Abstract
Carbonic anhydrase inhibitors of primary sulfonamide type, RSO2NH2, have clinical applications as diuretics, antiglaucoma, antiepileptic, antiobesity and antitumor drugs. Here we investigated inhibition of two human cytosolic isozymes, hCA I and II, with a series of secondary/tertiary sulfonamides, incorporating tosyl moieties (CH3C6H4SO2NR1R2). Most compounds inhibited both isoforms in low micromolar range, with inhibition constants between 0.181–6.01 μM against hCA I, and 0.209–0.779 μM against hCA II, respectively. These findings point out that substituted benzenesulfonamides may be used as leads for generating interesting CAIs probably possessing a distinct mechanism of action compared to primary sulfonamides. Indeed, classical RSO2NH2 inhibitors bind in deprotonated form to the Zn(II) ion from the CA active site and participate in many other favorable interactions with amino acid residues lining the cavity. The secondary/tertiary sulfonamides cannot bind to the zinc due to steric hindrance and probably are accommodated at the entrance of the active site, in coumarin binding-site.
Introduction
Carbonic anhydrase (CA, EC 4.2.1.1) inhibitors (CAIs) of the primary sulfonamide type, such as acetazolamide, ethoxzolamide or dichlorphenamide, are clinically used for decades, for various classes of diuretics and systemically acting antiglaucoma agentsCitation1–5. The CAs are involved in important physiological and pathological functions, such as pH and CO2 homeostasis, respiration and transport of CO2/HCO3− between metabolizing tissues and the lungs, ion secretion in different tissues/organs and biosynthetic reactions (such as gluconeogenesis, lipogenesis and ureagenesisCitation1–8). Among the sixteen isoenzymes described up to now in mammals, CA I and CA II are present at high concentrations in the cytosol of erythrocytes and the gastrointestinal tract, and CA II has the highest turnover rate of all isoformsCitation9–14.
Carbonic anhydrase inhibitors (CAIs) or activators have several medical applications, such as in the treatment of glaucoma, as diuretics, in the management of several neurological disorders, including epilepsy, possibly in the treatment of Alzheimer’s disease, whereas several agents are in clinical evaluations as antiobesity or antitumor drugs/diagnostic toolsCitation1–7.
A class of derivatives which showed very promising applications among the various CAIs reported by Supuran’s group in the last years, were the thioureas obtained from isothiocyanato sulfonamides (such as e.g. 4-isothiocyanatobenzenesulfonamide) and amines, hydrazines or amino acidsCitation15–17. Such compounds generally showed potent inhibitory activity against the cytosolic isozyme hCA II as well as the transmembrane, tumor-associated isozyme hCA IX, being thus interesting candidates for developing anti-glaucoma/antitumor therapies based on themCitation15–18. Many sulfonamide derivatives have been widely used as pro-drugs or drugs, as shown in for some clinically used/investigational primary sulfonamide CAIs. For instance, sulfadiazine is used as an antibiotic, sulfapyridine is mainly used for treatment of bacterial infections, acetazolamide is mainly used as an anti-glaucoma agentCitation1–5. However, all potent sulfonamide CAIs investigated till recently were of the primary type, i.e. RSO2NH2Citation1.
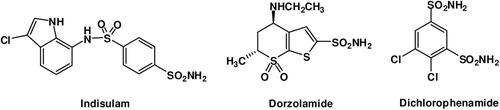
Figure 1. Some clinically used sulfonamides (dorzolamide and dichlorophenamide) or agents in clinical development (indisulam).
Our groups recently investigated the interaction of several CA isozymes with alternative chemotypes, such as salicylic acid derivatives, antioxidant phenolic compounds, organic nitrates, etcCitation9–13,Citation18. We would like to extend these earlier investigations to some secondary/tertiary sulfonamide derivatives, as this class of compounds was under investigated till recentlyCitation1,Citation3.
In this work we evaluated CA inhibition with tosyl pyrroles and other aromatic benzenesulfonamide derivatives which incorporate tosyl moieties. A series of 18 such compounds was evaluated for their ability to inhibit the dominant cytosolic human (h) isoforms, hCA I and II.
