Abstract
Earlier, we have reported the synthesis and anti-inflammatory evaluation of different 3-(4H-1,2,4-triazol-3-ylthio)-N-substituted propanamide. In this article, we are reporting the various tautomeric forms of the most active anti-inflammatory compound, 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide (6a) and their virtual screening by molecular docking using six principle tautomeric forms. Docking analysis suggested that compound 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide (6a) bound with COX-1 selectively and drug receptor complex was stabilized by tautomerism. Noticeably, hydroxy group formed by tautomerism appreciably improve the drug receptor interactions. It was also supervised that the compound 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide (6a) docked near the gate of COX-1 active site and might block the conversion of arachidonic acid to prostaglandin (PG) H2 in the active site of COXs. Moreover, we have carried out receptor based electrostatic analysis to clarify the electronic, steric and hydrophobic field requirement of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide (6a) to interact with COX -1 receptor.
Introduction
1,2,4-Triazoline-5-thione and their derivatives constitute an important class of organic compounds with diverse biological activities such as tuberculostaticCitation1,Citation2 analgesicCitation3 anti-inflammatoryCitation4–Citation6 antimicrobialCitation7,Citation8 and can also be used as herbicidesCitation9 or fungicidesCitation10.
Previously, we have reported the synthesis and anti-inflammatory activity of different 3-(4H-1,2,4-triazol-3-ylthio)-N-substituted propanamideCitation11. Nonsteroidal anti-inflammatory drugs (NSAIDs) are widely used as therapeutic agents for the management of pain and inflammation, and in addition, recently several evidences have been reported indicating them as a chemo preventive in colorectal cancerCitation12. Their mechanism of action is based on the blockage of the Cyclooxygenase enzymesCitation13 (COXs) by sterically hindering the entrance of the physiological binder arachidonic acid. The classical NSAIDs such as aspirin, ibuprofen, or flurbiprofen are nonselective and inhibit all the COXs isoforms. In the last few decades, the research has been focused mostly on selective inhibition of COXs.
Tautomerism by definition concerns all molecules which can readily interconvert into isomers by transfer of a chemical group. Tautomerism being very complex and is related to several phenomena: different types of migrating groups such as electrofuge or a nucleofuge, cationotropic/anionotropic properties, valence tautomerism, zwitterionic tautomerism, tautomerism related to migration of neutral groups in molecules and migration of bonds or ring-chain tautomerismCitation14–16.
Sadowski et al.Citation17 have addressed the issue of tautomery together with protonated molecules using a tautomer and protonation preprocessor for virtual screening. Subsequently, the top scored compounds and tautomers were compared to each other and revealed new characteristics of the ligand bindingCitation18. It can be postulated that when active site favors one particular tautomeric form over the other, this could reveal new lead structures and may be omitted in a classical, tautomerism-disregarded screening. Moreover, finding tautomeric hits may initiate new case studies of tautomery dependent ligand–protein interaction.
Stereochemistry of compound 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide 6a revealed characteristic ring chain and amide-amidic tautomerism. In order to investigate which tautomeric form of compound 6a predominantly involved in COX-1 inhibitory mechanism, its selectivity and stable interaction with COX-1 receptor, we have derived various tautomeric forms of compound 6a and carried out virtual screening of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide by docking analysis of six principal tautomeric forms of compound 6a with COX-1. Additionally, we have performed receptor based electrostatic analysis to get better insight of electronic and hydrophobic structural requirement of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide 6a that would be useful to get alert about the particular part of the field required for binding.
In this study, we aimed to understand structural features responsible for anti-inflammatory activity of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide 6a, to identify the types of interactions involved between six principal tautomeric forms of 6a with binding site of COX-1 receptor and to analyze hydrophobic, electronic and steric characteristics of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide 6a in receptor interaction.
