a heating rate of 10°C. min−1 under a nitrogen purge of 30 cm3. min−1. A Mettler Toledo DSC 822 equipped with a cooling chamber was operated while using liquid nitrogen as a purging and cooling medium. All the compounds in the amount of 3–5 mg were hermetically sealed in aluminum pans. DSC experiments were performed in the temperature range between −10 and 250°C with a 10°C. min−1 ramp in order to determine the melting temperature (Tm) of the novel pyrazole derivatives. Wide-angle XRD measurements were made using a Bruker-AXS D8 diffractometer at 298 K using Cu Kα radiation (λ = 1.54184 Å). The diffractometer was operated at 40 kV and 40 mA. Measurements were made in the 2θ range from θmin = 2° and θmax = 35° in steps of 0.05°. Elemental analyses were carried out on a Leco CHNS-932 instrument.
Synthesis
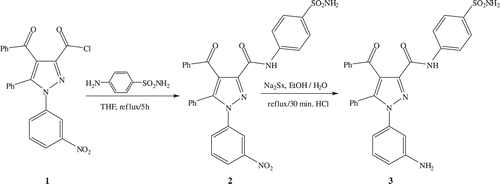
4-benzoyl-1-(3-nitrophenyl)-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (2)
Compound 1 (0.431 g, 1 mmol) was prepared starting from the compound 4-benzoyl-1-(3-nitrophenyl)-5-phenyl-1H-pyrazole-3-carboxylic acid as described in the literatureCitation18 and was dissolved in 20 mL dry THF. 4-aminobenzenesulfonamide (0.344 g, 2 mmol) was added to this solution and the mixture was refluxed for 5 h and solvent was evaporated. The crude product was washed with water and crystallized from 1-propanol. Yield: 92%; IR (ν cm−1): 3335 (NH), 3062 (Ar CH), 1694 and 1661 (C=O), 1591–1446 (Ar−C=C and C=N), 1349 and 1154 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.84 (s, 1H, CONH), 8.40 (s, 2H, SO2NH2), 8.29–7.26 (m, 18H, ArH); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 190.68 (benzoyl C=O), 159.45 (amide C=O), 147.99 (=C−NO2), 145.94, 144.01 and 120.15 (pyrazole C-3, C-5 and C-4), 141.36, 139.33, 139.14, 137.62, 133.75, 132.19, 130.78, 129.95, 129.28, 128.89, 128.81, 127.34, 126.68, 123.69, 122.49, 121.07; Anal Calcd for C29H21N5O6S: C, 61.37; H, 3.73; N, 12.34; S, 5.65. Found: C, 61.17; H, 3.66; N, 12.37; S, 5.73.
1-(3-aminophenyl)-4-benzoyl-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (3)
Compound 3, which contains an aromatic amine group, was prepared as described previouslyCitation18 and crystallized from methanol. (See indicating the synthesis of 2 and 3) Yield: 89%; IR (ν cm−1): 3354 (NH), 3062 (Ar CH), 1682 and 1660 (C=O), 1591–1448 (Ar−C=C and C=N), 1311 and 1154 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.75 (s, 1H, CONH), 7.84 (s, 2H, SO2NH2), 5.39 (s, 2H, NH2), 7.83–6.44 (m, 18H, ArH); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 191.07 (benzoyl C=O), 159.80 (amide C=O), 149.65 (=C-NH2), 145.06, 143.48 and 120.06 (pyrazole C-3, C-5 and C-4), 141.53, 139.43, 138.99, 137.71, 133.63, 129.53, 129.51, 129.18, 128.84, 128.66, 128.02, 126.67, 121.68, 114.49, 113.46, 111.39; Anal Calcd for C29H23N5O4S: C, 64.79; H, 4.31; N, 13.03; S, 5.96. Found: C, 64.51; H, 4.26; N, 13.18; S, 5.93.
Scheme 1. The synthesis of 2 and 3.
General procedure for the syntheses of compounds 4–8
To prepare compounds 4–8, 2 mL HCl were added to a 1 mmol solution of 3 in methanol and then this solution was cooled to 0–5°C on an ice bath. A solution of NaNO2 (1.2 mmol) in 2 mL water was slowly added to the former solution without exceeding 5°C. After dissolving an aromatic or β-dicarbonyl compound (1 mmol) in a sufficient amount of ethanol, the solution was cooled and added drop by drop into the already prepared diazonium salt solution. The resulting colored precipitate was filtered under vacuum and the crude product purified by crystallization from an appropriate solvent ().
Scheme 2. The synthesis of 4–9.