Materials and methods
Chemistry
Compounds 1–19 investigated here were reported earlierCitation3,Citation12.
Enzymology: CO2 hydration assay method
An applied photophysics stopped flow instrument has been used for assaying the CA-catalyzed CO2 hydration activityCitation19. Phenol red (at a concentration of 0.2 mM) has been used as indicator, working at the absorbance maximum of 557 nm, with 20 mM Hepes (pH 7.5) as buffer, and 20 mM Na2SO4 (for maintaining constant the ionic strength), following the initial rates of the CA-catalyzed CO2 hydration reaction for a period of 10–100 s. The CO2 concentrations ranged from 1.7 to 17 mM for the determination of the kinetic parameters and inhibition constants. For each inhibitor at least six traces of the initial 5–10% of the reaction have been used for determining the initial velocity, in triplicate measurements. The uncatalyzed rates were determined in the same manner and subtracted from the total observed rates. Stock solutions of inhibitor (0.1 mM) were prepared in distilled-deionized water and dilutions up to 0.01 nM were done thereafter with distilled-deionized water. Inhibitor and enzyme solutions were preincubated together for 15 min at room temperature prior to assay, in order to allow for the formation of the E-I complexCitation5,Citation19,Citation20. The curve-fitting algorithm allowed us to obtain the IC50 values, working at the lowest concentration of substrate of 1.7 mM, from which KI values were calculated by using the Cheng–Prusoff equationCitation21,Citation22.
Results and discussion
Diverse chemotypes of the primary sulfonamide one may show interest for the inhibition of the CAs, a superfamily of widely spread enzymes in organisms all over the tree of lifeCitation1. In a previous workCitation3 we have investigated some non-primary sulfonamides, such as derivatives 1–11, for their inhibition against some CA isoforms, by an esterase method with para-nitrophenyl acetate (NPA) as susbtrate. As NPA is not the physiologic CA substrate, and many isoforms show weak esterase activity, we re-evaluate here these compounds by monitoring their inhibition against the dominant isoforms hCA I and II, with a stopped-flow CO2 hydrase methodCitation19.
Recently, our groups also investigated the interactions of some methoxy-benzenesulfonamide derivatives and some of their substituted derivatives with different isozymes, such as hCA I, II and IXCitation22, evidencing several low micromolar/submicromolar inhibitors as well as the possibility to design isozyme selective CAIs. Indeed, the inhibition profile of various isozymes with this class of agents is very variable, with inhibition constants ranging from the millimolar to the submicromolar range for some methoxy-benzenesulfonamide or hydroxamate derivativesCitation22. All these data provide information on the fact that many non-primary sulfonamides may show significant CA inhibitory activity, and that the mechanism of action of such derivatives is different from that of the RSO2NH2derivativesCitation3,Citation22–29. It appeared thus of interest to extend the previous studiesCitation3, including in this research some benzenesulfonamides with clinical applications, such as sulfapyridine 18, sulfadiazine 19, as well as some of their substituted derivatives incorporating amino, cyano, methyl and nitro moieties as substituents at one of the aromatic ring in various different positions ().
Figure 2. Structures of tested compounds and the standard drug acetazolamide (AZA).
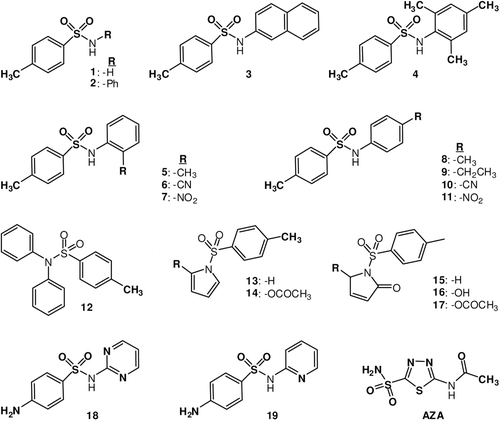
Tosyl pyrrole and benzenesulfonamides were synthesized by tosylation of pyrrole, aniline, o-methylaniline, p-methylaniline, trimethylaniline, diphenylamine, β-naphtylamine, p-ethylaniline, o-cyano-aniline, p-cyano-aniline, o-nitroaniline, p-nitroanilineCitation20. Inhibitory effects of compounds 1–19 and standard inhibitors on the enzyme activity were tested under in vitro conditions; KI values were calculated by using the Cheng–Prusoff equationCitation21 and are given in .