Materials and methods
Docking algorithms
Docking programmes “direct” and “unbiased” have their own advantages and disadvantages. Direct docking software such as DOCK has the benefit of speed but it has disadvantage of making assumptions about the potential energy landscape to save computational time. Unbiased methods such as Auto Dock, FTDOCK, and MOE-Dock have few assumptions about the potential energy landscape. Although in MOE 2009, computation time is longer but it is having extra advantage of potential energy landscape. Here we report the use of MOE-Dock by Chemical Computing Group Inc.Citation19, which has the advantage of flexible docking, integration with a graphical interface as well as with other modules such as analysis, molecular mechanics, and molecular dynamics.
Docking simulations
In MOE, London dG scoring is used as default setting to calculate the exact confirmation and configuration of the ligand for finding the best molecule with minimum binding energy and to develop potential drug molecules against the disease. The London dG scoring function estimates the free energy ΔG of binding of the ligand from a given pose. The functional form is a sum of terms:
Where C represents the average gain/loss of rotational and translational entropy; Eflex is the energy due to the loss of flexibility of the ligand (calculated from ligand topology only); fHB measures geometric imperfections of hydrogen bonds and takes a value in [0,1]; CHB is the energy of an ideal hydrogen bond; fM measures geometric imperfections of metal ligations and takes a value in [0,1]; CM is the energy of an ideal metal ligation; and Di is the desolvation energy of atom i. The difference in desolvation energies is calculated according to the formula
Where A and B are the protein and/or ligand volumes with atom i belonging to volume B; Ri is the solvation radius of atom i (taken as the OPLS-AA Van der Waals sigma parameter plus 0.5 Angstrom); and Ci is the desolvation coefficient of atom i. Atoms are categorized into ~12 atom types for the assignment of the Ci coefficients. MOE 2009.10 was run on a Windows XP based Pentium IV 2·66 GHz PC (with 2GB RAM).
Substrate docking
Structure of all the tautomers were derived in MOE programme by using Protomer option. Before molecular docking; the 3D structure of ligand was optimized. The pdb 3N8X (1) was subjected to energy and residue optimization by protonate 3D option in MOE programmeCitation20. The health of protein was ascertained by Ramchandran plot ().
Substrates were docked within the active site using the Monte Carlo docking procedure of MOE with repeated cycles of protein and substrate minimization. During the initial stage of the docking procedure, the side chains of the protein were fixed. The best ranking docking modes of the ligands were identified and energy minimized in the protein while allowing full side chain flexibility. Energy of conformation of different tautomeric forms of compound 6a is given in ().
Table 1. Table showing 30 conformations (as S. No.) of different tautomers generated by compound 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide (6a) with their respective energy of Conformation (E_con T).
Result and discussion
All six tautomers () were used as ligands in the binding interaction studies with the active site of the COX-1 receptor. Docking of these tautomers to the active site revealed that tauto 3 and 5 have zero energy of docked conformation while tauto 2 have the lowest energy of docked conformation followed by tauto 1, 6 and 4 ().
Figure 1. Structure of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropionamide 6a.
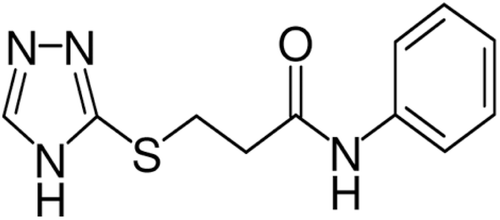
Figure 2. Six principal neutral possible tautomeric forms for compound 6a.
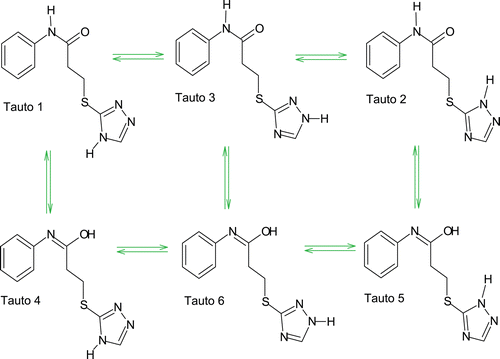
(The first line in table showed low energy of conformation for each tautomer which indicates most favorable conformation of ligand into the receptor pocket of COX-1). These results are parallel to the highest % inhibition observed experimentally for compound 6a. The lowest conformational energy of tauto 2 may be best conformation for compound 6a to interact with COX-1 receptor. Concurrently, hydroxy group of tauto 4 may be the important attribute for the ligand to interact with COX-1 receptor through hydrogen bonding.
