Abstract
Aminopeptidase N (APN/CD13) over expressed on tumour cells, plays a critical role in tumour invasion, metastasis and tumour angiogenesis. In this article, we described the design, synthesis and preliminary activity studies of novel 3-amino-2-hydroxyl-3-phenylpropanoic acid derivatives as APN inhibitors. The in vitro enzymatic inhibitions on APN from porcine kidney showed that compound 7e had the most potent inhibitory activity against APN with the IC50 value to 1.26 ± 0.01 μM, which is better than that of bestatin (IC50 = 2.55 ± 0.11 μM). In addition, compound 7e also showed better inhibitory activity against APN on human ovary clear cell carcinoma cell ES-2 than bestatin with the IC50 value to 30.19 ± 1.02 μM versus 60.61 ± 0.1 μM. Compound 7e could be used as the lead compound in the future for anti-cancer agent research.
Introduction
Aminopeptidase N (APN; EC 3.4.11.2) is a zinc-dependent type II membrane-bond ectopeptidase and belongs to the M1 family of MA clan of peptidaseCitation1,Citation2. This enzyme consists of 967 amino acids with a short N-terminal cytoplasmic domain, a single transmembrane part and a large cellular ectodomain containing the active siteCitation3,Citation4, which can preferentially release neutral or basic amino acids from the N-terminal of peptidesCitation5,Citation6. APN is widely distributed in the body of mammalian. It can be expressed on the surface of different cells such as myeloid progenitors and monocytes, epithelial cells of the intestine and kidney, synaptic membranes in the central nervous system, fibroblasts, endothelial cells, epithelial cellsCitation7–10 and so on. APN is highly expressed on tumour cells and plays a critical role in tumour invasion, metastasis and angiogenesis. Therefore, the design and synthesis of APN inhibitors may be a clinical significance for the discovery of anti-cancer agents.
Bestatin (AHPA-Leu), a well-known APN inhibitor, was first isolated from the culture filtrate of Streptomyces olivoreticuli in 1976Citation11, and now it is widely used in clinical practise for the treatment of adult acute non-lymphocytic leukemia. Since then, many APN inhibitors such as probestinCitation12, prebestinCitation12, AHPA-ValCitation13 and amatatinCitation14 have been developed. Judging from the structure of these compounds, we can see that they all contain the key unit 3-amino-2-hydroxy acyl scaffold, which is important for their biological activity for that it can chelate the Zn2+ in the active site of metal-dependent enzymesCitation15.
The interaction of bestatin with the active site of APN showed that the hydroxyl group and carbonyl group of bestatin belong to the zinc-binding group (ZBG) to chelate the Zn2+ in the active site of APN. The phenyl group can insert into the S1 pocket of APN and the Leu residue can insert into the S1′ pocket, while the free amino group can interact with Glu 350. In our previous work, we have designed a series of novel chloramphenicol amine derivatives as APN inhibitors, but the result was not satisfied, most compounds showed less activity than bestatinCitation16, the most active compound 13b is a pseudotripeptide with the IC50 value to 7.1 μM, which is still less than that of bestatin (IC50 = 3.00 μM). Recently, our group has synthesised a series of novel L-isoserine derivatives as APN inhibitors, most of these compounds showed moderate activities, the most active compound 14b (IC50 = 12.2 μM) showed still less inhibitory activity toward APN than that of bestatin (IC50 = 7.3 μM)Citation17,Citation18.
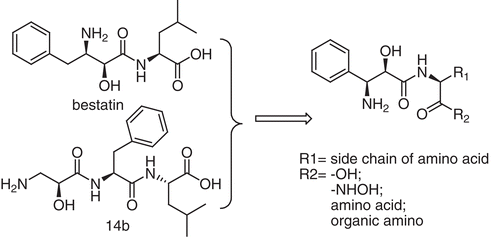
Taxol, a well-known anti-cancer drug, also has the 3-amino-2-hydroxy acyl scaffold ((2R, 3S)-3-amino-2-hydroxy-3-phenylpropanoic acid residue). Compared with L-isoserine, (2R, 3S)-3-amino-2-hydroxy-3-phenylpropanoic acid is much more similar with the residue of bestatin (2S, 3R)-3-amino-2-hydroxy-4-phenylbutyl acid except for the chirality and one carbon. There is a phenyl group in C3 of (2R, 3S)-3-amino-2-hydroxy-3-phenylpropanoic acid, which may be beneficial for APN interaction. So, we used bestatin and 14b as the lead compound, (2R, 3S)-3-amino-2-hydroxy-3-phenylpropanoic acid as the key scaffold, by coupling with different amino acids or other functional groups, and we got a series of novel bi-peptide and tri-peptide derivatives with 3-amino-2-hydroxy-3-phenylpropanoic acid scaffold. Considering the best ZBG hydroxamate group may be good for APN inhibitors, we also converted the carboxyl group of some compounds into hydroxamate group ().