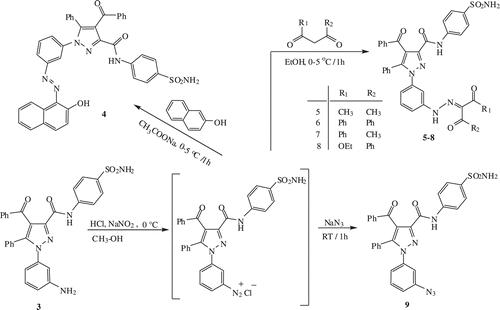
4-benzoyl-1-(3-((2-hydroxynaphthalen-1-yl) diazenyl) phenyl)-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (4)
This compound (4) was synthesized from diazonium solution of 3 (0.537 g, 1 mmol) prepared according to the general procedure and β-naphthol (0.144 g, 1 mmol). The product was purified by crystallization from acetic acid. Yield: 90%; IR (ν cm−1): 3324 (NH), 3060 and 3030 (Are CH), 1678 and 1660 (C=O), 1620–1449 (Ar−C=C and C=N), 1339 and 1151 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 15.58 (s, 1H, Ar-OH), 10.85 (s, 1H, CONH), 8.27 (s, 2H, SO2NH2), 7.94–6.85 (m, 24H, ArH); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 190.84 (benzoyl C=O), 171.30 (=C-OH), 159.63 (amide C=O), 145.62 (=C−N=N−), 145.27, 143.76 and 120.11 (pyrazole C-3, C-5 and C-4), 141.45, 141.31, 139.90, 139.08, 137.68, 133.70, 132.67, 130.71, 129.86, 129.82, 129.76, 129.56, 129.26, 129.22, 128.83, 128.11, 127.84, 126.68, 126.62, 124.54, 124.47, 122.35, 121.80, 119.66, 114.96; Anal Calcd for C39H28N6O5S: C, 67.62; H, 4.07; N, 12.13; S, 4.63. Found: C, 67.48; H, 4.06; N, 12.17; S, 4.71.
4-benzoyl-1-(3-((2-hydroxy-4-oxopent-2-en-3-yl) diazenyl) phenyl)-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (5)
This compound (5) was synthesized from diazonium solution of 3 (0.537 g, 1 mmol) prepared according to the general procedure and acetylacetone (0.102 mL, 1 mmol). The product was purified by crystallization from ethanol. Yield: 93%; IR (ν cm−1): 3333 and 3237 (NH), 3029 (Ar CH), 2971 (aliphatic CH), 1740 and 1658 (C=O), 1635–1461 (Ar−C=C and C=N), 1344 and 1158 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 13.75 (s, 1H, OH), 10.80 (s, 1H, CONH), 7.85 (s, 2H, SO2NH2), 7.83–7.21 (m, 18H, ArH), 2.44 (s, 3H, COCH3), 2.28 (s, 3H, HO−C−CH3); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 197.36 (acetyl C=O), 196.72 (HO−C−CH3), 190.84 (benzoyl C=O), 159.65 (amide C=O), 145.61 (=C−N=N−), 143.76, 142.71 and 120.10 (pyrazole C-3, C-5 and C-4), 141.46 (N−C=C−OH), 31.38 (HO−C−CH3), 26.62 (O=C−CH3), 139.71, 139.10, 137.66, 134.37, 133.70, 130.57, 129.75, 129.24, 128.83, 127.78, 126.70, 122.41, 122.22, 116.77, 113.54; Anal Calcd for C34H28N6O6S: C, 62.95; H, 4.35; N, 12.96; S, 4.94. Found: C, 62.79; H, 4.28; N, 13.02; S, 4.99.
4-benzoyl-1-(3-(2-(1,3-dioxo-1,3-diphenylpropan-2-ylidene)hydrazinyl)phenyl)-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (6)
This compound (6) was synthesized from diazonium solution of 3 (0.537 g, 1 mmol) prepared according to the general procedure and dibenzoylmethane (0.224 g, 1 mmol). The product was purified by crystallization from ethanol. Yield: 72%. IR (ν cm−1): 3427 and 3252 (NH), 3062 and 3027 (Ar CH), 1687 and 1669 (C=O), 1641–1447 (Ar−C=C and C=N), 1332 and 1158 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 11.72 (s, 1H, Ar-NH), 10.80 (s, 1H, CONH), 7.85 (s, 2H, SO2NH2), 7.96–6.99 (m, 28H, ArH); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 193.86, 190.75 and 190.66 (benzoyl C=O), 159.65 (amide C=O), 145.57 (=C−NH−N), 143.91, 143.72 and 120.11 (pyrazole C-3, C-5 and C-4), 143.61 (N=C-C=O), 141.46, 139.07, 137.61, 136.97, 136.01, 134.41, 133.70, 133.65, 133.27, 132.51, 130.26, 129.54, 129.28, 129.16, 128.77, 128.32, 127.61, 127.57, 126.69, 122.04, 120.27, 120.02, 115.35, 112.77; Anal Calcd for C44H32N6O6S: C, 68.38; H, 4.17; N, 10.87; S, 4.15. Found: C, 68.23; H, 4.12; N, 10.89; S, 4.15.