All the ligands showed reasonably low energy of docked conformation indicating that the docked conformers were in their most favorable conformations. The spatial arrangement of the most active tauto 4 bound to the active site of COX-1 is shown in .
Figure 3. Ramchandran Plot for pdb 3N8X (1) after energy and residue optimization.
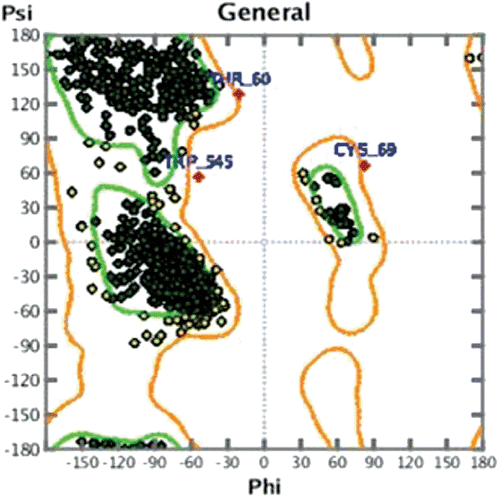
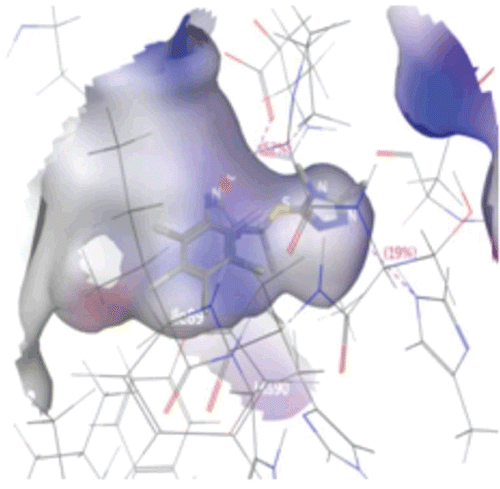
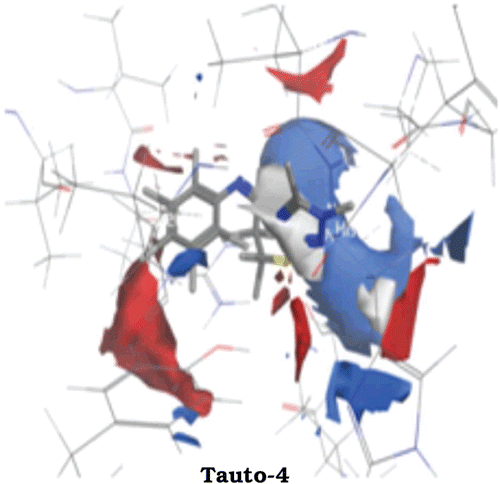
Figure 4. Connolly surface representations of the active site of COX-1 with the bound tauto-4, shown in Ball and Stick model. Connolly surface of the active site of the COX-1 is colored according to a charge spectrum: Blue colour for H-bonding region, white colour indicates lipophilic area while red colour is used to reveal mild polar area.
All the tautomers have shown characteristic interaction with COX-1 receptor as indicated by their energy of docked conformation and their hydrogen bonding pattern.
Interactions between tautomers and residues in COX-1
Hydrogen bond analysis was performed on the docked COX-1 and different tautomer complex to determine the possibility of hydrogen bonding or salt bridge formation between various tautomers and the active site of COX-1. The criteria for hydrogen bond interaction used, when the distance between the hydrogen and the heteroatom was within the range of 2.5–3.5 Å and the bond angle was at 109°–110°. Overall, these ligands exhibited binding interactions within the active site and the residues as suggested by Bazan and Fletterick (Bazan and Fletterick, 1989).
Interestingly, in our study, all the ligands were observed to form hydrogen bond with COX-1 active site residues except tauto 3 which is involved in strong hydrophobic interaction with COX-1.