Figure 1. Novel aminopeptidase N inhibitors with 3-amino-2-hydroxy-3-phenylpropanoic acid scaffold.
Results and discussion
Chemistry
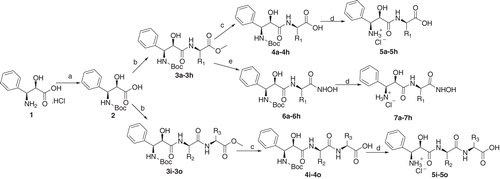
The target compounds were synthesised via the route outlined in and . The commercial (2R, 3S)-3-amino-2-hydroxyl-3-phenylpropanoic acid hydrochloride 1 was protected by (Boc)2O to get the key intermediate compound 2. Compound 2 can be coupled with different methyl ester of amino acids or methyl ester of dipeptides by classical EDCI/HOBt method to provide compounds 3a–3o. The esters of compounds 3a–3o were hydrolysed to carboxylic acid by NaOH/H2O in methanol, and then converted into the target compounds 5a–5o by deprotecting the Boc group. Compounds 7a–7h containing hydroxymate group were obtained from compounds 3a–3h by reacting with NH2OK in methanol and then cleaving the Boc group.
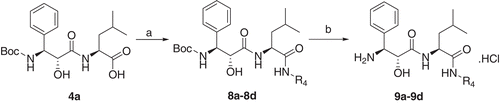
As shown in Scheme 2, compounds 9a–9d were obtained from compound 4a by coupling it with different substituted organic aromatic amines and then cleaving the Boc group.
Scheme 1. Reagents and conditions: (a) (Boc)2O, NaOH, THF; (b) EDCI, HOBT, DCM, NH2-AA-COOCH3; (c) NaOH, CH3OH; (d) 3N HCl-EtOAc; (e) NH2OK, CH3OH;
Scheme 2. Reagents and conditions: (a) EDCI, HOBT, DCM, NH2R4; (b) 3N HCl-EtOAc
Preliminary in vitro evaluation and SAR discussion
The newly synthesised 3-amino-2-hydroxyl-3-phenylpropanoic acid derivatives were assayed for the enzymatic inhibitions both on APN from porcine kidney (Microsomal; Sigma-Aldrich, St. Louis, MO, USA) and matrix metalloproteinases-2 (MMP-2). The results are listed in . Similar to APN, MMP-2 is also a zinc-dependent metalloproteinase, which is involved in the process of tumour invasion and metastasisCitation19. The difference between them is that MMP-2 is an endopeptidase, while APN is an exopeptidase. All the target compounds were assayed for the inhibitory activities on APN and MMP-2 to observe the selectivity.
Table 1. The structures and in vitro APN inhibitory activities of the target compounds.
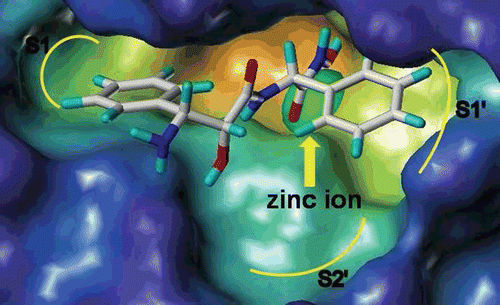
As shown in , the results showed that most compounds had moderate MMP inhibitory activity, which was consistent with our early report. But the result of APN inhibition was not satisfied, most compounds did not show expected APN inhibition, and some compounds (such as 5b, 5d, 5f and so on) even had no APN inhibitory activity except for the hydroxamic acid derivatives 7a–7h. Of all these compounds, compound 7e showed to be the best APN inhibitor with the IC50 value to 1.26 μM, which was much better than that of the positive control bestatin (IC50 = 2.55 μM). By analysing the results, we supposed that these compounds may not have the same interaction model with bestatin by using the hydroxyl group and carbonyl group to chelate the zinc ion but using the hydroxamic acid group, this hypothesis could be verified by the docking results (). If the phenyl group of 3-amino-2-hydroxyl-3-phenylpropanoic acid could form hydrophobic interaction with the S1 pocket as bestatin, the distance of the hydroxyl group and carbonyl group to the zinc ion may be unsuitable because of the lack of one carbon, this may explain why compounds 5a–5o and 9a–9d had poor APN inhibitory activity. But compounds 7a–7h showed very good APN inhibitory activity because of the introduction of the strong ZBG hydroxamic acid group.