4-benzoyl-1-(3-((3-hydroxy-1-oxo-1-phenylbut-2-en-2-yl)diazenyl)phenyl)-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (7)
This compound (7) was synthesized from diazonium solution of 3 (0.537 g, 1 mmol) prepared according to the general procedure and benzoylacetone (0.162 g, 1 mmol). The product was purified by crystallization from ethanol. Yield: 88%; IR (ν cm−1): 3271 (NH), 3063 (Ar CH), 2970 (aliphatic CH), 1661 (C=O), 1594–1449 (Ar-C=C and C=N), 1324 and 1158 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 13.77 (s, 1H, OH), 11.95 (s, 1H, Ar−NH−N=), 10.80 (s, 1H, CONH), 7.84 (s, 2H, SO2NH2), 7.82–7.05 (m, 23H, ArH), 2.40 (s, 3H, COCH3); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 196.32 (acetyl C=O), 195.06 (HO−C−CH3), 190.80 and 190.68 (benzoyl C=O), 159.61 (amide C=O), 145.49 (=C−N=N−), 143.91, 143.58 and 120.04 (pyrazole C-3, C-5 and C-4), 141.40 (N−C=C−OH), 30.31 (HO−C−CH3), 25.07 (O=C−CH3), 139.78, 139.47, 139.01, 137.58, 135.54, 134.65, 133.63, 130.24, 130.19, 129.63, 129.24, 129.16, 128.85, 128.78, 128.02, 127.79, 126.63, 122.06, 120.01, 115.01, 112.14; Anal Calcd for C39H33N6O6S: C, 65.90; H, 4.25; N, 11.82; S, 4.51. Found: C, 65.75; H, 4.18; N, 11.89; S, 4.55.
Ethyl-2-(2-(3-(4-benzoyl-5-phenyl-3-(4-sulfamoylphenylcarbamoyl)-1H-pyrazol-1-(phenyl) hydrazono)-3-oxo-3-phenylpropanoate (8)
This compound (8) was synthesized from diazonium solution of 3 (0.537 g, 1 mmol) prepared according to the general procedure and ethyl benzoylacetate (0.174 mL, 1 mmol). The product was purified by crystallization from ethanol. Yield: 88%; IR (ν cm−1): 3273 (NH), 3060 (Ar CH), 2980 (aliphatic CH), 1655 (C=O), 1593–1447 (Ar−C=C and C=N), 1310 and 1154 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 11.95 (s, 1H, Ar-NH), 10.81 (s, 1H, CONH), 7.84 (s, 2H, SO2NH2), 7.83–6.90 (m, 23H, ArH), 4.31 (q, 2H, OCH2CH3), 1.24 (t, 3H, CH3); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 190.82 and 189.09 (benzoyl C=O), 162.45 (ester C=O), 159.66 (amide C=O), 145.55 (=C−NH−N=), 143.67, 141.46 and 120.13 (pyrazole C-3, C-5 and C-4), 61.60 (OCH2CH3), 14.05 (CH3), 139.55, 139.07, 137.61, 137.10, 133.71, 132.75, 130.28, 129.98, 129.79, 129.57, 129.30, 129.18, 128.83, 128.75, 128.69, 126.70, 122.13, 122.04, 120.79, 120.05, 115.66, 112.88; Anal Calcd for C40H32N6O7S: C, 64.85; H, 4.35; N, 11.34; S, 4.33 Found: C, 64.59; H, 4.31; N, 11.43; S, 4.37.
1-(3-azidophenyl)-4-benzoyl-5-phenyl-N-(4-sulfamoylphenyl)-1H-pyrazole-3-carboxamide (9)
Sodium azide (0.065 g, 1 mmol) was dissolved in 2 mL distilled water and the solution was cooled to 0°C. Then, it was added drop-wise under continuous stirring into a diazonium salt solution of compound 3 (0.537 g, 1 mmol), which had been prepared in accordance with the general procedure. The pH was adjusted to 3–4 and after the solution had spent 1 h at room temperature; the resulting white precipitate was filtered under vacuum and dried. Yield: 87%; IR (ν cm−1): 3252 (NH), 3065 (Ar CH), 2162 (N3), 1668 (C=O), 1594–1449 (Ar−C=C and C=N), 1329 and 1158 (S=O); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 10.79 (s, 1H, CONH), 7.85 (s, 2H, SO2NH2), 7.79–7.20 (m, 18H, ArH); 13C NMR (500 MHz, DMSO-d6) δ (ppm): 190.80 (benzoyl C=O), 159.59 (amide C=O), 145.51 (=C−N3), 143.80, 141.42 and 120.11 (pyrazole C-3, C-5 and C-4), 140.56, 139.84, 139.07, 137.65, 133.69, 130.75, 129.85, 129.74, 129.24, 128.81, 127.69, 126.78, 126.68, 122.57, 122.18, 119.67, 116.95; Anal Calcd for C29H21N7O4S: C, 61.80; H, 3.76; N, 17.40; S, 5.69 Found: C, 61.68; H, 3.67; N, 17.45; S, 5.73.
