Abstract
DNA topoisomerases comprise a major aspect of basic cellular biology and are molecular targets for a variety of drugs like antibiotics, antibacterials and anticancer drugs. They act by inhibiting the topoisomerase molecule from relegating DNA strands after cleavage and convert the topoisomerases molecule into a DNA damaging agent. Though drugs of various categories acting through different mechanisms are available for the treatment, there are still problems associated with the currently available drugs. Therefore, Structural biologists, Structural chemists and Medicinal chemists all around the world have been identifying, designing, synthesizing and evaluating a variety of novel bioactive molecules targeting topoisomerase. This review summarizes types of topoisomerase and drug treating each class along with their structural requirement and activity. The emphasis has been laid in particular on the new potential heterocyles and the possible treatments as well as the current ongoing research status in the field of topoisomerase as dual targeting.
Keywords::
Table of Contents
1. Introduction00
1.1. Functions of DNA Topoisomerase
2. Classification
2.1. Eukaryotic topoisomerase
2.1.1. Type I Topoisomerase
2.1.2. Type II Topoisomerase
2.2. Prokaryotic topoisomerase
2.2.1. Bacterial topoisomerase I and II enzymes
2.2.2 Drug targeting bacterial topoisomerases
3. Topoisomerase as drug targets
3.1. Drug targeting DNA topoisomerase I
3.1.1. Camptothecin
3.1.2. CPT analogues
3.1.3. Homocamptothecin
3.1.4. Novel non-camptothecin topoisomerase I inhibitors
3.1.5. Benzoxanthone
3.1.6. 9-AC, CPT 11 and its derivatives
3.1.7. Indolocarbazole derivatives
3.1.8. Luotonin A
3.1.9. Furancoumarins
3.1.10. 7-Azaindenisoquinoline
3.2. Drug targeting DNA topoisomerase II
A. Different classes of compounds target topoisomerase II
ICRF 193 and HU-331
B. Intercalating topoisomerase-targeting drugs
Anthracene and its derivatives
Actinomycin and Ellipticin
C. Non-intercalating topoisomerase-targeting drugs
1. Epipodophyllotoxins
2. VP-16 and Etopside Phospate
3. Teniposide
4. Acridine
5. NK- 109
6. TOP-53
7. S16020-2
8. Merbarone
9. Voreloxin
10. Azaatoxin
3.3 Drug with heterocyclic moiety:
1. Substituted pyridines
2. Tetracyclic thienopyridones
3. Tetracyclic fluoroquinolones
4. Glycosylated diphyllin derivative
5. Natural product inhibitors
3.4 Dual targeting agents
1. Dual topoisomerase I/II inhibitors in cancer therapy.
2. Flavonol Glycoside
4. Conclusion
1. Introduction
DNA topoisomerases enzymes control DNA topology by cleaving and rejoining DNA strands and play an important role in the regulation of the physiological function of the genomeCitation1–3 as well as DNA processes such as replication, transcriptionCitation4–6, recombinationCitation7,Citation8, repairCitation9,Citation10, chromosome decondensation and sister chromatidCitation11–14. Beyond their normal functions, topoisomerases are important cellular targets especially in the treatment of human cancersCitation1,Citation2. Some of the most powerful anticancer drugs used clinically act by causing DNA disorders. Topoisomerase inhibitors block the ligation step of the cell cycle, generating single and double stranded breaks that harm the integrity of the genome and leading to apoptosis in proliferating cells and cell death.
1.1. Functions of DNA topoisomerases
DNA needs to be unwound in for the transcription/replication machinery so that it can be replicated or copied, respectively (accessing DNA). To solve the problems of winding due to double helical structure of DNA, topoisomerases attach to single or double stranded DNA, which makes single stranded nicks to relax the helix, and relive the topological tension (removing DNA supercoiling) 15. Topoisomerase II DNA-gyrase during takes energy from ATP hydrolysis to introduce tight supercoils into the DNA helix with a view to condense the chromosome (chromosome condensation)Citation16.
2.1. Eukaryotic topoisomerase
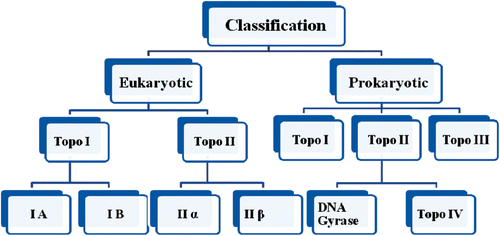
Eukaryotic topoisomerase are of two types: type I and type IICitation17 (). These enzymes are well represented in eukaryotes as well as in yeast, drosophila and mammalian cellsCitation18.
Figure 1. Classification of topoisomerases.
2.1.1. Type I topoisomerases
Type I topoisomerases cut one strand of double stranded DNA, relax the strand, and restrengthen the strands. They are further subdivided into two structurally and mechanistically characteristic topoisomerases: type IA and type IBCitation1,Citation2. Type IA topoisomerases change the linking number of a circular DNA strand by units of strictly 1. Type IB topoisomerases alter the linking number by multiples of 1 (nCitation19,Citation20).
Type IA topoisomerases have 4 domain I-IV. Toprim is included in domain I, whereas domain III and domain IV each consist of a helix-turn-helix domain in which the catalytic tyrosine exists. Domains III and IV have a flexible bridge of domain II connecting each otherCitation20. First, the single-stranded DNA binds with domain III and I. The DNA backbone breaks as a result of catalytic tyrosine action which leads in the formation of transient 5′ phosphotyrosine intermediate. The break is then separated, using domain II as an axis or the turning point, and a second duplex or strand of DNA is passed throughCitation21,Citation22. Topoisomerase type IB solves the problem of positively or negatively supercoiled DNA. Topo IB consists of an NTD, a capping lobe, a catalytic lobe, and a C-terminal domain. Relaxation is not an active process during the nicking or ligation steps; in the sense that energy in the form of ATP is not spentCitation23.
Topo V is the founding member of the type IC topoisomerase and works much like the type IB topoisomeraseCitation24,Citation25.
2.1.2. Type II topoisomerase
Type II topoisomerases is classified in two subclasses, type IIA and IIB. Type IIA topoisomerases involves the enzymes DNA gyrase, eukaryotic topoisomerase II, and bacterial topoisomerase IV. Type IIB topoisomerases are structurally and biochemically different, and comprise a single family member, topoisomerase VICitation27. Both isoforms share extensive sequence and amino acid homology. Type IIA topoisomerases make double-stranded breaks with four-base pair overhangs, while type IIB topoisomerases make double-stranded breaks with two base overhangs. In addition, type IIA topoisomerases are able to simplify DNA topology, while type IIB topoisomerases do notCitation26,Citation28.
2.2. Prokaryotic topoisomerases
Topoisomerases I (type I) enzymes that act as monomers are likely to be single chain polypeptides. On the contrary, the type II enzymes show a multimeric structure containing at least two active sites. DNA gyrase of Escherichia coli comprises subunits encoded by the gyrA and gyrB genes and is a heterotetramer of A2+B2 configuration, while the eukaryotic topoisomerase enzymes, which can be viewed as single chain chimeras of the two DNA gyrase subunits, are consequently homodimeric in structureCitation17.
2.2.1. Bacterial topoisomerase I and II enzymes
Prokaryotic topoisomerases: () Bacterial E. coli contains two types I and two type II enzymes. The two types of I enzymes, topoisomerase I (encoded by the topA/supX gene) and topoisomerase III (encoded by topB) though, removal of the topA gene requires that compensatory mutations occur in genes encoding one or other of the type II topoisomerases, DNA gyrase and topoisomerase IVCitation29,Citation30.
E. Coli Topoisomerase I and III. E. Coli topoisomerase I was published in 1971 and known as the omega proteinCitation31. The discovery of second type topoisomerase III was made in topA deletion strains. Topoisomerases I enzyme is a monomer of 97 kDaCitation32 (kilodaltons). Topoisomerases III has ability of coding for a 74 kDa protein. In the case of the type I topoisomerases, both topoisomerase I and topoisomerase III are active as dimers consisting of a single type of subunit encoded by the topA and topB genes, respectivelyCitation33–35.