Figure 2. The FlexX docking result of 7e with the active site of aminopeptidase N (PDB: 2DQM).
Comparing compounds 7a–7h, we can know that the APN inhibitory activities were different with the substitutions in R1. Compounds 7a and 7b with the Leu and Val in R1 had better APN inhibitory activity than the others (such as 7c, 7d, 7g and 7h), this may suggest that the Leu and Val in R1 is good for APN inhibition. Compound 7e with phenylglycine in R1 had the best APN inhibition with the IC50 value to 1.26 ± 0.01 μM. Compound 7f with phenylalanine in R1 also had good APN inhibition but not better than compound 7e with the IC50 value to 44.00 ± 2.32 μM. Comparing with compounds 7e and 7f, we can know that the length of the α-carbon atom to the aromatic ring may be important for APN inhibition. Compound 7g with a hydroxyl group in the para-position of the phenyl group had less inhibitory activity than compound 7f, this may suggest that the additional hydroxyl group may be unsuitable for APN interaction.
Compound 7e showed the most inhibitory activity toward APN. To investigate the interaction of these compounds with APN, the docking studies of the representative compound 7e were carried out via Sybyl/Sketch module and optimised using Powell’s method with the Tripos force field with convergence criterion set at 0.05 kcal/ (Å mol) and assigned with Gasteiger–Hückel method. The docking study performed using Sybyl7.3/FlexX module, the residues in a radius of 7.0 Å around Bestatin in the co-crystal structure (PDB code: 2DQM) were selected as the active site. Other docking parameters implied in the program were kept default. The docking result was shown in . As shown in , 3-amino-2-hydroxyl-3-phenylpropanoic acid scaffold of compound 7e inserted into the S1 pocket, and the hydroxymate group chelated with the zinc ion in the active site, while the phenyl group of L-phenylglycine moiety interacted with the S1′ pocket. For a further and detail understanding of the binding mode of 7e with APN, a two-dimensional picture was also created with the program LIGPLOT, which was shown in ; we can see that the phenyl group of 3-amino-2-hydroxyl-3-phenylpropanoic acid could form hydrophobic interaction with the S1 pocket formed by Arg825, Tyr376, Met260 and Tyr381. The free amino group could form hydrogen bond with Arg825 at the distance of 2.64 Å. The phenyl group of L-phenylglycine moiety could form hydrophobic interaction with the S1′ pocket formed by Arg293, Asp372 and His297. The two oxygen atoms of hydroxamic acid could chelate with the zinc ion of APN. Additionally, the hydroxyl group of hydroxamic acid could also form hydrogen bond with His297 and His301 at the distance of 2.70 Å and 2.84 Å.
Figure 3. The docking result of 7e with aminopeptidase N showed by LIGPLOT.
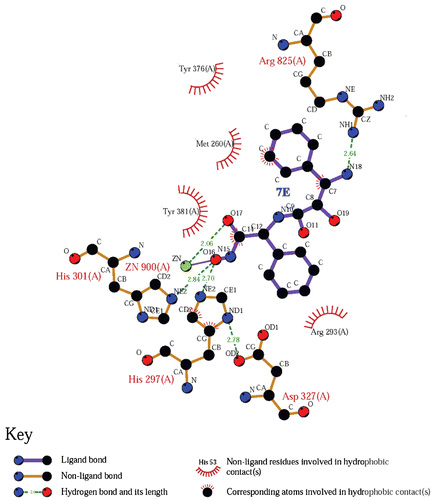
Although the computed information partially supported our assumption, the exact binding mode of the 3-amino-2-hydroxyl-3-phenylpropanoic acid derivatives with APN should be obtained from further X-ray crystal studies.