Biological activity evaluation
Hemolysate preparation
Erythrocytes were purified from fresh human blood, which was obtained from the University Hospital Blood Center of Erzurum Atatürk University. Following low-speed centrifugation (1500 rpm for 15 min) and removal of plasma and buffy coat, the red blood cells were isolated, washed twice with 0.9% NaCl and hemolyzed with 1.5 volumes of ice-cold water. Ghost and intact cells were then removed by high-speed centrifugation (20000 rpm for 30 min.) at 4°C and the pH of the hemolysate adjusted to 8.7 with solid Tris.
Purification of CA isozymes from human erythrocytes by affinity chromatography
A sepharose-4B l-tyrosine affinity chromatography column was prepared according to our previous studiesCitation19. The pH-adjusted human erythrocyte hemolysate (70 mL) was applied to the Sepharose 4B-l-tyrosine-sulfanylamide affinity column pre-equilibrated with 25-mM Tris-HCl/0.1 M Na2SO4 (pH = 8.7). The affinity gel was washed with 25 mM Tris-HCl/22 mM Na2SO4 (pH = 8.7). The human carbonic anhydrase isozymes (hCA-I and hCA-II) were eluted with 1.0 M NaCl/25 mM Na2HPO4 (pH = 6.3) and 0.1 M NaCH3COO/0.5 M NaClO4 (pH = 5.6), respectively. During purification procedures of hCA-I and hCA-II, the absorbency at 280 nm was measured to monitor protein elution by affinity chromatography. CO2-hydratase activity was determined in eluted fractions and the active fractions were collectedCitation20,Citation21.
Hydratase activity assay
CA activity was assayed by following the hydration of CO2 according to the method described by Wilbur and AndersonCitation20. CO2-Hydratase activity was calculated by using the equation, (t0−tc)/tc where t0 and tc are times for pH change of the nonenzymatic and the enzymatic reactions, respectively, and this was used to express enzyme unit (EU).
Esterase activity assay
CA enzymes catalyze some non-physiological reactions under in vitro conditionsCitation22. For instance, it was observed that the purified enzyme has esterase activity under in vitro conditionsCitation20.![]()
Esterase activity of human erythrocyte CA was assayed by following the change in absorbance at 348 nm of 4-nitrophenyl acetate to 4-nitrophenolate ion over a period of 3 min at 25°C using a spectrophotometer according to the method described in the literatureCitation23.
Quantitative protein determination
Quantitative protein determination was done by measuring the absorbance at 595 nm according to Bradford, using bovine serum albumin as a standardCitation24.
SDS polyacrylamide gel electrophoresis
The control of enzyme purity, using Laemmli’s procedure, was carried out in 3 and 8% acryl amide concentrations for running and stacking gel, respectively. The hCA-I and hCA-II isozyme samples were loaded into each slot of the stacking gel (slab gel dimensions: 16 × 18 cm). A voltage of 80 V was applied until the bromphenol blue reached the running gel. Then the voltage was increased to 200 V for 3–4 h. The gel was stabilized in a solution containing 50% propanol + 10% trichloroacetic acid (TCA) + 40% distilled water for 30 min. The staining was performed for about 2 h in a solution of 0.1% Coomassie Brilliant Blue R-250 + 50% methanol + 10% acetic acid. Finally, the washing was carried out in a solution of 50% methanol + 10% acetic acid + 40% distilled water until the protein bands were cleared.
In vitro inhibition studies
CA activity was assayed in inhibition studies by following the change in absorbance at 348 nm of 4-nitrophenylacetate (NPA) to 4-nitrophenylate ion over a period of 3 min at 25°C using a spectrophotometer according to the method described by Verpoorte et al.Citation23 The enzymatic reaction, in a total volume of 1 mL, contained 50 mM Tris-SO4 buffer (pH = 7.4), 3 mM 4-nitrophenylacetate and enzyme solution. A reference measurement was done by using the same cuvette without enzyme solution. The inhibitory effects of some synthesized sulfonamide derivatives on CA enzyme activity purified from human erythrocyte were tested on three samples at each concentration used. CA activities were measured in the presence of different substrate concentrations. Control activity in the absence of inhibitor was assumed to be 100%. For each compound, a percent activity versus inhibitor concentration graph was plotted. Ki values of the compounds were calculated by measuring enzyme activity at three different inhibitor concentrations with five different substrate concentrations. Lineweaver–Burk curves were used for the determination of Ki and inhibitor typeCitation25.