E. Coli Topoisomerases II- DNA Gyrase and Topoisomerases IV: Both of the type II topoisomerases of E. coli are essential for cell survival. Topoisomerase IV is the most recently discovered E. coli topoisomerase and is similar in structure to DNA gyrase. Both enzymes are tetramers composed of pairs of two types of sub-units. The GyrA and GyrB subunits of DNA gyrase (encoded by gyrA and gyrB, respectively) are similar in amino acid sequence to the ParC and ParE subunits (encoded by parC and parE) of topoisomerase IV, respectively. The type II enzymes all require ATP for catalysis, which is in contrast to type I topoisomerases. DNA gyrase actually has numerous roles in vivo, including general supercoiling homeostasis, the initiation phase of DNA replication, and chromosome partitioning, prior to the discovery of topoisomerase IV. DNA gyrase almost exactly has a general role both in removing supercoils and in introducing negative supercoils into DNA, combined with a more specialized role in removing knots and unlinking catenanes generated by recombination. Topoisomerase IV has a major role in catenanes generated by DNA replicationCitation36–39.
2.2.2. Drug targeted to bacterial topoisomerases
Antibiotics can be grouped under the broader title of chemotherapuetic agents that are capable to kill or inhibit the target pathogen with no too much harm to the host organism.
Aminocoumarin: The aminocoumarin antibiotics are known inhibitors of DNA Gyrase. Aminocoumarin antibiotics are known as inhibitor of DNA gyrase enzyme involved in the cell division in bacteria. The aminocumarin antibiotics comprise Novobiocin, (1), Albamycin, Clorobiocin, Coumermycin. Clorobiocin is a natural antibiotic differs from novobiocin in that a methyl group is present at 8th positionCitation39,Citation40.
Figure 2. Structures of antibiotics.
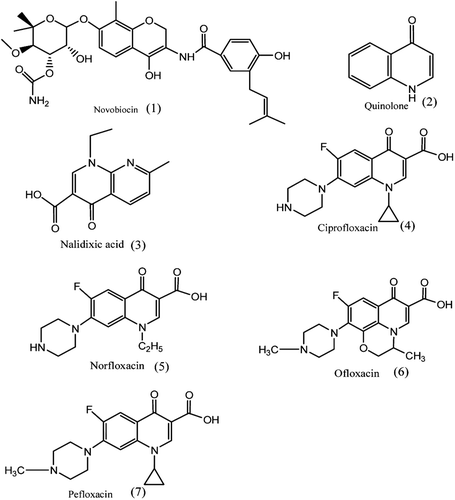
Quinolone: Quinolones, (2), are to potent synthetic chemotherapeutic antibacterials. Quinolone antibiotics inhibits DNA gyrase. The first generation of the quinolones is Nalidixic acid, (3), and development of generations of fluorinated quinolones led to compounds with broad antibacterial activity. Fluoroquinones represent an important class of antimicrobial such as Ciprofloxacin, Norfloxacin, Enoxacin, Pefloxacin, Ofloxacin, Lomefloxacin, Sparfloxacin shown in . Ciprofloxacin, a representative fluoroquinolone, is the most efficacious oral antibiotic in clinical useCitation39,Citation41,Citation42.
3. Drug-targeted chemotherapy: topoisomerases as drug targets
Drug targeting DNA topoisomerases: Anticancer drugs normally bind in reversible manner. When a piece of the cleaved DNA strand that has not been bound by the topoisomerase could be released, creating a permanent breakage in the DNA leading to cell death. Most of these inhibitors target either topoisomerase I or II, whereas some are capable of inhibiting both the enzymes actionCitation36. Two classes of antibiotic drugs: quinolonesCitation43 and coumarins targets bacterial gyraseCitation44 (topoisomerase II) and topoisomerase IV. These antibiotics operate by DNA replication inhibition in the bacteria responsible for variety of different diseases such as pneumonia, tuberculosis and malaria.
Topoisomerase inhibitors: Type I topoisomerase inhibitors include camptothecins: irinotecan and topotecanCitation45–47.
Type II inhibitors include amsacrine, etoposide phosphate and teniposide, aurintricarboxylic acidCitation48. These are semisynthetic derivatives of epipodophyllotoxins, alkaloids naturally occurring in the root of American Mayapple (Podophyllum peltatum), doxorubicin, idarubicin, epirubicin and mitoxantroneCitation49,Citation50.
3.1. Drug-targeting DNA topoisomerase I
3.1.1. Camptothecin
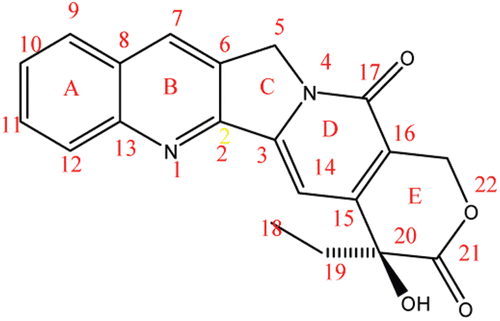
Camptothecin (CPT), (8), a quinoline alkaloid inhibits the DNA enzyme topoisomerases (topo I). CPT in mammalian cells has shown effective inhibitory action on nucleic acid synthesis and showed remarkable anticancer activity, but they suffer from drawbacks such as low solubility and adverse drug reaction. Because of these problems, medicinal chemists have developed numerous analogues of CPT, out of which two CPT analogues have been approved and are used in caner chemotherapyCitation51–54.
Figure 3. Camptothecin (8).
Apart from CPT, nucleus such as indolocarbazoles and indenoisoquinolines target human topoisomerase I (topo I). They bind with a brief topo I DNA covalent complex and shows inhibitory action on resealing of a single strand nick which the enzyme creates to aid superhelical tension in paired DNA. The topo I targeted drugs are S-phase specificCitation46,Citation55–57.
A, B, C, D and E ring modification: Substitutions of ethyl as well as chloromethyl at position 7 have increased the cytotoxicity. An increase in the length of carbon chain at 7th position resulted in increased lipophilic character. The presence of electron withdrawing groups like amino, nitro, bromo or chloro at position 9 and 10 and hydroxyl group at position 10 or 11, enhanced the activity. Methoxy group at both position 10 and 11 leads to inactiveCitation57,Citation58. The C and D rings have an important task in the antitumor activity. If molecules are replaced at any position of the C and D ring, it results in much less effective compound than parent compoundCitation58. The substitution of Cl, F or Br to hydroxyl group in the E ring results in no lose of activityCitation57. Extension of the lactone ring by one methylene unit results in homocamptothecin which shows improved activity. Another possible replacement is to insert a methylene unit between hydroxyl and lactone on the E ring which results in a seven membered β-hydroxylactone group. β-Hydroxylactone group is commonly known as homocamptothecin (hCPT)Citation57,Citation58.
Hexacyclic CPT analogues: Methylenedioxy or ethylenedioxy group connected between 10 and 11 position forms a 5- or 6-membered ring making them more water soluble derivates. An amino or chloro group at 9th position or chloromethyl group at 7th position shows greater cytotoxicity but low solubility in water. Lurtotecan is a compound with 10, 11-ethylenedioxy analogue with a 4-methylpiperazino-methylene at position 7 and shows a great potency in clinical researchesCitation80. A ring can also be formed between position 7 and 9, like position 10 and 11 and thus can lead to water soluble derivatives. When electron withdrawing groups are put in position 11 and methyl or amino groups at 10, these hexacyclic CPT becomes more active. Exatecan is an example of hexacyclic CPT that has a 6-membered ring over position 7 and 9, and is 10-methyl, 11-fluoro substituted. It is water-soluble and more potent than topotecanCitation57–59.
C and D ring modification: The C and D rings have an important role in the antitumour activity. If molecules are replacement at any position, it results in much less effective compound than parent compoundCitation58. Substitution at position 12 and 14 leads to inactive derivativeCitation57.