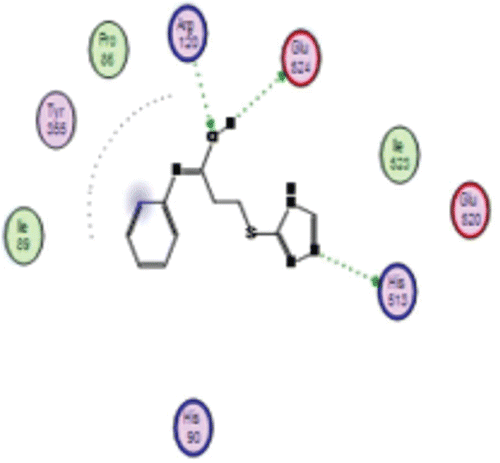
It was observed that among the various tautomers, tauto 1 interacted with residue, Arg (A) 120 through arene–cation interaction and forms hydrogen bond with Glu (A) 524. Percent of H-bonding was found to be 81%. Nitrogen atom of triazole nucleus acts as hydrogen bond donor and formed a hydrogen bond with Glu (A) 524. In tauto 2, protonated nitrogen atom of triazole nucleus acts as hydrogen bond donor and was involved in H-bond interaction with residue Glu (A) 524, whereas triazole ring interact with Arg (A) 120 through arene-cation interaction. Tauto 3 was not involved in hydrogen bonding but it interacted through hydrophobic and polar interaction with the residue Ser (A) 353, Met 522, Ile 523, Leu 352, Phe 518, Leu 384, Gly 526, Tyr 385, Ala 527, Trp 387 and Ser 530. Tauto 4 had shown interesting H-bond interaction with residues Arg (A) 120, Glu (A) 524 and His 513 (as shown in ). In this interaction, hydroxyl group formed by tautomerism was reasonably involved in hydrogen bonding interaction whereas nitrogen atom of triazole nucleus acts as hydrogen bond donor and form hydrogen bond with His 513. Hydroxyl group had shown strong hydrogen bonding with Arg (A) 120 in which percent of hydrogen bonding was found to be 64% with optimum interatomic distance of 1.6 Å, whereas Glu (A) 524 showed next strong H-bond interaction with the percent of H-bonding was 52% having interatomic distance of 2.4 Å while nitrogen revealed about 19 % H-bond interaction with residue His 513 with interatomic distance of 3.0 Å. Again in tauto 5, hydroxyl group formed by tautomerism was involved in H-bond interaction with the residue Glu 524 wherein hydroxyl group acts as hydrogen bond donor while triazole nucleus showed arene-cation interaction with the residue Arg 120. In this interaction percent of hydrogen bonding was found to be 70% with optimum interatomic distance of 2.8 Å between Glu 524 and hydroxyl group formed by tautomerism.
Figure 5. Showing 2D hydrogen bonding interaction of tauto-4 with COX-1.
In case of tauto 6, benzene nucleus was involved in arene-cation interaction with Arg 120 residue while protonated nitrogen of triazole acts as backbone hydrogen bond donor and form hydrogen bond with Pro 86 residue of COX-1.
In order to get useful results that may be valuable in future drug designing, we have carried out receptor based electrostatic analysis also.
In tauto1, blue colored region around triazole nucleus reveals the hydrogen bond donor region with slight white region near triazole ring suggest the requirement for any hydrophobic substituent that may enhance the activity as well as selectivity toward COX-1 whereas sulfur bridge don’t have any contribution in enhancing the hydrophobicity of tauto-1. Further aromatic substituents near amide linkage do not have any participation in hydrophobicity but substitution of any hydrogen bond donor may enhance activity of tautomer which is shown by blue region around aromatic ring.
In tauto 2 of compound 6a, receptor based electrostatic regions showed blue region around the protonated nitrogen of triazole nucleus reveals hydrogen bond donor nature of nitrogen and suggest the substitution of any H-bond donor may enhance the activity. Carbon atom in triazole ring may impart hydrophobicity to the tauto 2 of compound 6a.