Results and discussion
Structural and vibrational characterization
Compound 2 was obtained by amidizing compound 1 with benzene sulfonamide having aryl group instead of reacting with thiadiazole sulfonamide including heteroaryl group (see ). Then, compound 2 was reacted with β-dicarbonyl derivatives, β-naphtol and sodium azide compounds to yield the new pyrazole derivatives (see ). The 1H NMR results depict the successful synthesis of the molecules of interest. The resonances between ~8.00 ppm and ~6.40 ppm can be assigned to aromatic protonsCitation16 (with the exception of the aromatic peak at 8.29 ppm in the spectrum of 2 arising from the protons next to the nitro group). Isotropic chemical shifts of Ar-OH and NH appear as downfield shifts in 1H NMR spectra. The peaks of Ar-OH and NH are used to distinguish between possible tautomeric structures. For instance, compound 5 is stable only in enol-azo form since no peak belonging to Ar-NH is observed in 1H NMR spectrum. However, the spectrum of 7 has both Ar-NH and C-OH resonances. This proves that derivative 7 exists in both enol-azo and keto-azo forms. More interestingly, quantitative NMR reveals that in the case of 7, the relative intensities of C-OH and Ar-NH peaks are 1: 2.3. For this reason, molecule 7 is predominantly stable in the keto form as opposite to molecule 5 (see ).
Figure 2. Tautomeric structures for 5 and 7.
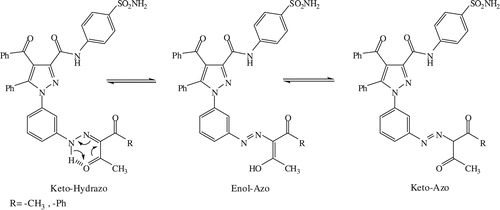
This is attributed to the difference in their chemical structures. In addition, hydrogen bonding plays an important role in the wide range of chemicalsCitation26. Hydrogen bonding of different systems has been studied by 1H NMR spectroscopyCitation26–28. If a proton is involved in hydrogen bonding, its electrons are shared by two electronegative elements. Thus, its electron density is decreased. Ultimately, the proton is deshielded and exhibits resonances at a lower fieldCitation27. The extent of intermolecular hydrogen bonding is affected by several factors such as solvent and solution concentration. In a non-polar solvent, the degree of hydrogen bonding increases as the concentration of the solution is increasedCitation27. We observed hydrogen bonding interaction as a slight down field shift in the 1H NMR spectra of 6, 7 and 8 (see Supporting Information Figure S1 showing the stack plot). A weaker hydrogen bonding interaction is depicted in the spectrum of 7. This is explained by the tautomeric structure of 7. In the literature, a down field shift of amide proton was reported from 6.7 to 7.6 ppm (~0.9 ppm) corresponding to an increase of the concentration of solution in deuterochloroformCitation27. The slight down field shift of 0.02 ppm in our case is due to the polar solvent DMSO-d6 decreasing the strength of possible hydrogen bonding.
In addition to the analysis of 1H NMR spectra, 13C resonance assignments have also been carried out16. The peaks of OH-C-CH3 and O=C-CH3 are observed around ~197.00 ppm while the peaks of Ph-C=O and O=C-NH are around 190.00 and 159.00 ppm, respectively. There are two interesting peaks at 171.30 ppm corresponding to the aromatic carbon HO-C belonging to 4 and at 162.45 ppm corresponding to the peak O=C-O of 8. There are three carbons located in the five-member ring having nitrogen as well. The resonances of two (C-3 and C-5) of the three carbons are between 148.00 and 141.40 ppm shifted down field with respect to benzene carbons, consistent with the values mentioned in the literatureCitation29. The occurrence of hydrogen bonding in 6, 7 and 8 is also in accordance with the observation of two O=C-NH resonances in 13C NMR spectra. Hydrogen bonding interaction deshields the carbon, leading to two distinct resonances. In addition, the carbon peaks in the spectrum of 7 arising from HO-C-CH3 and O=C-CH3 confirm the existence of the tautomeric structure of 7.
The FT-IR spectra of the compounds are assigned referring to our previous reports and literatureCitation16,Citation17,Citation30. Considering the reactions and the final products (see Schemes 1 and 2), one of the most important IR peaks is the one corresponding to the S=O bond. There are two peaks assigned to S=O; symmetric and asymmetric stretching. The peak of asymmetric stretch of S=O appears at a higher wave-number around ~1330 cm−1 while symmetric stretch is around ~1155 cm−1. The peaks at 2971, 2970 and 2980 cm−1 arising in the spectra of 5, 7 and 8, respectively are unique to aliphatic C-H stretchingCitation31. Both peaks corresponding to aliphatic C-H stretching confirm the existence of side groups -CH3 and -CH2-CH3 and differentiate the compounds 5, 7 and 8 from the rest of the derivatives. More interestinglyCitation17, the C-H stretching of aromatic groups indicates values shifted to frequencies of ~3060 cm−1. The peak appearing only in the spectrum of 9 at 2162 cm−1 is attributed to asymmetric stretching of −N3, the side group of 932. The IR data depict shifted downward wave-numbers and broad bands of the N-H stretching for each compoundCitation33. The broad band of N-H stretching shifted downward attributed to intermolecular hydrogen bondingCitation30,Citation33. The peaks for Ar-C=C and C=N appear as a region rather than a single peak, attributed to various motions such as in plane vibration of C=N. Thus, the FT-IR results confirm some of the NMR results, such as the existence of unique functional groups of the compounds, and provide an independent confirmation of the successful synthesis of the compounds.