E ring modifications: In the E ring, changing the hydroxyl group to Cl, F or Br stabilize the enzyme complex because of their polarizability and results in no lose of activityCitation57. Enlargement of the lactone ring by one methylene unit results in homocamptothecin (hCPT) with increased activity. Another possible replacement is to insert a methylene unit between hydroxyl and lactone on the E ring, which results in a 7-membered β-hydroxylactone group. β-Hydroxylactone group is commonly known as hCPT. Lactone gets very reactive as the hCPT’s hydroxyl has less ability to produce inductive effect on the carboxyl group. This appreciates the free hydroxyl group and topo I interaction optimally and the covalent complex that forms in its presence are found to be more stableCitation57,Citation58.
3.1.2. CPT analogue
3.1.3. HomocamptothecinsCitation60
hCPTs has showed effective action better plasma stability when compared with CPTs. Compound BN80765 and its difluoro analogue diflomotecan (DN80915) are powerfully provoking apoptosis in tumour cells. Diflomotecan, a 9,10-difluoro-hCPTs derivative is one of the new members of the “tecan” family. hCPTs have more potent inhibitory action against Topo I as compared to camptothecins in terms of biochemical assays. The limitation of hCPTs is that they get permanently inactivated when the lactone E ring is opened. Ketonic derivatives which are active Topo I poisons are the second novel family of lactone derivative of CPTCitation55.
3.1.4. Novel non-camptothecin topoisomerase I inhibitorsCitation55
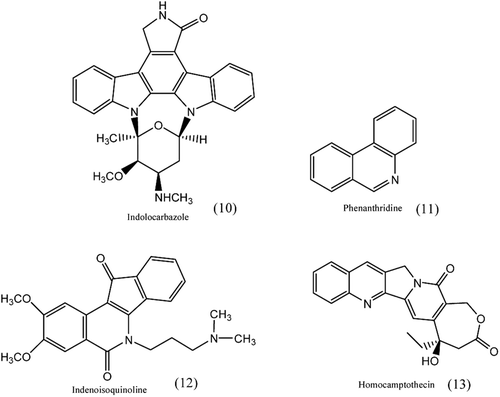
Non-camptothecin inhibitors which have structural difference from camptothecins include the indolocarbazoles, phenanthridines and indenoisoquinolinesCitation55,Citation61 as shown in . In recent times, three Topo I-DNA co-crystal structures: one with an indenoisoquinoline, one with an indolocarbazole and the other with the natural CPT bound to Topo I-DNA complexes have been observedCitation62.
Figure 4. Structure of non-camptothecin drugs.
3.1.5. Novel benzoxanthone analogues as non-camptothecin topoisomerase I inhibitorsCitation63
Structure modification of the side chain of the lead compound benzoxanthone resulted in moderate cytotoxicity against tumour cells. However, some derivatives have shown potent topoisomerase I (Topo I) inhibitory effect and indicated that these compounds have the potential for the development.
Emerging Topo I Inhibitor
3.1.6. 9-AC, CPT 11 and its derivatives
9-aminocamptothecin (9-AC) a water insoluble synthetic analogue of CPT, in preclinical studies showed effective against human tumour xenografts. Ongoing phase ICitation65 studies suggest that this drug has higher therapeutic index with prolonged administration.
CPT-11Citation64, (14), also known as irinotecan used for the treatment of lung, cervical and ovarian cancer. CPT-11 was recently approved in the USA for the treatment of colorectal cancerCitation45,Citation66. DX-8951f, (17), is a novel water-soluble derivative of CPT-11, and exhibits potent topo I inhibitory activity. DX8951f is undergoing clinical evaluation in the USA, Japan, and EuropeCitation45. BNP1350, structure is shown in is a lipophilic derivative of CPT-11 and has indicated promise of being active against head and neck tumoursCitation45.
Table 1. Eukaryotic and prokaryotic topoisomerases types and their function.
Table 2. Structure of CPT analogues.
Figure 5. Structures of novel topoisomerases I inhibitor.
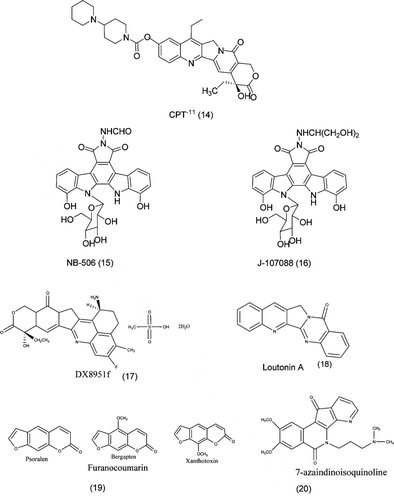
3.1.7. Indolocarbazole derivatives (NB-506 and J-107088)
NB-506, (15), an indolocarbazole derivative, is an effective topo I inhibitor and shows highly effective activity against tumour in vitro as well as in vivo. Cellular sensitivity is determined by cellular topo I activity and decreased topo ICitation67. Recently, a report suggested that an isomer of NB- 506 showed to be potent topo I inhibitor without an intercalating actionCitation68. J-107088, (16), is an indolocarbazole derivative having antitumour activity. Its antitumour activity is more effective than those of NB-506. J-107088 stabilizes the intracellular cleavable complex more strongly as compared to CPT-11. It shows highly effective antitumour action against multidrug-resistant cells, and an antimetastatic action has been demonstrated in a model of micrometastasis to the liverCitation69.
3.1.8. Luotonin A: topoisomerase poisoning activityCitation70
A series of A ring and E ring analogues of the natural product luotonin A, (18), a known topoisomerase I poison, was evaluated for growth inhibition in human carcinoma and leukemia cell lines. Rational design of structures was based on analogues of the related alkaloid camptothecin, which has been demonstrated to exert cytotoxic effects by the same mechanism of action.
3.1.9. Furanocoumarins: novel topoisomerase I inhibitors from Ruta graveolens L
Topoisomerase I inhibition from Ruta graveolensCitation71 were found to be potent and stabilization of DNA topoisomerase covalent complex was observed in all the tested extracts. The mechanism and affinity of topoisomerase inhibitors was determined by enzyme extract preincubation studies. The activity could be assigned to furanocoumarins, psoralen, bergapten and xanthotoxin, shown in (19) identifying them as novel, potent topoisomerase I inhibitors
3.1.10. 7-azaindenoisoquinolines: topoisomerase I inhibitors and potential anticancer agentsCitation72
A series of 7-azaindenoisoquinoline, (20), topoisomerase I (TopI) inhibitors have been investigated and the study showed that 7-azaindenoisoquinolines demonstrate improved water solubility without any decrease in TopI inhibitory activity or cytotoxicity. Analysis of the biological results reveals that smaller lactam ring substituents enable intercalation into both free DNA and TopI-DNA cleavage complex, whereas larger substituents only allow binding to the cleavage complex but not free DNA.
3.2. Drug targeting DNA topoisomerase II
A. Different classes of compounds target topoisomerase II
Drugs targeting topoisomerase II fall into two categories, Topo 2 poisons and Topo 2 catalytic inhibitors. These agents prepare lesions that include DNA strand breaks and protein covalently bound to DNA; they have been named Topo 2 poisons. Although second class of compounds inhibits Topo 2 catalytic activity it does not generate rise in the levels of Topo 2 covalent complexesCitation73,Citation74. Topo 2 poisons can be further sub-divided into intercalating and non-intercalating poisons. The intercalators are chemically diverse, and include doxorubicin and other anthracyclines, mitoxantrone, mAMSA, and a variety of other compounds that are not currently in clinical use such as amonafide and ellipticineCitation75–77.
Non-intercalating Topo 2 poisons include the epipodophyllotoxins etoposide and teniposide, and fluoroquinolones, which are mainly active against prokaryotic type II topoisomerases. Since non intercalating Topo 2 poisons do not interact strongly with DNA, it has been suggested that protein drug interactions play key roles in their ability to trap Topo 2 covalent complexes. Several classes of compounds have been described that inhibit Top 2 activity but do not increase DNA cleavageCitation78,Citation79.