Receptor based electrostatic regions in tauto 3 reveals some interesting facts. Blue region around triazole nucleus had shown a requirement for any hydrogen bond donor which may enhance the interaction between COX-1 and tauto 3. Compound 6a in 3rd tautomeric form did not show any hydrogen bonding interaction, it might interact with COX-1via hydrophobic interaction. Receptor based electrostatic regions reveals strong hydrophobic nature of aromatic ring which is represented by white surface around aromatic ring. Further blue region around aromatic ring suggest the requirement of any hydrogen bond donor for increasing the activity of compound 6a. Carbon chain which is connecting the sulfur atom and amide linkage may enhance the hydrophobicity of tauto 3 of compound 6a.
In tauto 4 of compound 6a, aromatic ring near amide link shows strong hydrophobic region indicated by white area around aromatic ring which may involved in hydrophobic interaction with COX-1. Further, red and blue region around aromatic ring suggests the substitution of any hydrogen bond donor as well as hydrogen bond acceptor may enhance the affinity of compound 6a towards COX-1 (as shown in ). Fourth tautomeric form of compound 6a gave rise to formation of one hydroxyl group near aromatic ring that form hydrogen bonding interaction with Cyclooxygenase residue, indicated by blue and red region around hydroxyl group. Presence of both blue and red region around hydroxyl group may suggest the dual nature, wherein oxygen atom acts as hydrogen bond acceptor and proton in hydroxyl group acts as hydrogen bond donor. Blue region around triazole ring reveals the hydrogen bonding nature of triazole nitrogen with slight hydrophobicity enhanced by carbon atom in triazole ring.
Figure 6. Receptor based electrostatic regions in the active site of COX-1. Blue = donor, Red = acceptor and white = hydrophobic regions.
Receptor based electrostatic analysis of tauto 5 reveals characteristic blue region covering the triazole nucleus of compound 6a suggest that substitution of any hydrogen bond donor group may enhance the affinity of ligand towards receptor. Noticeably, presence of sulfur bridge may enhance hydrophobic interaction with receptor which is indicated by white colored region. Especially, the Hydroxyl group formed by tautomerism imparting donor feature to the compound 6a, help in enhancing the affinity towards receptor. Further, presence of red colour contour around aromatic ring suggests that the substitution of any hydrogen bond acceptor enhances the hydrogen bond interaction as well as affinity towards receptor.
Electrostatic analysis of tauto 6 shows blue contour around triazole nucleus indicating the hydrogen bond donor nature of triazole nitrogen. Further Sulfur bridge connecting triazole ring shows characteristic white region indicating sulfur may be involved in enhancing the affinity of compound 6a towards the hydrophobic region of COX-1 receptor.
Evidently, the electrostatic feature map is an application of the Poisson-Boltzmann Equation (PBE) used for the prediction of electrostatically preferred locations of hydrophobic, H-bond acceptor and H-bond donor. It indicates various fields (electropositive region, electronegative region and hydrophobic region) on the surface of receptor which help us to design new ligand such as structural modification of lead by adding donor or acceptor features.
Selective inhibition of COX-1
In crystallization experimentsCitation21, it was reported that all COX-1 inhibitors interacted with the putative catalytic amino acid residue Tyr 385 and formed hydrogen bonds with Arg 120 and Tyr 355.
In mapping, it was observed that 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide docked near the gate of COX-1. Among all six principle tautomeric forms of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide, tauto 1 and 3 showed arene-cation interaction with Arg 120 and hydrogen bond interaction with Glu 524 residue of COX-1, whereas in tauto 4, hydroxyl group (formed by tautomerism) form hydrogen bond with Arg 120 and Glu 524 residue of COX-1. Further tauto 5 and 6 were interacted with COX-1 by arene-cation interaction and form hydrogen bond with Glu 524 and Pro 86.
Herein, different tautomeric forms of compound 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide interacted with COX-1 by hydrogen bonding, indicates involvement of tautomerism in drug receptor interaction which may give rise to stable drug receptor complex. Further, binding of compound 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide to Arg 120 and Glu 524 may block the gate of active site of COX-1 and interfere with the conversion of arachidonic acid to prostaglandin (PG) H2 in the active site of COX-1. It can be hypothesized that compound 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide binds with COX-1 in a selective manner wherein tautomerism seems to be the most important factor in selective inhibition of COX-1, particularly the hydroxyl group formed by tautomerism directly influence the selective inhibition of COX-1.