Thermal properties
DSC and XRD measurements were performed in order to determine both the melting temperatures and their correlations with possible crystalline structures of the compounds. Prior to melting temperature determination by DSC, the thermal stability of all compounds was investigated by TGA. All compounds were highly stable at elevated temperatures. No weight loss was observed up to 175°C in the nitrogen gaseous environment for all compounds during the TGA measurements. Compounds 8 and 9 started to decompose at 250 and 175°C, respectively, while the other compounds (2–7) were stable up to 300°C. The main degradation occurred between 300 and 600°C.
The synthesized compounds are in solid state and show endothermic melting transitions in their DSC thermograms above room temperature. The first heating scans (rates 10°C/min) were used for data analysis since some compounds started to decompose above 250°C. The corresponding melting temperatures are given in Supporting Information Table S1. Compounds 2–7 showed distinct endothermic melting peaks. The compounds 8 and 9, on the other hand, have broad exothermic peaks at elevated temperatures. Only one broad exothermic transition around 200°C was observed, which relates to the thermal curing reaction of compound 9. In the case of compound 8, the exothermic thermal curing starts at around 230 ± 3°C and has a very weak and broad endothermic melting peak at 123 ± 5°C. This indicates a comparatively higher thermal reactivity of compounds 8 and 9.
The origin of the endothermic peaks that can be an indication of the crystalline nature of compounds can be understood if we consider the relation between the crystal structure and melting behaviour. Thus, in order to investigate any superior ordering of all the compounds at the molecular level, XRD powder measurements were performed (see Supporting Information Figure S2). It seems that the chemical nature of functional groups attached to 4-benzoyl unit induce noticeable ordering effect in compounds 2–7, which is obvious from the appearance of DSC melting peaks and high order XRD diffraction peaks. It is interesting that, for 5 and 7, the peak position changes quite significantly, although their structure is very similar. For instance, there is only one phenyl unit and one CH3 on 5 while 7 has two CH3 groups attached to the 4-benzoyl unit. We also compared their XRD scans and observed that for compound 7 some of the high order crystal peaks either disappeared (2θ = 18.65°) or became weaker (2θ = 5.5 and 9.95°) in comparison to 5. Since two compounds (8 and 9) did not show any melting peak, their crystalline structures were also checked by using XRD. The lack of melting peaks was attributed to the presence of a large amorphous portion in compounds 8 and 9 that have a broad amorphous peak at 2θ = ~20° (see Figure S2).
Biological activity evaluation
CA isozymes (CA-I and II) were purified from human erythrocytes by sepharose-4B l-tyrosine affinity chromatography (see Supporting Information Table S2). The purification results for CA-I depicted a 40.5% yield, 524.2 specific activity and 69.4 purification fold. Those of CA-II were calculated as 43.6% yield, 5000 specific activity and 662.25 purification fold. This indicates that CA-II has a high specific activity and purification factor. In addition, the purity of enzymes was controlled by using SDS-PAGE. Purified enzymes show single bands around 29 kDa.
Then, the effects of these novel pyrazole carboxamide derivatives (2–9) on the esterase activity of purified isozymes were investigated. All compounds showed inhibitory effects on both CA-I and CA-II isozymes (see ), except compound 4. Compound 4 did not show any significant inhibition effect.
Table 1. Ki values and inhibition types of chemicals on CA-I and CA-II.
The effect of melatonin on CA enzyme from human erythrocytes and inhibitions of CA enzymes have been investigated with sulfamide and sulfamic acid derivativesCitation34,Citation35. Our compounds indicate inhibitory effects on CA. For example, 3, 5 and 9 depicted higher inhibitory effects than the other compounds on the activity of CA-I. 3 was the most powerful inhibitor according to the other compounds on the CA-I enzyme activity. 6 showed the weakest inhibition on the activity of CA-I. However, 3, 7 and 9 were more effective than the other compounds for CA-II enzyme activity. 8 was the one with the weakest inhibitory effect on the activity of CA-II. In a similar study, Kasımoğulları et al. (2009) pyrazole carboxylic acid amides of 5-amino-1,3,4-thiadiazole-2-sulfonamide 1 (inhibitor 1) were synthesized and investigated in vitro inhibition effects of this compounds on human CA-I and II isozymesCitation16. In another study, some pyrazole carboxylic acid derivatives of 5-amino-1,3,4-thiadiazole-2-sulfonamide (inhibitor 1) were synthesized from ethyl 3-(chlorocarbonyl)-1-(3-nitrophenyl)-5-phenyl-1H-pyrazole-4-carboxylate compound. The inhibition effects on hydratase and esterase activities of human erythrocyte carbonic anhydrase I and II isoenzymes of these compounds were investigated in vitro17. The structure-activity relationship can be drawn from the inhibition data. For instance, 3 and 9 with the highest inhibition have R moiety of small -NH2 and -N3 groups. This is consistent with one of our previous works where the highest inhibition was observed with the compound having−I as the R moeityCitation16. However, the compounds with lower inhibition such as 8 and 6 have bulky aromatic phenyl groups. 8 and 6 also show hydrogen bonding ability which seems to lower the inhibition effect. 5 and 7 have lower inhibition with respect to 3 and 9, and this is attributed to resonance structures of 5 and 7. In other words, the resonance structures of the molecules decrease the inhibitory effect. The obtained results of these studies indicate that even small changes in the nature of the R moiety cause large changes in inhibitory activity. Also, as discussed in literatureCitation6, hydrogen bonding ability and hydrophilic/hydrophobic nature of the compounds may affect the inhibition behaviour.