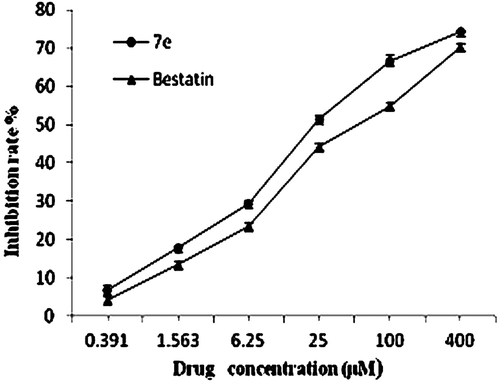
APN is highly expressed on the surface of human ovary clear cell carcinoma cell ES-2Citation20; to study the inhibitory activity of the target compounds toward human APN, we also tested the in vitro enzymatic inhibitions of compound 7e toward APN on ES-2. The results showed that compound 7e is also a dose-dependent APN inhibitor in the concentration range of 0.39–400 μM (). The IC50 value of compound 7e is 30.19 ± 1.02 μM, which is still better than that of bestatin (IC50= 60.61 ± 0.1 μM).
Figure 4. Inhibition of aminopeptidase N (APN)/CD13 activity on ES-2 induced by 7e or bestatin. OVCA cells (1 × 105) seeded in 96-well plates were exposed to various concentrations of 7e or bestatin at 37°C, and the substrate L-leucine-p-nitroanilide was added. APN/CD13 activity was estimated by measuring absorbance at 405 nm using a microplate reader after 60 min of incubation. The inhibition rate was calculated by comparing to the control (without drug exposure). The bars indicate means ± standard deviation (n = 3).
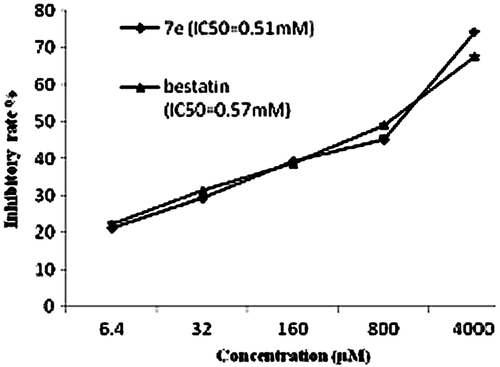
Additionally, the effects of compound 7e on the proliferation of human ovary clear cell carcinoma cell ES-2 compared with bestatin were further assessed by using MTT assay, which were shown in . The results showed that the anti-proliferative effects of compound 7e against ES-2 cell were slightly better than that of bestatin with the IC50 value of 0.51 ± 0.08 mM versus 0.57 ± 0.04 mM.
Figure 5. In vitro anti-proliferative activities of compound 7e and bestatin against ES-2 cells. The bars indicate means ± standard deviation (n = 3).
Conclusion
In summary, we have described the synthesis and properties of a series of novel (2R,3S)-3-amino-2-hydroxyl-3-phenylpropanoic acid derivatives as APN inhibitors. Because of the lack of one carbon as compared with 3-amino-2-hydroxyl-4- phenylbutyl acid, most of the target compounds did not show expected APN inhibitory activities, but the introduction of hydroxamate group to the compounds could greatly improve the inhibitory activities. The most effective compound 7e, not only had better inhibitory activity against APN both from porcine kidney and human but also showed slightly better anti-proliferative effects toward ES-2 cells than that of bestatin. Compound 7e could be used as a lead compound for further development of small molecular peptidomimetic APN inhibitors. Though the preliminary enzymatic results are not very encouraging, it still provides useful clues for further APN inhibitors exploitation.
Supplementary data
Supplementary data (Experimental details, and spectroscopic data for all the compounds and procedures for the activity assays were provided.).
Supplementary Material
Download PDF (350.5 KB)Declaration of interest
This work was supported by National High Technology Research and Development Program of China (863 project; Grant No. 2007AA02Z314), National Nature Science Foundation of China (No. 90713041 and No. 30772654) and National Science and Technology Major Project (Grant No. 2009ZX09103-118).