Conclusion
Earlier, we have reported anti-inflammatory activity of 3-(4H-1, 2, 4-triazol-3-ylthio)-N-phenylpropanamide but we were not convinced that the said compound elicit its anti-inflammatory activity either by inhibition of COX-1 or COX-2. In biological evaluation, Diclofenac sodium was used as standard drug and it was postulated that Diclofenac sodium showed selectivity for both isoforms of Cyclooxygenase. Subsequently to predict the mechanism of said compound by virtual screening, we have docked the most active compounds of the previously described series into the active site of COX-1 rather than COX-2.
In crystallization experiments, it was reported that all COX-1 inhibitors interacted with the putative catalytic amino acid residue Tyr 385 and formed hydrogen bonds with Arg 120 and Tyr 355. These amino acid residues serve as gate of active site of COX-1.Noticeably, in tauto 4, hydroxyl group (formed by tautomerism) established hydrogen bond with Arg 120 and Glu 524 residue of COX-1. Therefore, tauto 4 may be the most favourable tautomer as well as conformer of compound 3-(4H-1, 2, 4-triazol-3-ylthio)-N-phenylpropanamide that showed maximum interaction with the COX-1 receptor through hydroxyl group.
Subsequently receptor based electrostatic analysis revealed some important features necessary for anti-inflammatory activity. For tauto 1, addition of any hydrogen bond donor and hydrophobic substituent may enhance the anti-inflammatory activity while tauto 2 react with COX-1 through nonbonding interaction in which further substitution of any hydrophobic substituent may improve the drug receptor interactions. Electrostatic analysis of tauto 3 showed requirement of hydrogen bond donor and hydrophobic group that significantly influence the interaction of tauto 3 with COX-1. In case of tauto 4, hydrogen bond donor, hydrogen bond acceptor as well as hydrophobic substituents showed noteworthy input in drug receptor interaction. Next, sulfur bridge constitute hydrophobic core of tauto 5 and 6 which are engaged in hydrophobic interaction with COX-1 receptor while hydrogen bond donor are essential for both tauto 5 and 6 conventional for anti-inflammatory activity.
In the present study, it was concluded that interference in arachidonic acid binding channel might be possible mode of action of 3-(4H-1,2,4-triazol-3-ylthio)-N-phenylpropanamide, wherein tautomerism plays vital role in selective inhibition of COX-1.
Today, in the rational design of new COXs binders, medicinal chemists have to pay attention to the selectivity properties of the designed drugs, and a fine-tuning of the COX selectivity profile might be necessary to generate novel effective drugs with reduced side effects. The kinetics of selective and nonselective inhibitors is also different in general, and in recent years, much effort has been paid in elucidating the dynamic binding mechanism of COX inhibitors. Further studies are going on to elucidate the involvement of tautomerism in selective inhibition of COX-1.
Acknowledgement
We are thankful to Sumit O. Bajaj, C. Eugene Bennett Department of Chemistry, West Virginia University, Morgantown, West Virginia 26506-6045, USA for providing useful data and helpful discussions.
Declarations of interest
Authors declare no conflict of interest.
References
- Hoggarth E, Martin A.Studies in the chemotherapy of tuberculosis: Part-7. The oxidation and reduction products of thiosemicarbazoles. Brit J Pharrnacol 1951;6:454–458.
- Udupi RH, Puzushottaamachar P.Synthesis and biological activity of 3-pyridyl-4-(N-substitutedphenylcarbaxamido)-5-mercapto-1,2,4-triazole. Indian J Heterocycl Chem 2000;9:189–192.
- Tozkoparan B, Kupell E, Erden Y, Mevlut E.Preparation of 5-aryl-3-alkylthio-1,2,4-triazoles and corresponding sulfones with anti-inflammatory-analgesic activity. Bioorg Med Chem 2007;15:1808–1814.