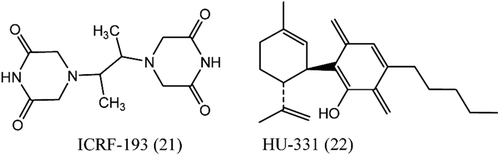
ICRF-193 (bisdioxopiperazine) and HU-331
Catalytic inhibitors which interfere with ATPase and DNA strand passage reactions leads to stabilization of the closed clamp form of the enzyme. ICRF-187, (21), a bisdioxopiperazineCitation73,Citation80. Targets Topo 2 along with novobiocinCitation81, merbaroneCitation82, and the anthracycline aclarubicin. Membrane has attracted interest because it is the only agent that has been found to inhibit Topo 2 cleavage of DNA but not affect protein: DNA bindingCitation83. HU-331, (22), synthesized from cannabidiol is a quinone anticarcinogenic drug. Cannabidiol, a cannabinoid showed a great efficacy against oncogenic human cells. HU-331 does not cause blockage in cell cycle, cell apoptosis or caspase activation. HU-331 shows inhibitory action against DNA topoisomerase II at nanomolar concentrations, but has no effective action on the action of DNA topoisomerase I. HU-331 has a very specific inhibitory action on topoisomerase II, compared with most known anticancer quinonesCitation84,Citation85.
Figure 6. Structure of ICRF-193 (bisdioxopiperazine) and HU-331.
B. Intercalating topoisomerase-targeting drugs
Anthracene and its derivatives
The anthracyclines, (23), are a fermentation product of Streptomyces peucetius var. caesius which is planar, hydrophobic tetracycline ring linked to a daunosamine sugar through a glycosidic linkageCitation86,Citation87. These compounds constitute anthracyclines also intercalate into double-stranded DNA to produce structural differences that interfere with DNA and RNA syntheses and are used for solid and hematologic malignanciesCitation88. Mitoxantrone is an anthracenedione that targets topoisomerase II and is the only agent of its class approved for clinical purpose. Mitoxantrone lacks the ability to form the quinone type free radicals thought to account for anthracycline cardiotoxicityCitation89. Glycoside substituents are absent in anthracenediones and is shown in . Unlike the anthracyclines, mitoxantrone has less possibility to participate in one electron reduction reactions and hence is less effective in forming free radicals90.Citation90.
Figure 7. Structures of intercalating topo targeting drugs.
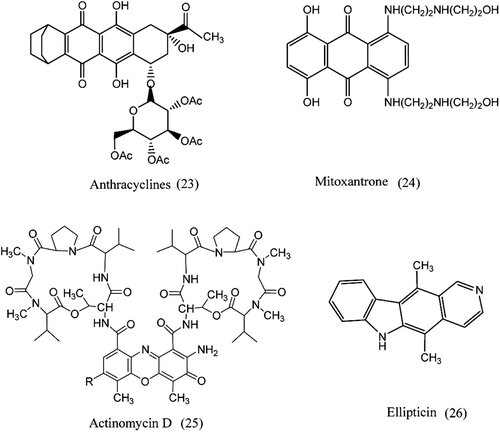
Actinomycin D (Dactinomycin; Cosmegen) and Ellipticin
The first antibiotics Actinomycins, (25), were isolated from the culture broth of StreptomycesCitation91. They consist of a planar phenoxazone actinocin ring joined to two peptide side chains. X-ray crystallographic and NMR studies showed that the interaction between actinomycin D and DNA takes place between the drug and deoxyguanosineCitation92. Ellipticin is an eukaryotic, (26), topoisomerase II inhibitors, thought to act as a poisons of the enzyme, intercalating into the DNA and increasing the cleavage reaction leading to an accumulation of cytotoxic DNA breaks in cellsCitation46.
C. Nonintercalating topoisomerase-targeting drugs
1. Epipodophyllotoxins
The epipodophyllotoxins, (27), were synthesized with a view to improve the activity of podophyllotoxinCitation67, an antimicrotubule agent. The epipodophyllotoxins etoposide (VP-16) and teniposide (VM-26) studies showed that cells treated with these drugs exhibited DNA strand breaks, topoisomerase II was not recognised as the target in the early 1980sCitation45,Citation93,Citation94. The pendant aromatic ring of epipodophyllotoxins is important for activity; changes in either the constituents of this ring or the stereochemical relationship between this ring and the tetracyclic ring system can hugely affect the topoisomerase poisoning activity of the drugs. The planar polycyclic component of epipodophyllotoxins stacks into the cleavage site of topoisomerase II–DNA complexesCitation93.
Figure 8. Structure of non-intercalating targeting drug.
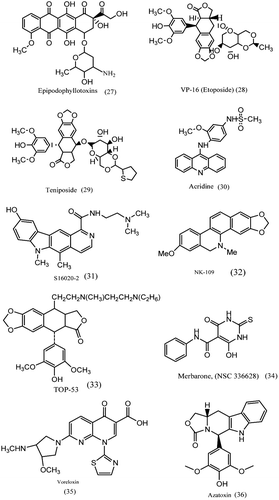
2. VP-16 (etoposide) and etoposide phosphate
Etoposide, (28), and teniposide are used on a large scale as a principle drug to treat various solid tumours. Etoposide being a typical topo II inhibitor stabilizes the cleavable complex formation with DNA topo II without an intercalating action. Etoposide with cisplatin exhibits a synergistic effect against lung cancer both in vitro and in vivo, and the modulatory effect of cisplatin on topo II activity is indicated to be the reason for the synergistic antitumour effectCitation45,Citation48,Citation93–95.
3. Teniposide
Teniposide, (29), is approved for the treatment of refractory childhood acute lymphocytic leukemia and gliomas. In teniposide, the methyl group present on the glucose moiety of etoposide is replaced by thiophene. This modification affects both the interaction with topoisomerase II and their clinical pharmacology. Teniposide (VM-26) has been used in the treatment of small cell lung cancer, malignant lymphoma, breast cancerCitation96.
4. Acridine: (mAMSA)
Acridine is eukaryotic topoisomerase II inhibitors, (30). Thought to act as poisons of the enzyme, stabilizing the covalent complex between DNA and enzyme leading to an accumulation of cytotoxic DNA breaks in cellsCitation97.
5. NK-109
The benzophenanthridine derivative NK109, (32), induces single and double strand breaks without the formation of cleavable complex. They showed high antitumour activity against etoposide resistant cells and was in phase I clinical evaluation in JapanCitation45.
6. TOP-53
TOP-53, (33), is a podophyllotoxin derivative and has potent topo II inhibition. TOP-53 is considerably more toxic to non-small cell lung cancer cells, is more active at generating chromosomal breaks, and displays improved cellular uptake and pharmacokinetics in animal lung tissuesCitation98.
7. S16020-2
S16020-2, (31), possesses the unique characteristic of inhibiting topo II in a strictly ATP dependent manner. Cells opposing this agent have been selected to check the cytotoxic mechanisms and very potent antitumour activity has been exhibitedCitation45.
8. Merbarone
Merbarone, (NSC 336628), (34), a derivative of barbituric acid and represents a sole class of antineoplastic agents. Merbarone exhibited activity against L1210 leukemia and other murine tumours. A brief study of the different steps in the catalytic cycle shows that merbarone has no effect on either DNA binding or ATP hydrolysis but it is a potent inhibitor of enzyme mediated DNA cleavage. More recently, colony formation assays show that both inhibitors, merbarone and ICRF-187 are able to significantly potentiate the cytotoxicity of the alkylating agent melphalanCitation48,Citation82.
9. Voreloxin
Voreloxin, (35), is a quinolone derivative in phase 2 clinical trials in AML and platinum resistant ovarian cancer. Voreloxin intercalates DNA and poisons topoisomerase II. The structural similarity between the prototype fluoroquinolone antibacterial like ciprofloxacin and voreloxin has helped in understanding the mechanistic studies with other classes of agents in clinical use that cause DNA damage by interfering with topoisomerase II functionCitation99.