References
- Look AT, Ashmun RA, Shapiro LH, Peiper SC. Human myeloid plasma membrane glycoprotein CD13 (gp150) is identical to aminopeptidase N. J Clin Invest 1989;83:1299–1307.
- Rawlings ND, Barrett AJ. MEROPS: the peptidase database. Nucleic Acids Res 1999;27:325–331.
- Olsen J, Cowell GM, Kønigshøfer E, Danielsen EM, Møller J, Laustsen L et al. Complete amino acid sequence of human intestinal aminopeptidase N as deduced from cloned cDNA. FEBS Lett 1988;238:307–314.
- Sjöström H, Norén O, Olsen J. Structure and function of aminopeptidase N. Adv Exp Med Biol 2000;477:25–34.
- Xu W, Li Q. Progress in the development of aminopeptidase N (APN/CD13) inhibitors. Curr Med Chem Anticancer Agents 2005;5:281–301.
- Zhang X, Xu W. Aminopeptidase N (APN/CD13) as a target for anti-cancer agent design. Curr Med Chem 2008;15:2850–2865.
- Breljak D, Gabrilovac J, Boranić M. Aminopeptidase N/CD13 and haematopoietic cells. Haema 2003; 6: 453–461.
- Dixon J, Kaklamanis L, Turley H, Hickson ID, Leek RD, Harris AL et al. Expression of aminopeptidase-n (CD 13) in normal tissues and malignant neoplasms of epithelial and lymphoid origin. J Clin Pathol 1994;47:43–47.
- Raynaud F, Bauvois B, Gerbaud P, Evain-Brion D. Characterization of specific proteases associated with the surface of human skin fibroblasts, and their modulation in pathology. J Cell Physiol 1992;151:378–385.
- Piela-Smith TH, Korn JH. Aminopeptidase N: a constitutive cell-surface protein on human dermal fibroblasts. Cell Immunol 1995;162:42–48.
- Umezawa H, Aoyagi T, Suda H, Hamada M, Takeuchi T. Bestatin, an inhibitor of aminopeptidase B, produced by actinomycetes. J Antibiot 1976;29:97–99.
- Aoyagi T, Yoshida S, Nakamura Y, Shigihara Y, Hamada M, Takeuchi T. Probestin, a new inhibitor of aminopeptidase M, produced by Streptomyces azureus MH663-2F6. I. Taxonomy, production, isolation, physico-chemical properties and biological activities. J Antibiot 1990;43:143–148.
- Chung MC, Lee HJ, Chun HK, Lee CH, Kim SI, Kho YH. Bestatin analogue from Streptomyces neyagawaensis SL-387. Biosci Biotechnol Biochem 1996;60:898–900.
- Rich DH, Moon BJ, Harbeson S. Inhibition of aminopeptidases by amastatin and bestatin derivatives. Effect of inhibitor structure on slow-binding processes. J Med Chem 1984;27:417–422.
- Kim H, Lipscomb WN. Structure and mechanism of bovine lens leucine aminopeptidase. Adv Enzymol Relat Areas Mol Biol 1994; 68: 153–213.
- Yang K, Wang Q, Su L, Fang H, Wang X, Gong J et al. Design and synthesis of novel chloramphenicol amine derivatives as potent aminopeptidase N (APN/CD13) inhibitors. Bioorg Med Chem 2009;17:3810–3817.
- Yang KH, Feng JH, Fang H, Zhang L, Gong JZ, Xu WF. Synthesis of a novel series of L-isoserine derivatives as aminopeptidase N inhibitors. J Enzyme Inhib Med Chem 2011; doi:10.3109/14756361003698147.
- Wang X, Zhang L, Yang K, Zhang C, Zhang J, Fang H et al. The effect of different species aminopeptidase N structure on the activity screening of aminopeptidase N inhibitor. Biol Pharm Bull 2010;33:1658–1665.
- Li Q, Fang H, Xu W. Novel 3-galloylamido-N’-substituted-2,6-piperidinedione-N-acetamide peptidomimetics as metalloproteinase inhibitors. Bioorg Med Chem Lett 2007;17:2935–2938.
- Wang XJ, Feng JH, Zhu HW, Fang H, Zhang J, Xu WF. Activity screening and structure-activity relationship of the hit compounds targeting APN/CD13. Fundam Clin Pharmacol 2010; 25: 217–228.