It is known that the activity of enzymes is due to the catalytic groups of the active sites. If a substrate interacts with the active site of an enzyme, its 3-D structure changes. In noncompetitive inhibition, inhibitors bond to the enzyme, except for its active site. Therefore, a noncompetitive inhibitor shows its inhibitory effect by decreasing turnover rate or catalytic activity of the enzyme (see Supporting Information Scheme S1).
The inhibition can be determined as a result of IC50 and Ki studies. If IC50 and Ki values of an inhibitor are smaller, its inhibition effect will be higher. So, it can be concluded that compounds 3 and 9, having the smallest Ki values on both isozymes, are the strongest inhibitors. For the other compounds, there are sterical interactions between the molecules and enzyme due to the group, 2-imino-1,3 diaryl (dialkyl) propane- 1,3 dione, bound to amino group. Therefore, these compounds have bigger Ki values and weaker inhibition effect on the isoenzymes.
Conclusion
We have reported the synthesis, characterization and biological activity evaluation of novel pyrazole derivatives. 1H, 13C NMR, FT-IR spectroscopy, TGA, DSC and XRD analysis results confirmed the successful synthesis of these compounds. There are strong relationships between chemical structures and properties of the compounds observed by means of NMR, DSC and XRD measurements. 6–8 showed hydrogen bonding interaction while 7 had also tautomeric structure. Moreover, all of these compounds showed inhibitory effects on the purified human CA isozymes. Compounds 3 and 9 had the highest inhibitory effects on both CA-I and CA-II isozymes. These current results show that these compounds possess strong inhibitory effects on CAs and may be used for the generation of antiglaucoma potent carbonic anhydrase inhibitors (CAIs).
Declaration of interest
This study was funded by The Scientific and Research Council of Turkey with Grant number 106T180. The facility of King Fahd University of Petroleum and Minerals (KFUPM) is used, and KFUPM is gratefully acknowledged for the support of this project. H.D. thanks Juergen Thiel and Max-Planck Institute for Polymer Research, Mainz, Germany for technical support. S.O. is grateful to German Science Foundation for the support of summer research stay at Osnabrück University/Germany with Grant number STE 1127/15-1.
References
- Wilkinson BL, Bornaghi LF, Houston TA, Innocenti A, Vullo D, Supuran CT et al. Carbonic anhydrase inhibitors: inhibition of isozymes I, II, and IX with triazole-linked O-glycosides of benzene sulfonamides. J Med Chem 2007;50:1651–1657.
- Yin Y, Cameron MD, Lin L, Khan S, Schroter T, Grant W, Pocas J et al. Discovery of potent and selective urea-based ROCK inhibitors and their effects on intraocular pressure in rats. ACS Med Chem Lett 2010;1:175–179.
- Kim CY, Whittington DA, Chang JS, Liao J, May JA, Christianson DW. Structural aspects of isozyme selectivity in the binding of inhibitors to carbonic anhydrases II and IV. J Med Chem 2002;45:888–893.
- Tarongoy P, Ho CL, Walton DS. Angle-closure glaucoma: the role of the lens in the pathogenesis, prevention, and treatment. Surv Ophthalmol 2009;54:211–225.
- Gokhale NH, Bradford S, Cowan JA. Catalytic inactivation of human carbonic anhydrase I by a metallopeptide-sulfonamide conjugate is mediated by oxidation of active site residues. J Am Chem Soc 2008;130:2388–2389.
- Hen N, Bialer M, Yagen B, Maresca A, Aggarwal M, Robbins AH et al. Anticonvulsant 4-aminobenzenesulfonamide derivatives with branched-alkylamide moieties: X-ray crystallography and inhibition studies of human carbonic anhydrase isoforms I, II, VII, and XIV. J Med Chem 2011;54:3977–3981.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–777.
- Chegwidden WR, Carter ND, Edwards YH (eds.) The Carbonic Anhydrases-New Horizons. Basel: Birkhauser Verlag 2000.
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Koike T, Kimura E, Nakamura I, Hashimoto Y, Shiro M. The first anionic sulfonamide-binding zinc(II) complexes with a macrocyclic triamine: chemical verification of the sulfonamide inhibition of carbonic anhydrase. J Am Chem Soc 1992;114:7388–7345.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229.
- Maren TH, Carbonic anhydrase: general perspective and advances in glaucoma research. Drug Dev Res 1987;10:255–276.