10. Azatoxin Is a mechanistic hybrid of the topoisomerase II-targeted anticancer drugs etoposide and ellipticineCitation100
Azatoxin, (36), a hybrid drug that fuses chemical structures from etoposide and ellipticine emerged as of topoisomerase II-targeted anticancer agents. Azatoxin were characterized to determine relationships between its mechanism of action and those of its parent compounds. Azatoxin, like etoposide, binds to DNA in a nonintercalative fashion. The competition studies indicate that azatoxin interacts with topoisomerase II in the enzyme domain utilized by etoposide and ellipticineCitation100.
3.3. Drug with heterocyclic moiety
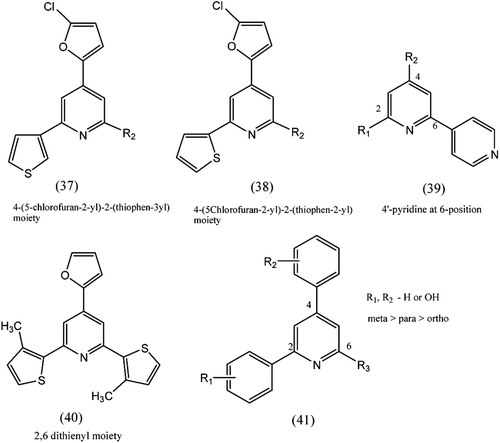
1. 2, 4, 6-trisubstitued pyridineCitation101–105
A series of pyridines substituted at 2, 4, and 6 positions with various 5- or 6-memberd heteroaromatics shows antitumour agents. Structure–activity relationship study revealed that 4-(5-chlorofuran-2-yl)-2-(thiophen-3-yl) moiety, (37), was important to display significant topo II inhibitory activity. 4-(5Chlorofuryl)-2-(thiophen-2-yl) moiety, (38), displayed moderate topo I and II inhibitory activity. When position 2,6 substituted with thienyl group, (40), exhibited strong topoisomerase I inhibitory activity. The substitution of hydroxyl group(s) increased topoisomerase I and II inhibitory activity in the order of meta > para > ortho position. Substitution of hydroxyl group on the para position showed better cytotoxicity. Structure activity relationship study indicates that 4′-pyridine at 6-position, (39), of central pyridine plays a key role in biological activity.
Figure 9. 2,4,6 trisubstitued pyridine moiety.
2. Tetracyclic thienopyridonCitation106
A series of 4-oxopyrido[3′,2′:4,5]thieno[3,2-b]indole-3-carboxylic acids, (42), was tested for in vitro antimicrobial as well as antiproliferative activity. Four compounds among the series exhibited very high potency against gram positive Bacillus subtilis and Bacillus megaterium and showed excellent activity against Gram positive bacilli and staphylococci and Gram negative Haemophilus influenzae. Two compounds were found to be inhibitors of DNA gyrase, but some compounds showed growth inhibition of MCF-7 breast tumour and A549 non-small cell lung cancer cells with IC50 1.6–2.8 μM and 2.6–6.9 μM, respectively. These compounds are promising as dual acting chemotherapeutics.
Figure 10. Structures of tetracyclic moieties.
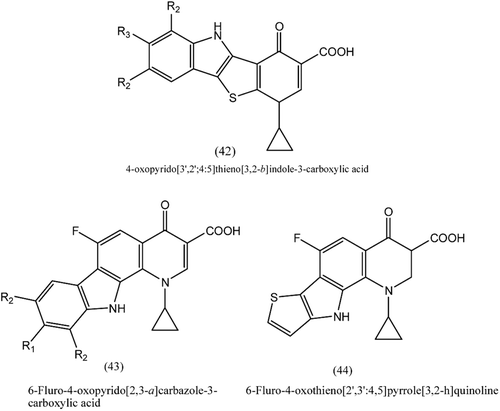
3. Tetracyclic fluoroquinolonesCitation107
A series of compounds belonging to 6-fluoro-4-oxopyrido [2,3-a]carbazole-3-carboxylic acids, (43), and 6-fluoro-4-oxothieno[2′,3′:4,5]pyrrolo[3,2-h]quinoline, (44), showed inhibitory activity on DNA gyrase and topoisomerase IV. The thieno isosteres was found to be most active antibacterial, where as the 9 fluoro derivative was potent against multidrug-resistant staphylococci. One of the derivatives was the most active anticancer against MCF-7 cells, with greater potency than ellipticine. The most active compounds in this series show promise as dual acting anticancer and antibacterial chemotherapeutics.
4. Glycosylated diphyllin derivatives as topoisomerase II inhibitorsCitation108
A novel glycosylated diphyllin (45) derivative showed potent anticancer activity by targeting topoisomerase IIα. The SAR study revealed that the sugar moiety on the diphyllin (46) is essential for the anticancer activity and equatorial C4′-OH on the sugar is superior to the axial one and a proper cyclic lipophilic group at the C4′ and C6′ of sugar might enhance the anticancer activity.
5. Natural products inhibitors
A natural products GUT-70, its synthetic analogue BNS-22, showed antiproliferative activity against human cancer cells. The BNS-22 belongs to the same cluster as ICRF-193 and shows DNA topoisomerase II (Topo2) catalytic inhibitor. BNS-22 does not affect DNA damage and antagonizes Topo2 poison-mediated DNA damage. Like ICRF-193, BNS-22 induces mitotic abnormalities and causing polyploidy in HeLa cellsCitation109. These results showed that BNS-22 targets Topo2 and acts as catalytic inhibitor. Resveratrol oligomers, Nepalensinol A, B and C, (47–49), from the stem of Kobresia nepalensis (Cyperaceae). C showed a potent inhibitory effect on topoisomerase II–stronger than etoposide (VP-16), Nepalensinol B, in particular, exhibited the most potent activity with an IC50 of 0.02 lg/mLCitation110. The flavones might interact with the DNA-topoisomerase I complex after their oxidation into quinones via autoxidation, enzymatic oxidation, or reactions with reactive oxygen speciesCitation111 ().
Table 3. Structures of flavones.
Figure 11. Structure of glycosylated diphyllin derivative.
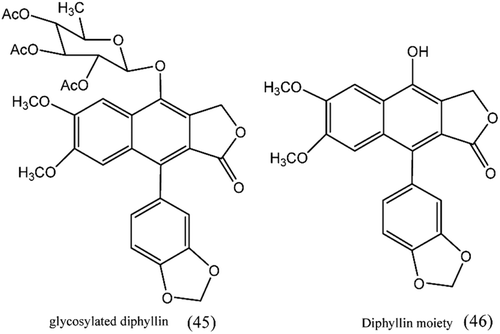
Figure 12. Structures of nepalensinol-topo II inhibitor.

3.4. Dual targeting agents
1. Dual topoisomerase I/II inhibitors in cancer therapyCitation112
Drugs bind to DNA by intercalation such as acridine DACA (50), benzopyridoindole intoplicine, indenoquinolinone TAS-103 (51), benzophenazine XR11576, and pyrazoloacridine NSC 366140 as show in (52) are the first class. The second category consists of hybridised molecules of different inhibitors belonging to topo I and topo II activity. The third category is less well defined as a structural class, but apparently identified structural motifs that are found in both topo I and II enzymes. These comprise a series of benzoisoquinolinium quaternary salts such as NK 109, and more interestingly altered versions of classical topo I or topo II inhibitors.
Figure 13. Dual targeting topoisomerases inhibitor.
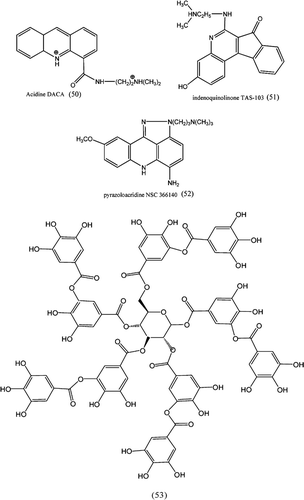
2. Flavonol glycosidesCitation113
Nine polyphenolic compounds derived from parts of Vicia faba and Lotus edulis of Leguminosae showed eukaryotic topoisomerases. The polyphenolic compounds, (53), exhibits catalytic inhibition of wheat germ topoisomerase I (IC50: 120–350 μM), human topoisomerase I (IC50): 110–260 μM), and human topoisomerase II (IC50: 240–600 μM) activities. The kaempherol glycoside with an acetyl group was found to be most powerful inhibitor. They also showed inhibition against human cancer cell lines MCF7, HeLa, and HepG2.