Table 1. hCA I and CA II inhibition data of the tested and reference compounds.
Data of show the following regarding inhibition of hCA I and II with compounds 1–19:
Against the slow cytosolic isozyme hCA I, compounds 2–12 and 14–17 behave as efficient inhibitors, with KI values in the range of 0.181–0.869 μM similar to the clinically used sulfonamide AZA (KI: 0.250 μM). Isoform hCA I was weakly inhibited by compound 13 which has a very different KI value (KI: 6.01 μM) from the structurally related derivative 14 (KI: 0.339 μM). Dorzolamide and dichlorophenamide were ineffective inhibitors for hCA I isoenzyme in this assay. The most powerful agent was compound 3, with KI values in the range of 0.181 μM. Derivatives 1, 18 and 19 were ineffective inhibitors for the isoenzyme, with inhibition constants in the range of 22.12–28.38 μM, similar to dorzolamide, a clinically used antiglaucoma agentCitation1.
Against the rapid cytosolic isozyme hCA II compounds 1–19 investigated here showed a moderate-weak inhibition, with KI values in the range of 0.276–0.779 μM (). Compound 13 was the weakest hCA II inhibitor, with a KI value of 0.779 μM, although it is not the bulkiest one among the investigated secondary/tertiary sulfonamides in the investigated series. It should be noted that the three clinically used/investigational compounds (AZA, indisulam, dorzolamide, dichlorophenamide, ) showed much more potent hCA II inhibitory activity compared to the secondary/tertiary sulfonamides investigated here, with KI-s in the range of 0.009–0.038 μM (). These findings clearly illustrate that a small variation in the structure of a sulfonamide CAI may have drastic consequences for the enzyme inhibitory activity and selectivity profile against various isozymes of such derivatives. Thus, all the secondary/tertiary sulfonamide derivatives 1–19 investigated here showed moderate hCA II inhibitory activity ().
Conclusions
We show here that compounds belonging to the secondary/tertiary sulfonamide class, incorporatring tosyl moeiteis, are inhibitors of hCA I and II in the submicromolar – micromolar range. These findings point out that substituted benzenesulfonamides may be used as leads for generating interesting CAIs probably possessing a distinct mechanism of action compared to the primary sulfonamides. Indeed, the classical RSO2NH2 inhibitors bind in deprotonated for to the Zn(II) ion from the CA active site and participate in many other favorable interactions with amino acid residues lining the cavity. The secondary/tertiary sulfonamides cannot bind to the zinc due to steric hindrance and probably are accommodated at the entrance of the active site, in the coumarin binding-siteCitation29–31. Indeed, coumarins and lactones/thiolactones were recently shown to be mechanism-based inhibitors, which bind in hydrolyzed state at the entrance of the active site cavity, without interference with the catalytic metal ionCitation29–32. As the entrance of the active site cavity is the most variable region of the various CA isoforms with therapeutic applicationsCitation1, this may lead to CAIs with a favourable inhibition profile and enhanced selectivity for the target over the offtarget isoforms.
Declaration of interest
Financial support for this research is provided in part by a 7th FP EU project (Metoxia, to CTS).
References
- Supuran CT. Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat Rev Drug Discov 2008;7:168–181.
- Hilvo M, Baranauskiene L, Salzano AM, Scaloni A, Matulis D, Innocenti A et al. Biochemical characterization of CA IX, one of the most active carbonic anhydrase isozymes. J Biol Chem 2008;283:27799–27809.