- Mullican M, Wilson M, David TC, Kostlan C, Schrier D, Dayer D.Design of 5(3,5-di-tert-butyl-4-hydroxyphenyl)-1,3,4-thiadiazole, -1,3,4-oxadiazole, and -1,2,4-triazole as orally active, nonulcerogenic anti-inflammatory agents. J Med Chem 1993;36:1090–1099.
- Palaska E, Gulay S, Kelicen P, Tugba N. Synthesis and anti-inflammatory of 1-acylthiosemicarbazides, 1,3,4-oxadiazoles, 1,3,4-thiadiazoles and 1,2,4-triazole-3-thiones. II Farmaco 2002;57:101–107.
- Labanauskas L, Udrenite E, Gaidelis P, Brukstus A.Synthesis of 5-(2,3,- and 4-methoxyphenyl)-4H-1,2,4-triazole-3-thiol derivatives exhibiting anti-inflammatory activity. II Farmaco 2004;59:255–259.
- Eweiss NF, Bahajaj AA, Elsherbini. Synthesis and antimicrobial activity of some 4-amino-5-aryl-1,2,4-triazole-3-thiones and their derivatives. J Heterocycl Chem 1986;23:1451–1458.
- Udupi RH, Ramchandran S, Srinivasulu N, Pasha T, Agrawal N, Ganesh V. Synthesis and biological evaluation of 3-substituted-4-(6′-methoxynapth-2′-yl) propionamido-5-mercapto-1, 2, 4-triazoles. Indian J Heterocycl Chem 2002;12:33–36.
- Holla BS, Sooryanarayana B, Shridlova K, Akberai PM., Studies on arylfuran derivatives Part, Xl. Synthesis, characterization and biological studies on some mannich bases. Farmaco 2000;55:338–344
- Abdelal AM, Gineinah MM, Nasr MN. 1,1-Bis-(1,2,4-triazolyl and 1,3,4-thiadiazolyl)ethane and phenylethane derivatives as potential antibacterial agents. Boll Chim Farm 1998;137:372–376.
- Manikrao AM, Fursule RA, Rajesh KS, Kunjwani HK, Sabale PM. Synthesis and biological screening of novel derivatives of 3-(N-substituted carboxamidoethylthio)-(4H)-1,2,4-triazoles. Indian J Chem 2010;49B:1642–1647.
- Cuzick J. Aspirin and non-steroidal anti-inflammatory drugs for cancer prevention: An international consensus statement. Lancet Oncol 2009;10:501–507.
- Smith WL, Borgeat P, Fitzpatrick FA. Biochemistry of Lipids, Lipoproteins and Membranes, eds DE Vance and J Vance Elsevier Science, Amsterdam; London 1991;297–3253.
- McNaught AD, Wilkinson A. IUPAC Compendium of Chemical Terminology, http://www.iupac.org/goldbook/T06252.pdf (accessed Aug 2003).
- Mockus J, Stobaugh RE. Chemical-abstracts-service chemical registry system.7.Tautomerism and Alternating Bonds. J ChemInf Comp Sci 1980;20:18–22.
- Repalin SV, Skorenko AV, Balakin KV, Nasonov AF, Lang SA, IvashchenkoAA,Savchuk NP. Advanced exact structure searching in large databases of chemical compounds. J ChemInfComputSci 2003;43:852–860.
- Sadowski J. A tautomer and protonation pre-processor for virtual screening. Abstr Pap Am Chem S 2002; 224:U504–U504.
- Pospisil P, Ballmer P, Folkers G, Scapozza L. Tautomerism of nucleobase derivatives and their score in virtual screening to thymidine kinase. Abstr Pap Am Chem S 2002;224:U501–U501.
- MOE (Molecular Operating Environment), Version 2009; Chemical Computing Group Inc.: Montreal, Canada, 2009.
- www.rcsb.com (Protein Data Bank.Pdb ID- 3N8X (1)).
- Kalgutkar AS, Crews BC, Rowlison CG, Seibest K, Marnett LJ. Aspirin-like molecules that covalently inactive COX-2. Science 1998;280:1268–1270.