4. Conclusion
The annual global morbidity and mortality figures attributed to the risk factors arising from cancer are on rise despite a battery of drugs available in the therapy. So it is important to explore new potential targets for their treatment apart from the already existing targets and therapy. Development of additional topoisomerase inhibitors with mechanisms of action distinct from existing drugs will probably be necessary to achieve the target. Several attractive pharmacologic targets have been identified, and agents that act on those targets, when used alone or in combination, should be very effective and dual. A brief introduction of topoisomerase, types and drugs currently used for its treatment and control is described along with their subtypes and physiological roles are discussed. Currently existing agents, detailed literature on the discovery and evaluation of a variety of chemically diverse structures on various subtypes of topoisomerase has been reviewed.
Acknowledgments
The authors gratefully acknowledge the contributions of Prof. M. N. Navale, President, and Dr. (Mrs.) S. M. Navale, Secretary, Sinhgad Technical Education Society, Pune, for constant motivation and encouragement.
Declaration of interest
The authors report no conflicts of interest.
References
- Wang JC. Cellular roles of DNA topoisomerases: A molecular perspective. Nat Rev Mol Cell Biol 2002;3:430–440.
- Wang JC. DNA topoisomerases. Annu Rev Biochem 1996;65:635–692.
- Fritz Boege. Analysis of eukaryotic DNA topoisomerases and topoisomerase-directed drug effects. Eur J Clin Chem Clin Biochem 1996;34:873–888.
- Merino A, Madden KR, Lane WS, Champoux JJ, Reinberg D. DNA topoisomerase I is involved in both repression and activation of transcription. Nature 1993;365:227–232.
- Kretzschmar M, Meisterernst M, Roeder RG. Identification of human DNA topoisomerase I as a cofactor for activator-dependent transcription by RNA polymerase II. Proc Natl Acad Sci USA 1993;90:11508–11512.
- Zhang H, Wang JC, Liu LF. Involvement of DNA topoisomerase I in transcription of human ribosomal RNA genes. Proc Natl Acad Sci USA 1988;85:1060–1064.
- Lim M, Liu LF, Jacobson-Kram D, Williams JR. Induction of sister chromatid exchanges by inhibitors of topoisomerases. Cell Biol Toxicol 1986;2:485–494.
- Shuman S. Recombination mediated by vaccinia virus DNA topoisomerase I in Escherichia coli is sequence specific. Proc Natl Acad Sci USA 1991;88:10104–10108.
- Yeh YC, Liu HF, Ellis CA, Lu AL. Mammalian topoisomerase I has base mismatch nicking activity. J Biol Chem 1994;269:15498–15504.
- Stevnsner T, Bohr VA. Studies on the role of topoisomerases in general, gene- and strand-specific DNA repair. Carcinogenesis 1993;14:1841–1850.
- Nitiss JL. Roles of DNA topoisomerases in chromosomal replication and segregation. Adv Pharmacol 1994;29A:103–134.
- Earnshaw WC, Mackay AM. Role of nonhistone proteins in the chromosomal events of mitosis. FASEB J 1994;8:947–956.
- Earnshaw WC, Heck MM. Localization of topoisomerase II in mitotic chromosomes. J Cell Biol 1985;100:1716–1725.
- Earnshaw WC, Halligan B, Cooke CA, Heck MM, Liu LF. Topoisomerase II is a structural component of mitotic chromosome scaffolds. J Cell Biol 1985;100:1706–1715.
- Jorge BS, Andrzej S. A topological view of the replicon. EMBO reports 2004;5:256–261.
- Losada A, Hirano T. Shaping the metaphase chromosome: Coordination of cohesion and condensation. Bioessays 2001;23:924–935.
- Paul MW, Ian DH. Structure and function of type 11 DNA topoisomerases. Biochem J 1994;303:681–695.
- D’Arpa P, Machlin PS, Ratrie H 3rd, Rothfield NF, Cleveland DW, Earnshaw WC. cDNA cloning of human DNA topoisomerase I: Catalytic activity of a 67.7-kDa carboxyl-terminal fragment. Proc Natl Acad Sci USA 1988;85:2543–2547.
- Juan CC, Hwang JL, Liu AA, Whang-Peng J, Knutsen T, Huebner K et al. Human DNA topoisomerase I is encoded by a single-copy gene that maps to chromosome region 20q12-13.2. Proc Natl Acad Sci USA 1988;85:8910–8913.
- Lima CD, Wang JC, Mondragón A. Three-dimensional structure of the 67K N-terminal fragment of E. coli DNA topoisomerase I. Nature 1994;367:138–146.
- Li TK, Liu LF. Tumor cell death induced by topoisomerase-targeting drugs. Annu Rev Pharmacol Toxicol 2001;41:53–77.
- David SG. The molecular perspective: DNA topoisomerases 2002;7:381–382.
- David FF, Clemencia P, Anca MS. New peptide inhibitors of type IB topoisomerases: Similarities and differences inhibitors of tyrosine recombinases. J Mol Biol 2006;363:891–907.
- Taneja B, Patel A, Slesarev A, Mondragón A. Structure of the N-terminal fragment of topoisomerase V reveals a new family of topoisomerases. EMBO J 2006;25:398–408.
- Malte W, Etienne W, Alfred ME. Combining EL4-B5-based B-cell stimulation and phage display technology for the successful isolation of human anti-Scl-70 autoantibody fragments. J Immun Method 2003;278:249–259.
- Willmore E, de Caux S, Sunter NJ, Tilby MJ, Jackson GH, Austin CA et al. A novel DNA-dependent protein kinase inhibitor, NU7026, potentiates the cytotoxicity of topoisomerase II poisons used in the treatment of leukemia. Blood 2004;103:4659–4665.
- Tan KB, Dorman TE, Falls KM, Chung TD, Mirabelli CK, Crooke ST et al. Topoisomerase II alpha and topoisomerase II beta genes: Characterization and mapping to human chromosomes 17 and 3, respectively. Cancer Res 1992;52:231–234.
- Liu LF, Liu CC, Alberts BM. Type II DNA topoisomerases: Enzymes that can unknot a topologically knotted DNA molecule via a reversible double-strand break. Cell 1980;19:697–707.
- Edward G, Karl D. Regulation of bacterial DNA supercoiling: Plasmid linking numbers vary with growth temperature (DNA topology/gel electrophoresis), Biochemistry 1984;81:4046–4050.
- Raji A, Zabel DJ, Laufer CS, Depew RE. Genetic analysis of mutations that compensate for loss of Escherichia coli DNA topoisomerase I. J Bacteriol 1985;162:1173–1179.
- Wang JC. Interaction between DNA and an Escherichia coli protein omega. J Mol Biol 1971;55:523–533.
- Tse-Dinh YC, Wang JC. Complete nucleotide sequence of the topA gene encoding Escherichia coli DNA topoisomerase I. J Mol Biol 1986;191:321–331.
- Yu CX, Zhu YC, Tse-Dinh Fesik, SW. Solution structure of the C-terminal single-stranded DNA-binding domain of E. coli topoisomerase I. Biochemistry 1995;34:7622–7628.
- David CH. Bacterial topoisomerases, anti topoisomerases, and anti topoisomerase resistance. Clin Inf Diseases 1998;27 (Suppl 1):S54–63.
- Changela A, DiGate RJ, Mondragón A. Structural studies of E. coli topoisomerase III-DNA complexes reveal a novel type IA topoisomerase-DNA conformational intermediate. J Mol Biol 2007;368:105–118.
- Joseph AH. DNA topoisomerases as anticancer drug targets: From the laboratory to the clinic. Curr Med Chem Anticancer Agents 2001;1:1–25.