- Alp C, Ozsoy S, Alp NA, Erdem D, Gültekin MS, Küfrevioglu OI et al. Sulfapyridine-like benzenesulfonamide derivatives as inhibitors of carbonic anhydrase isoenzymes I, II and VI. J Enzyme Inhib Med Chem 2011; doi:10.3109 /14756366.2011.617745.
- Fabrizi F, Mincione F, Somma T, Scozzafava G, Galassi F, Masini E et al. A new approach to antiglaucoma drugs: carbonic anhydrase inhibitors with or without NO donating moieties. Mechanism of action and preliminary pharmacology. J Enzyme Inhib Med Chem 2012;27:138–147.
- Maresca A, Supuran CT. ®-/(S)-10-camphorsulfonyl-substituted aromatic/heterocyclic sulfonamides selectively inhibit mitochondrial over cytosolic carbonic anhydrases. Bioorg Med Chem Lett 2011;21:1334–1337.
- Güzel O, Innocenti A, Scozzafava A, Salman A, Parkkila S, Hilvo M et al. Carbonic anhydrase inhibitors: synthesis and inhibition studies against mammalian isoforms I-XV with a series of 2-(hydrazinocarbonyl)-3-substituted-phenyl-1H-indole-5-sulfonamides. Bioorg Med Chem 2008;16:9113–9120.
- Durdagi S, Sentürk M, Ekinci D, Balaydin HT, Göksu S, Küfrevioglu ÖI et al. Kinetic and docking studies of phenol-based inhibitors of carbonic anhydrase isoforms I, II, IX and XII evidence a new binding mode within the enzyme active site. Bioorg Med Chem 2011;19:1381–1389.
- Neri D, Supuran CT. Interfering with pH regulation in tumours as a therapeutic strategy. Nat Rev Drug Discov 2011;10:767–777.
- Senturk M, Ekinci D, Goksu S, Supuran CT. Effects of dopaminergic compounds on carbonic anhydrase isozymes I, II, and VI. J Enzym Inhib Med Chem 2012; doi:10.3109 /14756366.2011.591290.
- Ekinci D, Cavdar H, Talaz O, Sentürk M, Supuran CT. NO-releasing esters show carbonic anhydrase inhibitory action against human isoforms I and II. Bioorg Med Chem 2010;18:3559–3563.
- Ekinci D, Ceyhun SB, Sentürk M, Erdem D, Küfrevioglu OI, Supuran CT. Characterization and anions inhibition studies of an a-carbonic anhydrase from the teleost fish Dicentrarchus labrax. Bioorg Med Chem 2011;19:744–748.
- Alp C, Ekinci D, Gültekin MS, Sentürk M, Sahin E, Küfrevioglu OI. A novel and one-pot synthesis of new 1-tosyl pyrrol-2-one derivatives and analysis of carbonic anhydrase inhibitory potencies. Bioorg Med Chem 2010;18:4468–4474.
- Ekinci D, Ceyhun SB, Aksakal E, Erdogan O. IGF and GH mRNA levels are suppressed upon exposure to micromolar concentrations of cobalt and zinc in rainbow trout white muscle. Comp Biochem Physiol C Toxicol Pharmacol 2011;153:336–341.
- Ceyhun SB, Sentürk M, Ekinci D, Erdogan O, Ciltas A, Kocaman EM. Deltamethrin attenuates antioxidant defense system and induces the expression of heat shock protein 70 in rainbow trout. Comp Biochem Physiol C Toxicol Pharmacol 2010;152:215–223.
- Casini A, Scozzafava A, Mincione F, Menabuoni L, Ilies MA, Supuran CT. Carbonic anhydrase inhibitors (Part 99): Water soluble 4-sulfamoylphenylthioureas as topical intraocular pressure lowering agents with long lasting effects. J Med Chem 2000;43:4884–4892.
- Cecchi A, Winum JY, Innocenti A, Vullo D, Montero JL, Scozzafava A et al. Carbonic anhydrase inhibitors: synthesis and inhibition of cytosolic/tumor-associated carbonic anhydrase isozymes I, II, and IX with sulfonamides derived from 4-isothiocyanato-benzolamide. Bioorg Med Chem Lett 2004;14:5775–5780.