- Lindskog S. Structure and mechanism of carbonic anhydrase. Pharmacol Ther 1997;74:1–20.
- Supuran CT, Scozzafava A. Carbonic anhydrase inhibitors. Curr Med Chem 2001;1:61–97.
- Bülbül M, Hisar O, Beydemir S, Ciftçi M, Küfrevioglu OI. The in vitro and in vivo inhibitory effects of some sulfonamide derivatives on rainbow trout (Oncorhynchus mykiss) erythrocyte carbonic anhydrase activity. J Enzyme Inhib Med Chem 2003;18:371–375.
- Kasimogullari R, Bülbül M, Arslan BS, Gökçe B. Synthesis, characterization and antiglaucoma activity of some novel pyrazole derivatives of 5-amino-1,3,4-thiadiazole-2-sulfonamide. Eur J Med Chem 2010;45:4769–4773.
- Kasimogullari R, Bülbül M, Günhan H, Güleryüz H. Effects of new 5-amino-1,3,4-thiadiazole-2-sulfonamide derivatives on human carbonic anhydrase isozymes. Bioorg Med Chem 2009;17:3295–3301.
- Sener A, Kasımogullari R, Sener MK, Bildirici I, Akcamur Y. Studies on the reactions of cyclic oxalyl compounds with hydrazines or hydrazones: synthesis and reactions of 4-benzoyl-1-(3-nitrophenyl)-5-phenyl-1H-pyrazole-3-carboxylic acid. J Heterocycl Chem 2002;39:869–875.
- Beydemir S, CIftçi M, Ozmen I, Buyukokuroglu ME, Ozdemir H, Küfrevioglu OI. Effects of some medical drugs on enzyme activities of carbonic anhydrase from human erythrocytes in vitro and from rat erythrocytes in vivo. Pharmacol Res 2000;42:187–191.
- Wilbur KM, Anderson NG. Electrometric and colorimetric determination of carbonic anhydrase. J Biol Chem 1948;176:147–154.
- Rickli EE, Ghazanfar SA, Gibbons BH, Edsall JT. Carbonic anhydrases from human erythrocytes. preparation and properties of two enzymes. J Biol Chem 1964;239:1065–1078.
- Shah GN, Hewett-Emmett D, Grubb JH, Migas MC, Fleming RE, Waheed A et al. Mitochondrial carbonic anhydrase CA VB: differences in tissue distribution and pattern of evolution from those of CA VA suggest distinct physiological roles. Proc Natl Acad Sci USA 2000;97:1677–1682.
- Verpoorte JA, Mehta S, Edsall JT. Esterase activities of human carbonic anhydrases B and C. J Biol Chem 1967;242:4221–4229.
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 1976;72:248–254.
- Lineweaver H, Burk D. The determination of enzyme dissociation constants. J Am Chem Soc 1934;56:658–666.
- Kim H-D, Ishida H. Model compounds study on the network structure of polybenzoxazines. Macromolecules 2003;36:8320–8329.
- Cheuk KKL, Lam JWY, Chen J, Lai LM, Tang BZ. Amino acid-containing polyacetylenes: synthesis, hydrogen bonding, chirality transcription, and chain helicity of amphiphilic poly(phenylacetylene)s carrying l-leucine pendants. Macromolecules 2003;36:5947–5959.
- Syud FA, Stanger HE, Gellman SH. Interstrand side chain–side chain interactions in a designed beta-hairpin: significance of both lateral and diagonal pairings. J Am Chem Soc 2001;123:8667–8677.
- Nishimura N, Banno M, Maki A, Nishiyama Y, Maeba I. Synthesis of 3-β-d-ribofuranosylpyrazole-1-carboxamide. Carbohydr Res 1998;307:211–215.
- Kaushal AM, Chakraborti AK, Bansal AK. FTIR studies on differential intermolecular association in crystalline and amorphous states of structurally related non-steroidal anti-inflammatory drugs. Mol Pharm 2008;5:937–945.
- Stauffer SR, Katzenellenbogen JA. Solid-phase synthesis of tetrasubstituted pyrazoles, novel ligands for the estrogen receptor. J Comb Chem 2000;2:318–329.
- Travers MJ, Eldenburg EL, Gilbert JV. Low-temperature matrix isolation studies of BCl(N3)2: infrared spectra and photolysis processes. J Phys Chem A 1999;103:9661–9668.
- Unterberg C, Gerlach A, Schrader T, Gerhards M. Clusters of a protected amino acid with pyrazole derivatives: β-sheet model systems in the gas phase. Eur Phys J 2002;20:543–550.
- Beydemir S, Gülçin I. Effects of melatonin on carbonic anhydrase from human erythrocytes in vitro and from rat erythrocytes in vivo. J Enzyme Inhib Med Chem 2004;19:193–197.
- Scozzafava A, Banciu MD, Popescu A, Supuran CT. Carbonic anhydrase inhibitors: inhibition of isozymes I, II and IV by sulfamide and sulfamic acid derivatives. J Enzym Inhib 2000;15:443–453.