- Kato J, Suzuki H, Ikeda H. Purification and characterization of DNA topoisomerase IV in Escherichia coli. J Biol Chem 1992;267:25676–25684.
- Peng H, Marians KJ. Escherichia coli topoisomerase IV. Purification, characterization, subunit structure, and subunit interactions. J Biol Chem 1993;268:24481–24490.
- Maxwell A. DNA gyrase as a drug target. Trends Microbiol 1997;5:102–109.
- Schmutz E, Mühlenweg A, Li SM, Heide L. Resistance genes of aminocoumarin producers: Two type II topoisomerase genes confer resistance against coumermycin A1 and clorobiocin. Antimicrob Agents Chemother 2003;47:869–877.
- Nelson JM, Chiller TM, Powers JH, Angulo FJ. Fluoroquinolone-resistant Campylobacter species and the withdrawal of fluoroquinolones from use in poultry: A public health success story. Clin Infect Dis 2007;44:977–980.
- Ivanov DV, Budanov SV. Ciprofloxacin and antibacterial therapy of respiratory tract infection. Antibiot Khimioter 2006;51:29–37.
- Fisher LM, Pan XS. Methods to assay inhibitors of DNA gyrase and topoisomerase IV activities. Methods Mol Med 2008;142:11–23.
- Gore J, Bryant Z, Stone MD, Nöllmann M, Cozzarelli NR, Bustamante C. Mechanochemical analysis of DNA gyrase using rotor bead tracking. Nature 2006;439:100–104.
- Yasuhiro K, Kazuto N. Mechanisms of action of cancer chemo therapeutic agents: Topoisomerase inhibitors. John Wiley & Sons, Ltd. 2002:2–7.
- Joyce M, Nirmala A. Samundeeswari, Deepa Sankar, P. Natural plant resources in anti-cancer therapy-A review. Research in Plant Biology 2011;1:01–14.
- Chalet Tan. Topoisomerases inhibitor. PHA 557. HemeOnc, 2009.
- Benchokroun Y, Couprie J, Larsen AK. Aurintricarboxylic acid, a putative inhibitor of apoptosis, is a potent inhibitor of DNA topoisomerase II in vitro and in Chinese hamster fibrosarcoma cells. Biochem Pharmacol 1995;49:305–313.
- Devendra ST. Topoisomerase II inhibitors in cancer treatment. Int J Pharm Sci Nanotechnology 2011;3:1174–1179.
- Ferrer S, Marce B, Bellosillo N, Villamor F. Activation of mitochondiral apoptotic pathway in mantel cell lymphoma: High sensitivity to mitoxantrone in cases with functional DNA-damage response genes. Oncogene 1991;23:8941–8949.
- Felix CA. Leukemias related to treatment with DNA topoisomerase II inhibitors. Med Pediatr Oncol 2001;36:525–535.
- Takimoto CH, Calvo E, Pazdur R, Wagman LD, Camphausen KA, Hoskins WJ (eds.) “Principles of Oncologic Pharmacotherapy” in Cancer Management: A Multidisciplinary Approach, 11th edn. 2008.
- Wall ME, Wani MC, Cook CE, Palmer KH, McPhail AI, Sim GA. Plant antitumor agents. I. The isolation and structure of camptothecin, a novel alkaloidal leukemia and tumor inhibitor from camptotheca acuminate. J Am Chem Soc 1966;88:3888–3890.
- Mi Z, Malak H, Burke TG. Reduced albumin binding promotes the stability and activity of topotecan in human blood. Biochemistry 1995;34:13722–13728.
- Barth, Stephan, Briviba, Esselen. In vivo bioassay to detect irinotecan-stabilized DNA/topoisomerase I complexes in rats. Biotechnology Journal Wiley-VCH 2009:2–26.
- Yves P. Camptothecins and topoisomerase I: A Foot in the Door, p. 1.
- Adams DJ. The impact of tumor physiology on camptothecin-based drug development. Curr Med Chem Anticancer Agents 2005;5:1–13.
- Zunino F, Dallavalleb S, Laccabuea D, Berettaa G, Merlinib L, Pratesi G. Current status and perspectives in the development of camptothecins. Curr Pharm Des 2002;8:2505–2520.
- Ulukan H, Swaan PW. Camptothecins: A review of their chemotherapeutic potential. Drugs 2002;62:2039–2057.
- Chung MK, Han SS, Kim JC. Evaluation of the toxic potentials of a new camptothecin anticancer agent CKD-602 on fertility and early embryonic development in rats. Regul Toxicol Pharmacol 2006;45:273–281.
- Christian B. Homocamptothecins: Potent topoisomerase I inhibitors and promising anticancer drugs. Critical Reviews in Oncology/Hematology 2003;45:91–108.
- Meng LH, Liao ZY, Pommier Y. Non-camptothecin DNA topoisomerase I inhibitors in cancer therapy. Curr Top Med Chem 2003;3:305–320.
- Antony S, Jayaraman M, Laco G, Kohlhagen G, Kohn KW, Cushman M, Pommier Y. Progress in nucleic acid research and molecular biology. Cancer Res 2003;63:7428.
- Cheng P, Zhu L, Guo W, Liu W, Yao J, Dong G, Zhang Y, Zhuang C, Sheng C, Miao Z, Zhang W. Synthesis of novel benzoxanthone analogues as non Camptothecin. J Enzyme Inhib Med Chem 2011;20:1–5.
- Gerrits CJ, de Jonge MJ, Schellens JH, Stoter G, Verweij J. Topoisomerase I inhibitors: The relevance of prolonged exposure for present clinical development. Br J Cancer 1997;76:952–962.
- Moore MJ, Siu LL, Oza A et al. 9-aminocamptothecin (9AC) given by 24 hour infusion once weekly. A phase I pharmacokinetic (PK) and pharmacodynamic (PD) study. Proc Am Soc Clin Oncol 1997;16:200a.
- Reginald B., Ewesuedo, Mark J., Ratain. Topoisomerase I inhibitors. The Oncologist 1997;2:359–364.
- Kanzawa F, Nishio K, Kubota N, Saijo N. Antitumor activities of a new indolocarbazole substance, NB-506, and establishment of NB-506-resistant cell lines, SBC-3/NB. Cancer Res 1995;55:2806–2813.
- Ren J, Bailly C, Chaires JB. NB-506, an indolocarbazole topoisomerase I inhibitor, binds preferentially to triplex DNA. FEBS Lett 2000;470:355–359.
- Ohkubo M, Nishimura T, Honma T, Nishimura I, Ito S, Yoshinari T et al. Synthesis and biological activities of NB-506 analogues: Effects of the positions of two hydroxyl groups at the indole rings. Bioorg Med Chem Lett 1999;9:3307–3312.
- Kassoum N, Conxiang CZ, Peter R, Guzzo R, Jason H, Denise P, Thomas DF. Synthesis and topoisomerase poisoning activity of A-ring and E-ring substituted luotonin A derivatives. Bioorg Med Chem, 2007;15:4237–4246.
- Renuka D, Nutan M. Furanocoumarins: Novel topoisomerase I inhibitors from Ruta graveolens L. Bioorg Med Chem 2009;17:7052–7055.
- Kiselev E, DeGuire S, Morrell A, Agama K, Dexheimer TS, Pommier Y et al. 7-azaindenoisoquinolines as topoisomerase I inhibitors and potential anticancer agents. J Med Chem 2011;54:6106–6116.
- John LN. Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 2009;9:338–350.
- Stacie J. Froelich-Ammon and Neil Osheroff. Topoisomerase poisons: Harnessing the dark side of enzyme mechanism. J Biol Chem 270:21429–21432.
- Nitiss JL, Beck WT. Antitopoisomerase drug action and resistance. Eur J Cancer 1996;32A:958–966.
- Gewirtz DA. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem Pharmacol 1999;57:727–741.
- Ferreira AL, Matsubara LS, Matsubara BB. Anthracycline-induced cardiotoxicity. Cardiovasc Hematol Agents Med Chem 2008;6:278–281.