- Innocenti A, Casini A, Alcaro MC, Papini AM, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors: the first on-resin screening of a 4-sulfamoylphenylthiourea library. J Med Chem 2004;47:5224–5229.
- Bayram E, Senturk M, Kufrevioglu OI, Supuran CT. In vitro inhibition of salicylic acid derivatives on human cytosolic carbonic anhydrase isozymes I and II. Bioorg Med Chem 2008;16:9101–9105.
- Khalifah RG. The carbon dioxide hydration activity of carbonic anhydrase. I. Stop-flow kinetic studies on the native human isoenzymes B and C. J Biol Chem 1971;246:2561–2573.
- Park KK, Lee JJ, Ryu J. Photo-Fries rearrangement of N-arylsulfonamides to aminoaryl sulfone derivatives. Tetrahedron 2003;59:7651–7676.
- Cheng Y, Prusoff WH. Relationship between the inhibition constant (K1) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol 1973;22:3099–3108.
- Nuti E, Orlandini E, Nencetti S, Rossello A, Innocenti A, Scozzafava A et al. Carbonic anhydrase and matrix metalloproteinase inhibitors. Inhibition of human tumor-associated isozymes IX and cytosolic isozyme I and II with sulfonylated hydroxamates. Bioorg Med Chem 2007;15:2298–2311.
- Briganti F, Pierattelli R, Scozzafava A, Supuran CT. Carbonic anhydrase inhibitors. Part 37. Novel classes of isozymes I and II inhibitors and their mechanism of action. Kinetic and spectroscopic investigations on the native and cobalt-substituted enzymes. Eur J Med Chem 1996;31:1001–1010.
- Sentürk M, Gülçin I, Dastan A, Küfrevioglu OI, Supuran CT. Carbonic anhydrase inhibitors. Inhibition of human erythrocyte isozymes I and II with a series of antioxidant phenols. Bioorg Med Chem 2009;17:3207–3211.
- Sentürk M, Gülçin I, Beydemir S, Küfrevioglu OI, Supuran CT. In vitro inhibition of human carbonic anhydrase i and ii isozymes with natural phenolic compounds. Chem Biol Drug Des 2011;77:494–499.
- Cavdar H, Ekinci D, Talaz O, Saraçoglu N, Sentürk M, Supuran CT. a-Carbonic anhydrases are sulfatases with cyclic diol monosulfate esters. J Enzyme Inhib Med Chem 2012;27:148–154.
- Pastorekova S, Parkkila S, Pastorek J, Supuran CT. Carbonic anhydrases: current state of the art, therapeutic applications and future prospects. J Enzyme Inhib Med Chem 2004;19:199–229.
- Ceyhun SB, Sentürk M, Yerlikaya E, Erdogan O, Küfrevioglu OI, Ekinci D. Purification and characterization of carbonic anhydrase from the teleost fish Dicentrarchus labrax (European seabass) liver and toxicological effects of metals on enzyme activity. Environ Toxicol Pharmacol 2011;32:69–74.
- Maresca A, Temperini C, Vu H, Pham NB, Poulsen SA, Scozzafava A et al. Non-zinc mediated inhibition of carbonic anhydrases: coumarins are a new class of suicide inhibitors. J Am Chem Soc 2009;131:3057–3062.
- Maresca A, Supuran CT. Coumarins incorporating hydroxy- and chloro-moieties selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II. Bioorg Med Chem Lett 2010;20:4511–4514.
- Maresca A, Scozzafava A, Supuran CT. 7,8-disubstituted- but not 6,7-disubstituted coumarins selectively inhibit the transmembrane, tumor-associated carbonic anhydrase isoforms IX and XII over the cytosolic ones I and II in the low nanomolar/subnanomolar range. Bioorg Med Chem Lett 2010;20:7255–7258.
- Carta F, Maresca A, Scozzafava A, Supuran CT. 5- and 6-Membered (thio)lactones are prodrug type carbonic anhydrase inhibitors. Bioorg Med Chem Lett 2012;22:267–270.