- Roca J, Ishida R, Berger JM, Andoh T, Wang JC. Antitumor bisdioxopiperazines inhibit yeast DNA topoisomerase II by trapping the enzyme in the form of a closed protein clamp. Proc Natl Acad Sci USA 1994;91:1781–1785.
- Tanabe K, Ikegami Y, Ishida R, Andoh T. Inhibition of topoisomerase II by antitumor agents bis(2,6-dioxopiperazine) derivatives. Cancer Res 1991;51:4903–4908.
- Patrick C, Joëlle R, Joseph S. Catalytic inhibition of topoisomerase II by a novel rationally designed ATP-competitive purine analogue. BMC Chem Biol 2009;9:1.
- Collins A. Topoisomerase II can relax; novobiocin is a mitochondrial poison after all. Bioessays 1990;12:493–494.
- Drake FH, Hofmann GA, Mong SM, Bartus JO, Hertzberg RP, Johnson RK et al. In vitro and intracellular inhibition of topoisomerase II by the antitumor agent merbarone. Cancer Res 1989;49:2578–2583.
- Chène P, Rudloff J, Schoepfer J, Furet P, Meier P, Qian Z et al. Catalytic inhibition of topoisomerase II by a novel rationally designed ATP-competitive purine analogue. BMC Chem Biol 2009;9:1.
- Natalya MK, Michael S, Esther P, Ruth R, Eduard B, Mordechai C, Raphael M. HU-331, a novel cannabinoid-based anticancer topoisomerase II inhibitor. Mol cancer Ther 2007;6:173–183.
- Natalya MK, Michael S, Maximilian P, Gergana M, Ronen B, Raphael M. A cannabinoid anticancer quinone, HU-331, and less is more powerful than cardiotoxic doxorubicin: A comparative in vivo study. J Pharmacol Exp Ther 2007;322:646–653.
- Zunino F, Gambetta R, DiMarco A, Luoni G, Zaccara A. Effects of the stereochemical configuration on the interaction of some daunomycin derivatives with DNA. Biochem Biophys Res Commun 1976;69:744–750.
- Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C et al. Adriamycin, 14-hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol Bioeng 1969;11:1101–1110.
- Sinha BK, Katki AG, Batist G, Cowan KH, Myers CE. Differential formation of hydroxyl radicals by adriamycin in sensitive and resistant MCF-7 human breast tumor cells: Implications for the mechanism of action. Biochemistry 1987;26:3776–3781.
- Faulds D, Balfour JA, Chrisp P, Langtry HD. Mitoxantrone. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic potential in the chemotherapy of cancer. Drugs 1991;41:400–449.
- Crespi MD, Ivanier SE, Genovese J, Baldi A. Mitoxantrone affects topoisomerase activities in human breast cancer cells. Biochem Biophys Res Commun 1986;136:521–528.
- Waksman SA, Woodruff HB. Bacteriostatic and bactericidal substances produced by a soil actinomyces. Proc Soc Exp Biol Med 1940;45:609.
- Yu C, Tseng YY. NMR study of the solution conformation of actinomycin D. Eur J Biochem 1992;209:181–187.
- Eric HR, William NH. (2000).Chapter 49 Anthracyclines and DNA Intercalators/Epipodophyllotoxins/DNA Topoisomerases, Cancer Medicine 5th edition. Hamilton, Ontario, Canada: BC Decker Inc.
- Gordaliza M, García PA, del Corral JM, Castro MA, Gómez-Zurita MA. Podophyllotoxin: Distribution, sources, applications and new cytotoxic derivatives. Toxicon 2004;44:441–459.
- Amy MW, Ryan PB, Murrell G, Sungjo C. Topoisomerase II-drug interaction domains: Identification of substituents on etoposide that interact with the enzyme. Biochemistry 2007;46:8217–8225.
- Jinzhong L, Wantao C, Ping Z, Ningyi L. Topoisomerase II trapping agent teniposide induces apoptosis and G2/M or S phase arrest of oral squamous cell carcinoma. World J Surg Oncol 2006;4:41.
- John DB, Eng SH. Two specific topoisomerase I inhibitors prevent replication of human cytomegalovirus DNA: An implied role in replication of the viral genome. J Virolog 1988;4797–4800.
- Jo Ann WB, Susan DC, Teruhiro U. DNA topoisomerase II as the target for the anticancer drug TOP-53: Mechanistic basis for drug action. Biochemistry 2001;40:712–718.
- Rachael EH, David ES, Jo Ann WB. Voreloxin is an anticancer quinolone derivative that intercalates DNA and poisons topoisomerase II, PLoS ONE 2010;5:1–10.
- Susan DC, Timothy LM, Neil O. Azatoxin is a mechanistic hybrid of the topoisomerase II-targeted anticancer drugs etoposide and ellipticine. Biochemistry 1997;36:13095–13101.
- Pritam T, Radha K, Hoyoung C, Jae HC, Eung-Seok L et al. Synthesis of 2-(thienyl-2-yl or -3-yl)-4-furyl-6-aryl pyridine derivatives and evaluation of their topoisomerase I and II inhibitory activity, cytotoxicity, and structure activity relationship. Bioorg Med Chem 2010;18:2245–2254.
- Arjun B, Pritam T, Radha K, Hoyoung C, Jae Hun Choi Eung-Seok Lee et al. 2,6-Dithienyl-4-furyl pyridines: synthesis, topoisomerase I and II inhibition, cytotoxicity, structure–activity relationship, and docking study. Bioorg Med Chem Lett 2010;20:42–47.
- Pritam T, Radha K, Uttam T, Yurngdong J, Eung-Seok L et al. 2-Thienyl-4-furyl-6-aryl pyridine derivatives: synthesis, topoisomerase I and II inhibitory activity, cytotoxicity, and structure–activity relationship study. Bioorg Med Chem 2010;18:377–386.
- Radha K, Pritam T, Mi JK, Tae CJ, Jung MN, Eung-Seok L et al. Synthesis, topoisomerase I and II inhibitory activity, cytotoxicity, and structure activity relationship study of hydroxylated 2,4-diphenyl-6-aryl pyridines. Bioorg Med Chem 2010;18:3066–3077.
- Arjun B, Pritam T, Radha K, Chong-Soon L, Eung-Seok L et al. 2,4,6-Trisubstituted pyridines: synthesis, topoisomerase I and II inhibitory activity, cytotoxicity, and structure–activity relationship. Bioorg Med Chem 2007;15:4351–4359.
- Al-Trawneh SA, El-Abadelah MM, Zahra JA, Al-Taweel SA, Zani F, Incerti M et al. Synthesis and biological evaluation of tetracyclic thienopyridones as antibacterial and antitumor agents. Bioorg Med Chem 2011;19:2541–2548.
- Al-Trawneh SA, Zahra JA, Kamal MR, El-Abadelah MM, Zani F, Incerti M et al. Synthesis and biological evaluation of tetracyclic fluoroquinolones as antibacterial and anticancer agents. Bioorg Med Chem 2010;18:5873–5884.
- Shi DK, Zhang W, Ding N, Li M, Li YX. Design, synthesis and biological evaluation of novel glycosylated diphyllin derivatives as topoisomerase II inhibitors. Eur J Med Chem 2012;47:424–431.
- Kawatani M, Takayama H, Muroi M, Kimura S, Maekawa T, Osada H. Identification of a small-molecule inhibitor of DNA topoisomerase II by proteomic profiling. Chem Biol 2011;18:743–751.
- Masashi Y, Ken-Ichiro H, Hiroshi H, Shogo I, Takuji H, Ken T et al. Stilbenoids of Kobresia nepalensis (Cyperaceae) exhibiting DNA topoisomerase II inhibition, Phytochemistry 2006;67:307–313.
- Bensasson RV, Zoete V, Jossang A, Bodo B, Arimondo PB, Land EJ. Potency of inhibition of human DNA topoisomerase I by flavones assessed through physicochemical parameters. Free Radic Biol Med 2011;51:1406–1410.
- Denny WA, Baguley BC. Dual topoisomerase I/II inhibitors in cancer therapy. Curr Top Med Chem 2003;3:339–353.